

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Diabetic neuropathy (DN) is the most common form of neuropathy in developed countries and may affect about half of all patients with diabetes (DM), contributing to substantial morbidity and mortality and resulting in a huge economic burden. DN encompasses multiple different disorders involving proximal, distal, somatic, and autonomic nerves. It may be acute and self-limiting or a chronic, indolent condition. DN may progress insidiously or present with clinical symptoms and signs that may mimic those seen in many other diseases. The proper diagnosis therefore requires a thorough history, clinical and neurological examinations, and exclusion of secondary causes. Distal peripheral neuropathy (DPN) is the most common manifestation and is characteristically symmetric, glove and stocking distribution and a length-dependent sensorimotor polyneuropathy. It develops on a background of long-standing chronic hyperglycemia superimposed upon cardiovascular risk factors. Diagnosis is mainly based on a combination of symptoms and signs and occasionally neurophysiological tests are required. Apart from optimizing glycemic control and cardiovascular risk factor management, there is no approved treatment for the prevention or reversal of DPN. Even tight glycemic control at best limits the progression of DPN in patients with type 1 DM, but not to the same extent in type 2 DM. It has been estimated that between 3 and 25% of persons with DM might experience neuropathic pain. Painful DPN can be difficult to treat, and is associated with reduced quality of life, poor sleep, depression, and anxiety. Pharmacotherapy is the mainstay symptomatic treatment for painful DPN. The reported prevalence of diabetic autonomic neuropathy (DAN) varies widely (7.7 to 90%) depending on the cohort studied and the methods used for diagnosis, and can affect any organ system. Cardiovascular autonomic neuropathy (CAN) is significantly associated with overall mortality and with morbidity, including silent myocardial ischemia, coronary artery disease, stroke, DN progression, and perioperative complications. Cardiovascular reflex tests are the criterion standard in clinical autonomic testing. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Diabetic neuropathy (DN) is the most common and troublesome complication of diabetes mellitus, leading to the greatest morbidity and mortality resulting in a huge economic burden for diabetes care (1,2). It is the most common form of neuropathy in the developed world, accounting for more hospitalizations than all the other diabetes related complications combined. It is the primary risk factor for complications such as foot ulceration, which is responsible for 50-75% of non-traumatic amputations (3). In the United Kingdom, the cost of managing diabetic foot disease is greater than the combined cost of three of the four most common cancers – breast, lung and prostate (4,5). DN is a set of clinical syndromes that affect distinct regions of the nervous system, singly or combined. It may be silent and go undetected while exercising its ravages; or it may present with clinical symptoms and signs that, although nonspecific and insidious with slow progression also mimics those seen in many other diseases.
SCOPE OF THE PROBLEM
Diabetic neuropathy results in a variety of syndromes and can be subdivided into focal/multifocal neuropathies, including diabetic amyotrophy, and symmetric polyneuropathies, including sensorimotor polyneuropathy (DPN). The latter is the most common type. The Toronto Diabetic Neuropathy Expert Group defined DPN as a symmetrical, length-dependent sensorimotor polyneuropathy attributable to metabolic and microvascular alterations as a result of chronic hyperglycemia exposure (diabetes) and cardiovascular risk covariates (6). Its onset is generally insidious, and without treatment the course is chronic and progressive. The loss of small fiber-mediated sensation results in the loss of thermal and pain perception, whereas large fiber impairment results in loss of touch and vibration perception. Sensory fiber involvement may also result in “positive” symptoms, such as paresthesias and pain, although up to 50% of neuropathic patients are asymptomatic. DPN can be associated with the involvement of the autonomic nervous system, i.e., diabetic autonomic neuropathy (7,8) and in its cardiovascular form is associated with at least a three-fold increased risk for mortality (9,10). Cardiac autonomic dysfunction in patients with diabetes is strongly associated with major cardiovascular events and mortality (11).
Painful DPN which occurs in up to 34% of patients with diabetes is defined as ‘pain as a direct consequence of abnormalities in the peripheral somatosensory system in people with diabetes’ (12). Persistent neuropathic pain interferes significantly with quality of life (QOL), impairing sleep and recreation; it also significantly impacts emotional well-being, and is associated with – if not the cause of – depression, anxiety, loss of sleep, and noncompliance with treatment (13). Painful DPN can pose a significant clinical management challenge and if poorly managed can lead to mood and sleep disturbances. Hence, recognition of psychosocial problems that co-exist with neuropathic pain is critical to the management of painful DPN. For many patients, optimal management of chronic pain may require a multidisciplinary team approach with appropriate behavioral therapy, as well as input from a broad range of healthcare professionals (14).
CLASSIFICATION OF DIABETIC NEUROPATHIES
Figure 1 and Table 1 describe the classification first proposed by PK Thomas (15) and modified in a recent Position Statement by the American Diabetes Association (16).

Figure 1.
Classification of diabetic neuropathy
Table 1.
Classification of Diabetic Neuropathies
Cardiovascular
Abnormal pupillary function |
| B. Mononeuropathy (mononeuritis multiplex) (atypical forms) Isolated cranial or peripheral nerve (e.g., Cranial Nerve III, ulnar, median, femoral, peroneal) Mononeuritis multiplex (if confluent may resemble polyneuropathy) |
| C. Radiculopathy or polyradiculopathy (atypical forms) Radiculoplexus neuropathy (a.k.a. lumbosacral polyradiculopathy, proximal motor amyotrophy) Thoracic radiculopathy |
| D. Nondiabetic neuropathies common in diabetes Pressure palsies Chronic inflammatory demyelinating polyneuropathy Radiculoplexus neuropathy Acute painful small-fiber neuropathies (treatment-induced) |
NATURAL HISTORY OF DIABETIC NEUROPATHIES (DN)
The natural history of DPN remains poorly understood, as there are few prospective studies that have examined this. The main reason for this is the lack of standardized methodologies for the diagnosis of DPN. Unlike diabetic retinopathy and nephropathy, the lack of simple, accurate and readily reproducible methods of measuring neuropathy is a major challenge. Furthermore, the methods currently used are not only subjective and reliant on the examiner’s interpretation but tend to diagnose DPN when it’s already well established. Nevertheless, it appears that the most rapid deterioration of nerve function occurs soon after the onset of type 1 diabetes; then within 2-3 years there is a slowing of the progress with a shallower slope to the curve of dysfunction (17). In contrast, in type 2 diabetes, slowing of nerve conduction velocities (NCVs) may be one of the earliest neuropathic abnormalities and often is present even at diagnosis. In fact, there is accumulating evidence that indicates that the risk of DPN is increased even in patients with prediabetes. In a large population study conducted in Augsburg, Southern Germany, the prevalence of DPN was 28% in subjects with known diabetes, 13% in impaired glucose tolerance (IGT), 11% among those with impaired fasting glucose and 7% in those with normal glucose tolerance (18). After diagnosis, slowing of NCV generally progresses at a steady rate of approximately 1 m/sec/year, and the level of impairment is positively correlated with duration of diabetes. Moreover, nerve conduction velocities remained stable with intensive therapy but decreased significantly with conventional therapy (19,20). In a long term follow up study of type 2 diabetes patients (9), electrophysiologic abnormalities in the lower limb increased from 8% at baseline to 42% after 10 years; in particular, a decrease in sensory and motor amplitudes (indicating axonal destruction) was more pronounced than the slowing of the NCVs. However, there now appears to be a decline in this rate of evolution. It appears that host factors pertaining to general health, management of risk factors and nerve nutrition are changing/improving. This is particularly important when doing studies on the treatment of DPN, which have always relied on differences between drug treatment and placebo, and have apparently been successful because of the decline in function occurring in placebo-treated patients (21). Recent studies have pointed out the changing natural history of DPN with the advent of therapeutic lifestyle change and the use of statins and ACE inhibitors, which have slowed the progression of DPN and drastically changed the requirements for placebo-controlled studies (22,23). It is also important to recognize that DPN is a disorder wherein the prevailing abnormality is loss of axons that electrophysiologically translates to a reduction in amplitudes and not conduction velocities; therefore, changes in NCV may not be an appropriate means of monitoring progress or deterioration of nerve function. Moreover, small, unmyelinated nerve fibers are affected early in DM and are not assessed in NCV studies. Other methods such as quantitative sensory testing, autonomic function testing, skin biopsy with quantification of intraepidermal nerve fibers (IENF), or corneal confocal microscopy are necessary to identify these patients. These techniques will be discussed in greater depth later in this chapter.
Although, the true prevalence is unknown and reports vary, it is estimated to be 30% with a range between 6-54% of patients with diabetes (24). It largely depends on the criteria and sensitivity of the diagnostic tests used to define neuropathy, the population (e.g., hospital/community or urban/rural), or the country surveyed and even the etiology of diabetes (24,25). Eleven to 13% of patients reported DN using a questionnaire based survey (26,27); 42-54% were found to have neuropathy when more sensitive measures such as nerve conduction studies were used (28,29). Neurologic complications occur equally in type 1 and type 2 diabetes mellitus and additionally in various forms of acquired diabetes (30).
The major morbidity associated with somatic neuropathy is foot ulceration, the precursor of gangrene and limb loss. Neuropathy increases the risk of amputation 1.7 fold; 12 fold if there is deformity (itself a consequence of neuropathy), and 36 fold if there is a history of previous ulceration (31). For more than a decade now, it has been recognized that a limb is lost to diabetes every 30 seconds worldwide (32). According to the International Diabetes Federation (IDF), lower-limb amputations are ten times more common in people with diabetes than in people without diabetes (32, 33). Each week in England there is about 169 amputations in people with diabetes and almost all of these individuals have DN (34). Amputation is not only devastating in its impact on the individual and their family, but also leads to loss of independence and livelihood. In low-income countries, the financial costs can be equivalent to 5.7 years of annual income, potentially resulting in financial ruin for individuals and their families (35). DN also places a substantial financial burden on health-care systems and society in general.
MODIFIABLE RISK FACTORS FOR DPN INCIDENCE AND PROGRESSION
In both type 1 and 2 diabetes, chronic hyperglycemia has a key role in the pathogenesis of DPN (36). The benefit of glucose lowering is, however, more pronounced in type 1 diabetes (78% relative risk reduction) (37) compared to type 2 (5-9% relative risk reduction) (38). In fact, the benefit of intensive glucose lowering is greatest in younger patients at early stages of the disease. This treatment effects becomes weaker once nerve damage is established. The relationship between glycemic control and DPN in type 2 diabetes is less clear cut. Even when trials have shown that tighter glucose control might have a modest beneficial effect in preventing progression of DPN in type 2 diabetes, such as the Action to Control Cardiovascular Risk in Diabetes (ACCORD) study (39), confusion has arisen when it was reported that a self-reported history of DPN at baseline was associated with an increased risk of mortality with intensive glycemic treatment (40). This highlights the differences between the pathogenesis of DPN in type 1 and 2 diabetes and emphasizes the point that many people with type 2 diabetes develop DPN despite adequate glucose control. The presence of other risk factors, weight gain and multiple comorbidities may have significant roles to play. Although hyperglycemia and duration of diabetes play an important role in DPN, other risk factors have been identified. The EURODIAB Prospective Complications study in type 1 diabetes demonstrated that the incidence of DPN is associated with other potentially modifiable cardiovascular risk factors, including hypertriglyceridemia, hypertension, obesity and smoking (41). More recently, data from the ADDITION study also implicated similar cardiovascular risk factors in the pathogenesis of DPN in type 2 diabetes (26).
PATHOGENESIS OF DIABETIC NEUROPATHIES
Despite considerable research, the pathogenesis of diabetic neuropathy remains undetermined (42). This is one reason why, despite several clinical trials, there has been relatively little progress in the development of disease-modifying treatments (43). Historically, a number of causative factors have been identified including persistent hyperglycemia, microvascular insufficiency, oxidative and nitrosative stress, defective neurotrophism, and autoimmune-mediated nerve destruction. Figure 2 summarizes our current view of the pathogenesis of DPN (44). Detailed discussion of the different theories is beyond the scope of this Chapter and there are several excellent recent reviews (45).

Figure 2.
Pathogenesis of diabetic neuropathies. Ab, antibody; AGE, advance glycation end products; C’, complement; DAG, diacylglycerol; ET, endothelin; EDHF, endothelium-derived hyperpolarizing factor; GF, growth factor; IGF; insulin-like growth factor; NFkB, nuclear factor kB; NGF, nerve growth factor; NO, nitric oxide; NT3, neurotropin 3; PKC, protein kinase C; PGI2, prostaglandin I2; ROS, reactive oxygen species; TRK, tyrosine kinase.
CLINICAL PRESENTATION
The spectrum of clinical neuropathic syndromes described in patients with diabetes mellitus includes dysfunction of almost every segment of the somatic peripheral and autonomic nervous system (16). Each syndrome can be distinguished by its pathophysiologic, therapeutic, and prognostic features.
Focal and Multifocal Neuropathies
Focal neuropathies comprise focal limb neuropathies and cranial neuropathies.
Focal limb neuropathies are usually due to entrapment, and mononeuropathies must be distinguished from these entrapment syndromes (Table 2) (46). Mononeuropathies often occur in the older population; they have an acute onset, are associated with pain, and have a self-limiting course resolving in 6–8 weeks. Mononeuropathies can involve the median (5.8% of all diabetic neuropathies), ulnar (2.1%), radial (0.6%), and common peroneal nerves (47). Cranial neuropathies in patients with diabetes are extremely rare (0.05%) and occur in older individuals with a long duration of diabetes (48). The commonest cranial neuropathy is the third nerve palsy and patients present with acute onset unilateral pain in the orbit or sometimes with a frontal headache. There is typically ptosis and ophthalmoplegia, although the pupillary response to light is usually spared. Recovery occurs usually over three months (48). The clinical onset and time-scale for recovery, and the focal nature of the lesions on the third cranial nerve, on post-mortem studies suggested an ischemic etiology. It is important to exclude any other cause of third cranial nerve palsy (aneurysm or tumor) by CT or MR scanning, where the diagnosis is in doubt. Fourth, sixth and seventh cranial nerve palsies have also been described in patients with diabetes, but the association with diabetes is not as strong as that with third cranial nerve palsy.
Table 2.
Distinguishing Characteristics of Mononeuropathies, Entrapment Syndromes and Distal Symmetrical Polyneuropathy
| Feature | Mononeuropathy | Entrapment syndrome | Neuropathy |
|---|---|---|---|
| Onset | Sudden | Gradual | Gradual |
| Pattern | Single nerve but may be multiple | Single nerve exposed to trauma | Distal symmetrical poly neuropathy |
| Nerves involved | CN III, VI, VII, ulnar, median, peroneal | Median, ulnar, peroneal, medial and lateral plantar | Mixed, Motor, Sensory, Autonomic |
| Natural history | Resolves spontaneously | Progressive | Progressive |
| Treatment | Symptomatic | Rest, splints, local steroids, diuretics, surgery | Tight Glycemic control, Pregabalin, Duloxetine, Antioxidants, “Nutrinerve”, Research Drugs. |
| Distribution of Sensory loss | Area supplied by the nerve | Area supplied beyond the site of entrapment | Distal and symmetrical. “Glove and Stocking” distribution. |
CN, cranial nerves; NSAIDs, non-steroidal anti-inflammatory drugs
Entrapment Syndromes
These start slowly and will progress and persist without intervention. A number of nerves including the median, ulnar, radial, lateral femoral cutaneous, fibular, and plantar nerves are vulnerable to pressure damage in diabetes. The etiology is multifactorial involving metabolic and ischemic factors, impaired reinnervation, and even obesity. Carpal tunnel syndrome occurs three times as frequently in people with diabetes compared with healthy populations (49) and is found in up to one third of patients with diabetes. Its increased prevalence in diabetes may be related to repeated undetected trauma, metabolic changes, or accumulation of fluid or edema within the confined space of the carpal tunnel. The diagnosis is confirmed by electrophysiological studies. Treatment consists of rest, aided by placement of a wrist splint in a neutral position to avoid repetitive trauma. Anti-inflammatory medications and steroid injections are sometimes useful. Surgery should be considered if weakness appears and medical treatment fails (50). It consists of sectioning the volar carpal ligament or unentrapping the nerves in the ulnar canal or the peroneal nerve at the head of the fibula and release of the medial plantar nerve in the tarsal tunnel amongst others. A more detailed review of other peripheral nerves vulnerable to entrapment in anatomically constraint channels are discussed elsewhere (51).
Proximal Motor Neuropathy (Diabetic Amyotrophy) and Chronic Demyelinating Neuropathies
For many years proximal neuropathy has been considered a component of DN. Its pathogenesis was ill understood (52), and its treatment was neglected with the anticipation that the patient would eventually recover, albeit over a period of some 1-2 years and after suffering considerable pain, weakness and disability. The condition has a number of synonyms including diabetic amyotrophy and femoral neuropathy. It can be clinically identified based on the occurrence of these common features: 1) primarily affects those aged 50 to 60 years old with type 2 diabetes; 2) onset can be gradual or abrupt; 3) presents with severe pain in the thighs, hips and buttocks, followed by significant weakness of the proximal muscles of the lower limbs with inability to rise from the sitting position (positive Gower's maneuver); 4) can start unilaterally and then spread bilaterally; 5) often coexists with distal symmetric polyneuropathy; and 6) is characterized by muscle fasciculation, either spontaneous or provoked by percussion. Pathogenesis is not yet clearly understood although immune-mediated epineural microvasculitis has been demonstrated in some cases. Despite limited evidence of efficacy some immunosuppressive therapy is recommended using high dose steroids or intravenous immunoglobulin (53). Close monitoring and appropriate management of blood glucose is advised if high dose steorids are used (54). The condition can occur secondary to a variety of causes unrelated to diabetes, but which have a greater frequency in patients with diabetes than the general population. Hence, it is important to exclude other causes such as chronic inflammatory demyelinating polyneuropathy (CIDP), monoclonal gammopathy, circulating GM1 antibodies, and inflammatory vasculitis (55,56). In the classic form of diabetic amyotrophy, axonal loss is the predominant process (57). Electrophysiologic evaluation reveals lumbosacral plexopathy (58). In contrast, if demyelination predominates and the motor deficit affects proximal and distal muscle groups, the diagnoses of CIDP, monoclonal gammopathy of unknown significance, and vasculitis should be considered (59,60). The diagnosis of these demyelinating conditions is often overlooked although recognition is very important because unlike DN, they are sometimes treatable. Furthermore, they occur 11 times more frequently in patients with diabetes than nondiabetic patients (61,62). Biopsy of the obturator nerve have revealed deposition of immunoglobulin, demyelination and inflammatory cell infiltrate of the vasa nervorum (63). Cerebrospinal fluid (CSF) protein content is high and lymphocyte count increased. Treatment options include: intravenous immunoglobulin for CIDP (64), plasma exchange for MGUS, steroids and azathioprine for vasculitis, and withdrawal of drugs or other agents that may have caused vasculitis. It is important to divide proximal syndromes into these two subcategories, because the CIDP variant responds dramatically to intervention (65), whereas amyotrophy runs its own course over months to years. Until more evidence is available, they should be considered separate syndromes.
Diabetic Truncal Radiculoneuropathy
Diabetic truncal radiculoneuropathy affects middle-aged to elderly patients and has a predilection for male sex (16). Acute onset of pain is the most important symptom and it occurs in a girdle-like distribution over the lower thoracic or abdominal wall. It can be uni- or bilaterally distributed. Motor weakness is rare but there may be local bulging of the muscle. Patchy sensory loss may be present and other causes of nerve root compression should be excluded. Resolution generally occurs within 4-6 months (16).
Rapidly Reversible Hyperglycemic Neuropathy
Reversible abnormalities of nerve function may occur in patients with recently diagnosed or poorly controlled diabetes. These are unlikely to be caused by structural abnormalities, as recovery soon follows restoration of euglycemia. Rapidly reversible hyperglycemic neuropathy usually presents with distal sensory symptoms, and whether these abnormalities result in an increased risk of developing chronic neuropathies in the future remains unknow (8).
Generalized Symmetric Polyneuropathy
ACUTE SENSORY NEUROPATHY
Acute sensory (painful) neuropathy is considered by some authors a distinctive variant of distal symmetrical polyneuropathy (66). The syndrome is characterized by severe pain, cachexia, weight loss, depression and sexual dysfunction. It occurs predominantly in male patients and may appear at any time in the course of both type 1 and type 2 diabetes. It is self-limiting and invariably responds to simple symptomatic treatment (67). Conditions such as Fabry's disease, amyloidosis, HIV infection, heavy metal poisoning (such as arsenic), and excess alcohol consumption should be excluded. Autonomic nervous system involvement can also occur and can be very disabling.
Patients report unremitting burning, deep pain and hyperesthesia especially in the feet. Other symptoms include sharp, stabbing, lancinating pain; “electric shock” like sensations in the lower limbs that appear more frequently during the night; paresthesia; tingling; coldness, and numbness. Signs are usually absent with a relatively normal clinical examination, except for allodynia (exaggerated response to non-noxious stimuli) during sensory testing and, occasionally, absent or reduced ankle reflexes. There are no motor signs and little or no abnormality on nerve conduction studies.
Acute sensory neuropathy is usually associated with poor glycemic control but may also appear after sudden improvement of glycemia. Most commonly associated with the onset of insulin therapy, being termed "insulin neuritis", it can also occur with oral hypoglycemic treatment. Patients present with severe neuropathic pain and/or autonomic symptoms with or without an acute worsening of retinopathy. Although the pathologic basis has not been determined, one hypothesis suggests that changes in blood glucose flux produce alterations in epineural blood flow, leading to ischemia; proinflammatory cytokines from activation of microglia have also been implicated (68). Hence, rapid glycemic changes in patients with uncontrolled diabetes increases the risk of this complication and should be avoided. A 2-3% (10-42mmol/mol) decrease in HbA1c over 3 months is associated with a 20% absolute risk of developing this complication. The risk exceeds 80% with a decreased in HbA1c of >4% (20mmol/mol) (69). A study using in vivo epineural vessel photography and fluorescein angiography demonstrated abnormalities of epineural vessels including arteriovenous shunting and proliferating new vessels in patients with acute sensory neuropathy (68). Other authors relate this syndrome to diabetic lumbosacral radiculoplexus neuropathy (DLRPN) and propose an immune mediated mechanism (70).
The key in the management of this syndrome is achieving and maintaining blood glucose stability (71). Most patients also require medication for neuropathic pain. The natural history of this disease is resolution of symptoms within one year.
CHRONIC SENSORIMOTOR NEUROPATHY OR DISTAL SYMMETRIC POLYNEUROPATY (DPN)
The most common form of neuropathy in diabetes is a distal symmetrical polyneuropathy. It occurs in both type 1 and type 2 DM with similar frequency and may already be present at the time of diagnosis of type 2 DM (18). Sensory symptoms include numbness (‘dead feeling’), paraesthesia, and neuropathic pain (hyperalgesia, allodynia, deep aching, burning and sharp stabbing sensations). Patients do occasionally present paradoxically with a painful/painless leg i.e. painful neuropathic symptoms in the presence of severe sensory loss (72). Symptoms begin in the toes before progressing in a stocking and then a glove distribution as the disease progresses. Unsteadiness or sensory ataxia leading to increased falls risk occurs in advanced neuropathy loss of proprioception, foot deformity, and abnormal muscle sensory function (73). In the absence of painful symptoms, the onset of DPN is insidious whereby patients remain completely asymptomatic and signs discovered by a detailed neurological examination. Unfortunately, DPN is often already established or well advanced when identified by bedside clinical examination.
It is critically important to annually (at least) examine the feet of patients with diabetes as loss of protective sensation is the strongest risk factor for diabetic foot ulceration. On physical examination, a symmetrical stocking like distribution of sensory abnormalities in both lower limbs is usually seen. In more severe cases, hands may be involved. All sensory modalities can be affected, particularly vibration, touch and position perceptions (large Aα/β fiber damage); and pain, with abnormal heat and cold temperature perception (small thinly myelinated Aδ and unmyelinated C fiber damage, see Figure 3, 4 and 5; Table 3). Deep tendon reflexes may be absent or reduced, especially in the lower extremities, although this may occur with advancing age in the absence of neuropathy. When DPN is established, small muscle wasting of the foot and extensor halluces longus may be seen but severe weakness is rare and should raise the possibility of a non-diabetic etiology of the neuropathy. High arching of the foot, clawing of the toes with prominent metatarsal heads also become apparent – increasing the risk ulceration (74). A thorough assessment of patient’s footwear is essential. A poor fit, abnormal wear from internal pressure areas and foreign objects found in footwear are common causes of trauma leading to foot ulceration (75).

Figure 3.
Clinical presentation of small and large fiber neuropathies. Aα fibers are large myelinated fibers, in charge of motor functions and muscle control. Aα/β fibers are large myelinated fibers too, with sensory functions such as perception to touch, vibration, and position. Aδ fibers are small myelinated fibers, in charge of pain stimuli and cold perception. C fibers can be myelinated or unmyelinated and have both sensory (warm perception and pain) and autonomic functions (blood pressure and heart rate regulation, sweating, etc.)

Figure 4.
Clinical manifestations of small fiber neuropathies

Figure 5.
Nerve fibers of the skin and their functions
Table 3.
Subtypes of Neuropathies
Clinical Manifestations of Small Fiber Neuropathies:
|
Clinical Manifestations of Large Fiber Neuropathies
|
DIAGNOSIS OF DIABETIC NEUROPATHIES
Diabetic peripheral neuropathy can be diagnosed by the bedside with careful clinical examination of the feet and legs using simple tools within a few minutes. The basic neurological assessment comprises the general medical and neurological history, inspection of the feet, and neurological examination of sensation using simple semi-quantitative bed-side instruments such as the 10g Semmes-Weinstein monofilament, Neuropen (76) (to assess touch/pressure), NeuroQuick (77) or Tiptherm (78) (temperature), calibrated Rydel-Seiffer tuning fork (vibration), pin-prick (pain), and tendon reflexes (knee and ankle) (Table 4). In addition, assessment of joint position and motor power should also be assessed. The Rydel Seiffer tuning fork is a 128 Hz tuning fork which allows quantifiable assessment of vibration perception in the feet of diabetic patients. When vibrating, two triangles appear on the graduated scale of 0–8 which join together as the amplitude decreases. The normal range for the graduated tuning fork on the dorsal distal joint of the great toe is ≥5/8 scale units in persons 21-40 years old, ≥4.5/8 in those 41-60 years old, ≥4/8 in individuals 61-71 years old, and ≥3.5/8 scale units in those 72-82 years old (79). In resource, limited settings the simple Ipswich Touch Test can be performed by lightly touching the tips of the first, third and fifth toes (80). It is recommended that a simple foot examination to detect loss of protective foot sensation, pedal pulses, and foot deformity is performed from the diagnosis of type 2 diabetes, 5-years after the diagnosis of type 1 diabetes and annually thereafter (81,82,16). This is performed in order to determine the risk of foot ulceration and prompt early referral for foot protection, regular podiatry or specialist input.
Table 4.
Examination - Bedside Sensory Tests
| Sensory Modality | Nerve Fiber | Instrument | Associated Sensory Receptors |
| Vibration | Aβ (large) | 128 Hz Tuning fork | Ruffini corpuscle mechanoreceptors |
| Pain (pinprick) | C (small) | Neuro-tips | Nociceptors for pain and warmth |
| Pressure | Aβ, Aα (large) | 1 g and 10 g Monofilament | Pacinian corpuscle |
| Light touch | Aβ, Aα (large) | Wisp of cotton | Meissner’s corpuscle |
| Cold | Aδ (small) | Cold tuning fork | Cold thermoreceptors |
A consensus definition of DPN has been proposed by the Toronto Diabetic Neuropathy Expert Group, see below (6). In a clinical context, the diagnosis of ‘possible’ or ‘probable’ DPN is normally sufficient without the need for specialist investigations. For research purposes further tests are needed for a diagnosis of ‘confirmed’ DPN’, ‘Subclinical’ DPN or small fiber neuropathy.
Toronto Classification of DPN (6)
- 1.
Possible DSN: The presence of symptoms or signs of DPN may include the following: symptoms–decreased sensation, positive neuropathic sensory symptoms (e.g., “asleep numbness,” prickling or stabbing, burning or aching pain) predominantly in the toes, feet, or legs; or signs–symmetric decrease of distal sensation or unequivocally decreased or absent ankle reflexes.
- 2.
Probable DPN: The presence of a combination of symptoms and signs of neuropathy including any 2 or more of the following: neuropathic symptoms, decreased distal sensation, or unequivocally decreased or absent ankle reflexes.
- 3.
Confirmed DPN: The presence of an abnormality of nerve conduction and a symptom or symptoms, or a sign or signs, of neuropathy confirm DPN. If nerve conduction is normal, a validated measure of small fiber neuropathy (with class 1 evidence) may be used. To assess for the severity of DPN, several approaches can be recommended: for e.g., the graded approach outlined above; various continuous measures of sum scores of neurologic signs, symptoms or nerve test scores; scores of functions of activities of daily living; or scores of predetermined tasks or of disability.
- 4.
Subclinical DPN: The presence of no signs or symptoms of neuropathy are confirmed with abnormal nerve conduction or a validated measure of small fiber neuropathy (with class 1 evidence). Definitions 1, 2, or 3 can be used for clinical practice and definitions 3 or 4 can be used for research studies.
- 5.
Small fiber neuropathy (SFN): SFN should be graded as follows: 1) possible: the presence of length-dependent symptoms and/or clinical signs of small fiber damage; 2) probable: the presence of length-dependent symptoms, clinical signs of small fiber damage, and normal sural nerve conduction; and 3) definite: the presence of length-dependent symptoms, clinical signs of small fiber damage, normal sural nerve conduction, and altered intraepidermal nerve fiber density (IENFD) at the ankle and/or abnormal thermal thresholds at the foot (Figure 4).
The following findings should alert the physician to consider causes for DPN other than diabetes and referral for a detailed neurological work-up: 1.) pronounced asymmetry of the neurological deficits, 2.) predominant motor deficits, mononeuropathy, or cranial nerve involvement, 3.) rapid development or progression of the neuropathic impairments, 4.) progression of the neuropathy despite optimal glycemic control, 5.) symptoms from the upper limbs, 6.) family history of non-diabetic neuropathy, and 7.) diagnosis of DPN cannot be ascertained by clinical examination.
Conditions Mimicking Diabetic Neuropathy
An atypical pattern of presentation of symptoms or signs, i.e., the presence of relevant motor deficits, an asymmetrical or proximal distribution, or rapid progression, always requires referral for electrodiagnostic testing. Furthermore, in the presence of such atypical neuropathic signs and symptoms other forms of neuropathy should be sought and excluded. A good medical history is essential to exclude other causes of neuropathy: a history of trauma, cancer, unexplained weight loss, fever, substance abuse, or HIV infection suggests that an alternative source should be sought. Screening laboratory tests may be considered: serum B12 with its metabolites, folic acid, thyroid function, full blood count, metabolic profile, and serum free light chains (16).
There are a number of conditions that can be mistaken for painful DPN: intermittent claudication in which the pain is exacerbated by walking; Morton’s neuroma, in which the pain and tenderness are localized to the intertarsal space and are elicited by applying pressure with the thumb in the appropriate intertarsal space; osteoarthritis/inflammatory arthritis, in which the pain is confined to the joints, made worse with joint movement or exercise, and associated with morning stiffness that improves with ambulation; radiculopathy in which the pain originates in the shoulder, arm, thorax, or back and radiates into the legs and feet; Charcot neuropathy in which the pain is localized to the site of the collapse of the bones of the foot, and the foot is hot rather than cold; plantar fasciitis, in which there is shooting or burning in the heel with each step and there is exquisite tenderness in the sole of the foot; and tarsal tunnel syndrome in which the pain and numbness radiate from beneath the medial malleolus to the sole and are localized to the inner side of the foot. These contrast with the pain of DPN which is bilateral, symmetrical, covering the whole foot and particularly the dorsum, and is worse at night interfering with sleep.
Scored Clinical Assessment Tools for Diabetic Peripheral Neuropathy
Scored Clinical assessments provide standardized, quantitative, and objective measures to assess for both the severity of symptoms and the degree of neuropathic deficits. These tools which have been subjected to strict validation studies, are sufficiently reproducible but require some minimal training. The most widely used instruments include: the Michigan Neuropathy Screening Instrument Questionnaire (MNSIQ, 15-item self-administered questionnaire), Michigan Neuropathy Screening Instrument (MNSI, MNSIQ plus a structured clinical examination), Michigan Diabetic Neuropathy Score (neurological assessment coupled with nerve conduction studies) (83), Toronto Clinical Neuropathy Score (TCNS, composite score of neuropathy symptoms sensory exam and reflexes) (84), modified TCNS (composite score of neuropathy symptoms and signs) (85), Neuropathy Disability Score (neuropathy signs, including reflexes) (86), Neurological Disability Score (neurological examination of cranial nerves, and upper and lower limbs) (87), the Neuropathy Symptom Score (assessment of sensory, motor and autonomic neuropathy symptoms) (87), and the Neuropathy impairment score (NIS) for neuropathic deficits (impairments) (87). A number of instruments have also been used to assess neuropathic pain and these include: the Neuropathy Total Symptom Score-6 (NTSS-6; measures frequency and intensity of neuropathic symptoms) (88), PainDETECT (patient administered 10-item questionnaire) (89), DN4 (Doleur Neuropathique en 4 Questions; 7 sensory descriptors and 3 clinical signs) (90) and the Neuropathic Pain Symptom Inventory (NPSI; self-administered 12-item questionnaire evaluating different symptoms of neuropathic pain) (91).
Objective Devices for the Diagnosis of Neuropathy
Nerve conduction studies are the current ‘gold’ standard for the diagnosis of DN. This robust measure also predicts foot ulceration and mortality. However, they are time consuming, labor intensive, costly, and impractical in routine clinical care.
Skin biopsy has become a widely used tool to investigate small caliber sensory nerves including somatic unmyelinated intraepidermal nerve fibers (IENF), dermal myelinated nerve fibers, and autonomic nerve fibers in peripheral neuropathies and other conditions (92). Different techniques for tissue processing and nerve fiber evaluation have been used. For diagnostic purposes in peripheral neuropathies, the current recommendation is to perform a 3-mm punch skin biopsy at the distal leg and quantification of the linear density of IENF in at least three 50-µm thick sections per biopsy, fixed in 2% PLP or Zamboni's solution, by bright-field immunohistochemistry or immunofluorescence with anti-protein gene product (PGP) 9.5 antibodies (93). Quantification of IENF density appeared more sensitive than sensory nerve conduction study or sural nerve biopsy in diagnosing SFN.
Quantitative sensory testing (QST) enables more accurate assessment of sensory deficits - also those related to small fiber function - by applying controlled and quantified stimuli and standardized procedures. Moreover, assessment of thermal thresholds can be a helpful tool in the diagnostic pathway of small fiber polyneuropathy (16).
Point of Care Devices for the Diagnosis of DN
Significant progress has been made to develop point-of-care (POC) devices that are capable of diagnosing early, subclinical neuropathy. Papanas et al have recently comprehensively reviewed these devices (94). Therefore, we will briefly outline the following devices: the NeuroQuick 77, NeuroPAD (95), NC-Stat DPN-Check (96), Corneal Confocal Microscopy (CCM) (97,98), and Sudoscan (99,100).
DPN CHECK
The DPN-Check is a novel, user-friendly, handheld POC devices that performs a sural nerve conduction study in three minutes (Figure 6). It is an acceptable proxy to standard nerve conduction studies which are time-consuming, expensive, and often require patients to be seen in specialist’s clinics. The DPN check has been demonstrated to have excellent reliability with an inter- and intra-observer intraclass correlation coefficients of between 0.83 and 0.97 for sensory nerve action potentials respectively (101). It also has good validity with 95% sensitivity and 71% specificity when compared against reference standard nerve conduction study (101) for the diagnosis of DN.

Figure 6.
DPN Check device
As detailed above, nerve conduction studies are only an assessment of large nerve fiber function. DPN, on the other hand, usually involves both small and large nerve fibers, with some evidence suggesting small nerve fiber involvement early in its natural history (102,103). Small nerve fibers constitute 80-91% of peripheral nerve fibers and control pain perception, autonomic and sudomotor function. Although intraepidermal nerve fiber density measurement from lower limb skin biopsy is considered the gold standard for the diagnosis of small fiber neuropathy (104,92) it is invasive and hence not suitable for routine screening. However, a number of POC devices have been developed to assess small fiber dysfunction. These include:
NEUROQUICK
Thinly myelinated Aδ and unmyelinated C-fibers are small caliber nerves that mediate thermal sensation and nociceptive stimuli. Quantitative sensory testing of thermal discrimination thresholds is a non-invasive test used to examine impaired small nerve fiber function. NeuroQuick is a handheld device for quantitative bedside testing of cold thermal perception threshold. It allows near patient assessment of small fiber dysfunction avoiding the use of time-consuming and expensive quantitative sensory testing equipment in a laboratory. To date, one published clinical validation study has been performed in a diabetic population which suggests it is a valid and reliable screening tool for the assessment of small fiber dysfunction (77). Use of NeuroQuick was more sensitive in detecting early DPN compared to the traditional bedside screening tests such as the tuning fork or elaborate thermal testing (77). However, it is a psychophysical test that relies on the cognition/attention of the patient. Furthermore, the coefficients of variation for repeated NeuroQuick measurements ranged between 8.5% and 20.4% (77). Further studies are required to demonstrate whether the NeuroQuick is a useful screening tool to detect small fiber dysfunction in DPN.
NEUROPAD
This is a 10-minute test which measures sweat production on the plantar surface of the foot (Figure 7). It is based on a color change in a cobalt compound from blue to pink which produces a categorical output with modest diagnostic performance for DPN compared to electrophysiological assessments. If the patch remains completely or partially blue within 10 min, the result is considered abnormal (105). No training is required to administer Neuropad, nor does it require responses from the patient. Therefore, this method of assessment may be more suitable for screening in community settings and those with cognitive or communication difficulties who have to respond to other methods of assessment. A number of clinical validation studies (95, 106) have been conducted which demonstrates low sensitivity for large fiber neuropathy (50-64%) but much higher sensitivity for small fiber neuropathy (80%) 107. Neuropad has also shown good reproducibility with intra- and inter-observer coefficient of variation between 4.1% and 5.1% (108).

Figure 7.
NeuroPAD
CORNEAL CONFOCAL MICROSCOPY
Corneal confocal microscopy (CCM, Figure 8) is a noninvasive technique used to detect small nerve fiber loss in the cornea which correlates with both increasing neuropathic severity and reduced IENFD in patients with diabetes (103,109). A novel technique of real-time mapping permits an area of 3.2 mm2 to be mapped with a total of 64 theoretically non-overlapping single 400 µm2 images (110). There have been a number of clinical validation studies including one 3.5-year prospective study in T1DM which demonstrated relatively modest to high sensitivity (82%) and specificity (69%) of CCM for the incipient DPN (98). It has good reproducibility for corneal nerve fiber length measurements with intra- and inter-observer intraclass correlation coefficients of 0.72 and 0.73 respectively. Currently, CCM is used in specialist centers, but would suit widespread application given its easy application for patient follow-up. However, large, multicenter, prospective studies are now required to confirm that corneal nerve changes unequivocally reflect the complex pathological processes in the peripheral nerve. Moreover, the establishment of a normative database and technical improvements in automated fiber measurements and wider-area image analysis may be useful to increase diagnostic performance.

Figure 8.
Examples of corneal nerve fiber density in a patient with no diabetic neuropathy on the left and with established diabetic neuropathy on the right.
CONTACT HEAT EVOKED POTENTIALS
Contact Heat Evoked Potentials (CHEPS) has been studied in healthy controls, newly diagnosed and established patients with diabetes, and patients with the metabolic syndrome. It does appear that CHEPS is capable of detecting small fiber neuropathy in the absence of other indices, and that CHEPS correlates with quantitative sensory perception and objective tests of small fiber structure (intraepidermal nerve fiber density) (111) and function (cooling detection threshold and cold pain) (112) .
SUDOSCAN
Sudoscan®, an instrument capable of detecting chloride ion flux in response to a very low current (Figure 9), is an objective and quantitative sudomotor function test with promising sensitivity and specificity in the investigation of DPN (113). The entire evaluation takes only 2 minutes and can be done in an ambulatory setting. A measurement of electrochemical skin conductance (ESC) for the hands and feet, that are rich in sweat glands, is generated from the derivative current associated with the applied voltage. Sensitivity and specificity of foot ESC for classifying DPN were 87.5% and 76.2%, respectively. The area under the ROC curve (AUC) was 0.85 (99).

Figure 9.
SUDOSCAN test of sudomotor function being performed
SUMMARY OF POINT OF CARE DEVICES
In summary, the sensitivity of point of care devices seems acceptable and perhaps a combination of devices may be used in the future for detecting DPN. However, there is high heterogeneity and patient selection bias in most of the studies. Further studies are needed to evaluate the performance of point of care devices against Wilson criteria for screening of undiagnosed DPN at the population level. Prospective studies of hard endpoints (e.g., foot ulcerations and lower limb amputations) are also necessary to ensure that the benefits of screening are important for patients. The cost-effectiveness of implementing screening using these devices also needs to be carefully appraised. Point of care devices provide rapid, non-invasive tests that could be used as an objective screening test for DPN in busy diabetic clinics, ensuring adherence to current recommendation of annual assessment for all patients with diabetes that remains unfulfilled.
Summary of Clinical Assessment of DPN
Symptoms of neuropathy can vary markedly from one patient to another. For this reason, a number of symptom screening questionnaires with similar scoring systems have been developed. These questionnaires are useful for patient follow-up and to assess response to treatment. A detailed clinical examination is the key to the diagnosis of DPN. The latest position statement of the American Diabetes Association recommends that all patients with diabetes be screened for DPN at diagnosis in type 2 DM and 5 years after diagnosis in type 1 DM. DPN screening should be repeated annually and must include sensory examination of the feet and ankle reflexes (16). One or more of the following can be used to assess sensory function: pinprick (using the Waardenberg wheel or similar instrument), temperature, vibration perception (using 128-Hz tuning fork) or 10-g monofilament pressure perception at the distal halluces. For this last test a simple substitute is to use 25 lb strain fishing line cut into 4 cm and 8 cm lengths, which translate to 10 and 1 g monofilaments respectively (114). The most sensitive measure has been shown to be the vibration detection threshold, although sensitivity of 10-g Semmes-Weinstein monofilament to identify feet at risk varies from 86 to 100% (115,116). Combinations of more than one test have more than 87% sensitivity in detecting DPN (117). Longitudinal studies have shown that these simple tests are good predictors of foot ulcer risk (118). Numerous composite scores to evaluate clinical signs of DN, such as the Neuropathy Impairment Score (NIS) are currently available. These, in combination with symptom scores, are useful in documenting and monitoring neuropathic patients in the clinic (119). Feet should always be examined in detail to detect ulcers, calluses, and deformities, and footwear must be inspected at every visit. However, these simple bedside tests are crude and detect DN very late in its natural history. Even the benefits gained by standardising clinical assessment using scored clinical assessments such as the Michigan Neuropathy Screening Instrument (MNSI) (120), the Toronto Clinical Neuropathy Score (TCNS) (84,85) and the United Kingdom Screening Test (UKST) (86), remain subjective, heavily reliant on the examiners’ interpretations (121). Bedside tests used to aid diagnosis of neuropathy such as the 10g monofilament (122), the Ipswich Touch Test (80), and vibration perception threshold using the tuning fork (123) are not only reliant on patients’ subjective response but are mainly utilised to identify the loss of protective foot sensation and risk of ulceration (124). As such, these tests tend to diagnose DPN when it is already well-established (125). Late diagnosis hampers the benefits of early identification which includes a focus on early, intensified diabetes control, and the prevention of neuropathy-related sequelae. Conversely, the situation is different for the detection of diabetic retinopathy using digital camera-based retinal photography (126) or diabetic kidney disease using blood and urine tests. These developments led to the institution of a robust annual screening program that has led to significant reduction in blindness, such that retinopathy is no longer the commonest cause of blindness in working age adults (127) and reductions in end stage renal failure (128). Unfortunately, by the time neuropathy is detected using these crude tests, it is often very well established and consequently impossible to reverse or even to halt the inexorable neuropathic process.
In the clinical research settings nerve conduction studies, quantitative sensory testing, and skin biopsy is used to identify and quantify early, subclinical neuropathy. Multiple studies have proven the value of Quantitative Sensory Testing (QST) measures in the detection of subclinical neuropathy (small fiber neuropathy), the assessment of progression of neuropathy, and the prediction of risk of foot ulceration (117,129,130). These standardized measures of vibration and thermal thresholds also play an important role in multicenter clinical trials as primary efficacy endpoints. A consensus subcommittee of the American Academy of Neurology stated that QST receive a Class II rating as a diagnostic test with a type B strength of recommendation (131).
The use of electrophysiologic measures (nerve conduction velocity, NCV) in both clinical practice and multicenter clinical trials is recommended (6, 132). In a long term follow-up study of type 2 patients with diabetes (28) NCV abnormalities in the lower limbs increased from 8% at baseline to 42% after 10 years of disease. A slow progression of NCV abnormalities was seen in the Diabetes Control and Complication Trial (DCCT). The sural and peroneal nerve conduction velocities diminished by 2.8 and 2.7 m/s respectively, over a 5-year period (21). Furthermore, in the same study, patients who were free of neuropathy at baseline had a 40% incidence of abnormal NCV in the conventionally treated group versus 16% in the intensive therapy treated group after 5 years. However, the neurophysiologic findings vary widely depending on the population tested and the type and distribution of the neuropathy. Patients with painful, predominantly small fiber neuropathy have normal studies. There is consistent evidence that small, unmyelinated fibers are affected early in DM and these alterations are not diagnosed by routine NCV studies (45). Therefore, other methods, such as QST, autonomic testing, or skin biopsy with quantification of intraepidermal nerve fibers (IENF) are needed to detect these patients (22,133,134). Nevertheless electrophysiological studies play a key role in ruling out other causes of neuropathy and are essential for the identification of focal and multifocal neuropathies (46,8).
Intraepithelial Nerve Fiber Density
The importance of the skin biopsy as a diagnostic tool for DPN is increasingly being recognized (45, 135). This technique quantitates small epidermal nerve fibers through antibody staining of the pan-axonal marker protein gene product 9.5 (PGP 9.5). Though minimally invasive (3-mm diameter punch biopsy), it enables a direct study of small fibers, which cannot be evaluated by NCV studies. It has led to the recognition of the small nerve fiber syndrome as part of IGT and the metabolic syndrome (Figure 10). When patients present with the “burning foot or hand syndrome”, evaluation for glucose tolerance and the metabolic syndrome (including waist circumference, blood pressure, and plasma triglyceride and HDL-C levels) becomes mandatory. Therapeutic life style changes (136) can result in nerve fiber regeneration, reversal of the neuropathy, and alleviation of symptoms (see below).

Figure 10.
Intraepidermal nerve fiber loss in small vessel neuropathy. Loss of cutaneous nerve fibers that stain positive for the neuronal antigen protein gene product 9.5 (PGP 9.5) in metabolic syndrome and diabetes.
It is widely recognized that neuropathy per se can affect the quality of life (QOL) of patients with diabetes. A number of instruments have been developed and validated to assess QOL in DPN. The NeuroQoL measures patients’ perceptions of the impact of neuropathy and foot ulcers (137). The Norfolk QOL questionnaire for DPN is a validated tool addressing specific symptoms and the impact of large, small, and autonomic nerve fiber functions (138). The tool has been used in clinical trials and is available in several validated language versions. It was tested in 262 subjects (healthy controls, controls with diabetes, and DPN patients): differences between DN patients and both diabetes and healthy controls were significant (p<0.05) for all item groupings (small fiber, large fiber, and autonomic nerve function; symptoms; and activities of daily living (ADL). Total QOL scores correlated with total neuropathy scores. The ADL, total scores, and autonomic scores were also greater in controls with diabetes compared to healthy controls (p<0.05), suggesting that diabetes per se impacts some aspects of QO (137).
The diagnosis of DPN is mainly a clinical one with the aid of specific diagnostic tests according to the type and severity of the neuropathy. However other non-diabetic causes of neuropathy must always be excluded, depending on the clinical findings (B12 deficiency, hypothyroidism, uremia, CIDP, etc.) (Figure 11).

Figure 11.
A diagnostic algorithm for assessment of neurologic deficit and classification of neuropathic syndromes: B12, vitamin B12; BUN, blood urea nitrogen; CHEPS, Contact Heat Evoked Potentials CIDP, chronic inflammatory demyelinating polyneuropathy; EMG, electromyogram; Hx, history; MGUS, monoclonal gammopathy of unknown significance; NCV, nerve conduction studies; NIS, neurologic impairment score (sensory and motor evaluation); NSS, neurologic symptom score; QAFT, quantitative autonomic function tests; QST, quantitative sensory tests; Sudo, sudomotor function testing.
Central Nervous System Involvement
Hitherto considered a disease of the peripheral nervous system, there is now mounting evidence of central nervous system (CNS) involvement in DN (Figure 12). Several magnetic resonance imaging studies provide valuable insight into CNS alterations in DN. From the spinal cord (139,140) to the cerebral cortex, structural (141), biochemical (142,143), perfusion (144), and functional changes (145,146) have been described. Although the initial injury may occur in the peripheral nerves, concomitant changes within the CNS may have a crucial role in the pathogenesis and determining clinical phenotype and even treatment response in painful DN.
Central nervous system involvement was first recognized in the 1960’s when post-mortem autopsy studies of patients with advanced diabetes found evidence of spinal cord atrophy, demyelination, and axonal loss (147,148). These findings were largely dismissed as being secondary to poor diabetes control and infection (e.g., syphilis) rather than DN. Indeed, the pathological abnormalities in the spinal cord were reported in isolation and not examined in the context of DN related peripheral nerve changes. Subsequent studies performed in the late 70’s and 80’s utilized advances in somatosensory evoked potentials and demonstrated central (brain and spinal cord) slowing in humans with DN (149) and rodent models (150). With the advent and accessibility of demonstrated magnetic resonance imaging in the 90’s and early 00’s, investigators were able to demonstrate clear spinal cord involvement in the form of cervical cord atrophy not only in patients with established DN (140) but also in those with early subclinical DN (139). Subsequent studies have sought to apply advances multimodal magnetic resonance imaging to gain unique insights into brain involvement, particularly brain regions involved with somatosensory and nociception in DN – e.g. primary somatosensory cortex (141) and the thalamus (142). Accompanying the reduction in cervical spine volume is a reduction in primary somatosensory cortical volume in both painful and painless DN (141). Proton magnetic resonance spectroscopy studies have demonstrated evidence of thalamic neuronal dysfunction in painless but not in painful DN – indicating that preservation of thalamic neuronal function may be a prerequisite for the perception of pain in DN (142). In addition, there was also an increase in thalamic vascularity (144), altered thalamic-cortical functional connectivity (146), and a reorganization of the primary somatosensory cortex in patients with painful DN (146). Thus, the involvement of the central nervous system in DN has opened a whole new area of further research and has great potential for future patient stratification and development of new therapeutic targets.

Figure 12.
Multimodal magnetic resonance imaging studies of the central nervous system in diabetic neuropathy.
Risk Factors for Diabetic Polyneuropathies
Diabetic neuropathy is the end results of a culmination of several etiologically linked pathophysiological processes – some not fully understood. Although hyperglycemia and duration of diabetes play an important role in DN, other risk factors have been identified. The EURODIAB Prospective Complications study demonstrated that the incidence of DN is associated with other potentially modifiable cardiovascular risk factors, including hypertriglyceridemia, hypertension, obesity and smoking (41). In the Look AHEAD study in patients with type 2 diabetes, there was a greater increase in neuropathic symptoms (but not neuropathic signs) in the control cohort (diabetes support and education program) compared to the cohort receiving intensive diet and exercise lifestyle intervention programmed focused on weight loss (151).
TREATMENT OF DIABETIC POLYNEUROPATHIES
Treatment of DN should be targeted towards a number of different aspects: firstly, treatment of specific underlying pathogenic mechanisms; secondly, treatment of symptoms and improvement in QOL; and thirdly, prevention of progression and treatment of complications of neuropathy.
Targeting Risk Factors
GLYCEMIC AND METABOLIC CONTROL
Several long-term prospective studies that assessed the effects of intensive diabetes therapy on the prevention and progression of chronic diabetic complications have been published. The large randomized trials such as the Diabetes Control and Complications Trial (DCCT) and the UK Prospective Diabetes Study (UKPDS) were not designed to evaluate the effects of intensive diabetes therapy on DPN, but rather to study the influence of such treatment on the development and progression of the chronic diabetic complications (152,153). Thus, only a minority of the patients enrolled in these studies had symptomatic DPN at entry. Studies in patients with type 1 diabetes show that intensive diabetes therapy retards but does not completely prevent the development of DPN. In the DCCT/EDIC cohort, the benefits of former intensive insulin treatment persisted for 13-14 years after DCCT closeout and provided evidence of a durable effect of prior intensive treatment on DPN and cardiac autonomic neuropathy (“hyperglycemic memory”) (154,155).
In contrast, in patients with type 2 diabetes, who represent the vast majority of people with diabetes, the results were largely negative. The UKPDS showed a lower rate of impaired vibration perception threshold (VPT) (VPT >25 V) after 15 years for intensive therapy (IT) vs. conventional therapy (CT) (31% vs. 52%). However, the only additional time point at which VPT reached a significant difference between IT and CT was the 9-year follow-up, whereas the rates after 3, 6, and 12 years did not differ between the groups. Likewise, the rates of absent knee and ankle reflexes as well as the heart rate responses to deep breathing did not differ between the groups (153). In the ADVANCE study including 11,140 patients with type 2 diabetes randomly assigned to either standard glucose control or intensive glucose control, the relative risk reduction (95% CI) for new or worsening neuropathy for intensive vs. standard glucose control after a median of 5 years of follow-up was −4 (−10 to 2), without a significant difference between the groups (156). Likewise, in the VADT study including 1,791 military veterans (mean age, 60.4 years) who had a suboptimal response to therapy for type 2 diabetes, after a median follow-up of 5.6 years no differences between the two groups on intensive or standard glucose control were observed for DPN or microvascular complications (157). In the ACCORD trial (39), intensive therapy aimed at HbA1c <6.0% was stopped before study end because of higher mortality in that group, and patients were transitioned to standard therapy after 3.7 years on average. At transition, loss of sensation to light touch was significantly improved on intensive vs standard diabetes therapy. At study end after 5 years, MNSI score >2 and loss of sensation to vibration and light touch were significantly improved on intensive vs standard diabetes therapy. However, because of the premature study termination and the aggressive HbA1c goal, the neuropathy outcome in the ACCORD trial is difficult to interpret.
In the Steno 2 Study (158), intensified multifactorial risk intervention including intensive diabetes treatment, angiotensin converting enzyme (ACE)-inhibitors, antioxidants, statins, aspirin, and smoking cessation in patients with microalbuminuria showed no effect on DPN after 7.8 (range: 6.9-8.8) years and again at 13.3 years, after the patients were subsequently followed for a mean of 5.5 years. However, the progression of cardiac autonomic neuropathy (CAN) was reduced by 57%. Thus, there is no evidence that intensive diabetes therapy or a target-driven intensified intervention aimed at multiple risk factors favorably influences the development or progression of DPN as opposed to CAN in patients with type 2 diabetes. However, the Steno study used only vibration detection, which measures exclusively the changes in large fiber function.
DYSLIPIDEMIA
Observational and cross-sectional studies have demonstrated, to varying degrees, an association between lipids and DPN (159). The strongest evidence, however, is for the association of elevated levels of triglycerides and DPN (160). In a study of patients with T2DM there was a graded relationship between triglyceride levels and the risk of lower-limb amputations (160). Likewise, another study demonstrated that hypertriglyceridemia was an independent risk factor of loss of sural (myelinated) nerve fiber density and lower limb amputations (161). In addition to hypertriglyceridemia, low-level of HDL cholesterol is reported to as an independent risk factor for DPN (159). However, clinical studies investigating the effects of statins on the development of DPN are far from conclusive. This is partly because several large statin studies that included patients with diabetes did not report data on the development of microvascular disease (162,163) let alone DPN. The Freemantle Diabetes Study, an observational study with cross-sectional and longitudinal analysis, suggested that statin or fibrate therapy may protect against DPN in T2DM (164). Two subsequent, relatively small, randomized clinical studies have reported improvements in nerve conduction parameters of DPN following 6 to 12 weeks of statin treatment (165,166). The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study has since, demonstrated that fibrates are beneficial in preventing microvascular complications (retinopathy and nephropathy) and non-traumatic lower limb amputations but DPN outcomes have not been reported (167). Subsequently, a patient registry study from Denmark, found that the use of statins before diagnosis of incident diabetes was protective against the development of DPN (168). In summary, whether lipid lowering treatment reduces the risk of DPN —a possibility raised by these data—will need to be addressed in other studies preferably in randomized controlled trials.
HYPERTENSION
An association between hypertension and DPN has been demonstrated in several observational studies in both T2DM (169,170) and T1DM (171). There is some preliminary evidence from relatively small randomized control trials with improvements in DPN based on clinical and nerve conduction parameters following antihypertensive treatment with angiotensin converting enzyme (ACE) inhibitors (172) and calcium channel blockers (173). However, the significance of this relationship is uncertain as several large intervention studies targeting hypertension (26) studies failed to show a reduction in DPN despite clear benefits in renal and retinal complications (174). One possible explanation is that the methods used in these intervention studies to diagnose/quantify DPN lacked the necessary sensitivity or reliability to diagnose/quantity DPN let alone examine differences between study groups. The heterogeneity in effect size estimates for this outcome in many of these studies supports this view. Another possible explanation for this finding could be the strengthening of guidelines for diabetes care and the more widespread routine use antihypertensive treatment.
OBESITY
Several studies have revealed an association between obesity and polyneuropathy even in the presence of normoglycemia (175,176) The prevalence of polyneuropathy, however, increases in obese patients with prediabetes and diabetes (177). Subsequent studies appear to demonstrate that adopting a healthy lifestyle incorporating a balanced diet, regular aerobic and weight-resistance physical activities may reverse the process, particularly if they are undertaken at an early stage of DPN (136,178,179). A randomized control study of a 2.5-hour, weekly supervised treadmill exercise and dietary intervention program aimed at normalizing body mass index or losing 7% baseline body weight in T2DM demonstrated significant improvement in markers (intraepithelial nerve fiber density and regenerative capacity) of DPN (180). However, once DPN is established, restoration of normal weight did not show significant improvement.
Targeting Underlying Pathophysiological Mechanisms
OXIDATIVE STRESS
Several studies have shown that hyperglycemia causes oxidative stress in tissues that are susceptible to complications of diabetes, including peripheral nerves. Figure 2 presents our current understanding of the mechanisms and potential therapeutic pathways for oxidative stress-induced nerve damage. Studies show that hyperglycemia induces an increased presence of markers of oxidative stress, such as superoxide and peroxynitrite ions, and that antioxidant defense moieties are reduced in patients with diabetic peripheral neuropathy (181). Therapies known to reduce oxidative stress are therefore recommended. Therapies that are under investigation include aldose reductase inhibitors (ARIs), α-lipoic acid, γ-linolenic acid, benfotiamine, and protein kinase C (PKC) inhibitors.
Advanced glycation end-products (AGE) are the result of non-enzymatic addition of glucose or other saccharides to proteins, lipids, and nucleotides. In diabetes, excess glucose accelerates AGE generation that leads to intra- and extracellular protein cross-linking and protein aggregation. Activation of RAGE (AGE receptors) alters intracellular signaling and gene expression, releases pro-inflammatory molecules, and results in an increased production of reactive oxygen species (ROS) that contribute to diabetic microvascular complications. Aminoguanidine, an inhibitor of AGE formation, showed good results in animal studies but trials in humans have been discontinued because of toxicity (182). Benfotiamine is a transketolase activator that reduces tissue AGEs. Several independent pilot studies have demonstrated its effectiveness in diabetic polyneuropathy. The BEDIP 3-week study used a 200 mg daily dose, and the BENDIP 6-week study used 300 and 600 mg daily doses; both studies demonstrated subjective improvements in neuropathy scores in the groups receiving benfotiamine, with a pronounced decrease in reported pain levels (183). In a 12-week study, the use of benfotiamine plus vitamin B6/B12 significantly improved nerve conduction velocity in the peroneal nerve along with appreciable improvements in vibratory perception. An alternate combination of benfotiamine (100 mg) and pyridoxine (100 mg) has been shown to improve diabetic polyneuropathy in a small number of patients with diabetes (184,185). The use of benfotiamine in combination with other antioxidant therapies such as α-Lipoic acid (see below) are commercially available.
ARIs reduce the flux of glucose through the polyol pathway, inhibiting tissue accumulation of sorbitol and fructose. In a 12-month study of zenarestat a dose dependent improvement in nerve fiber density was shown (186). In a one year trial of fidarestat in Japanese patients with diabetes, improvement of symptoms was shown (187), and a 3 year study of epalrestat showed improved nerve function (NCV) as well as vibration perception (188). Epalrestat is marketed only in Japan and India. Newer ARIs are currently being explored, and some positive results have emerged (189), but it is becoming clear that these may be insufficient per se and combinations of treatments may be needed.
Gamma-Linolenic acid can cause significant improvement in clinical and electrophysiological tests for neuropathy (190). Alpha-Lipoic acid or thioctic acid has been used for its antioxidant properties and for its thiol-replenishing redox-modulating properties. A number of studies show its favorable influence on microcirculation and reversal of symptoms of neuropathy (191,192). A meta-analysis including 1,258 patients from four randomized clinical trials concluded that 600 mg of i.v. α-lipoic acid daily significantly reduced symptoms of neuropathy and improved neuropathic deficits (193). The SYDNEY 2 trial showed significant improvement in neuropathic symptoms and neurologic deficits in 181 diabetes patients with 3 different doses of α-lipoic acid compared to placebo over a 5-week period (194). The long-term effects of oral α-lipoic acid on electrophysiology and clinical assessments were examined during the NATHAN-1 study. The study showed that 4 years of treatment with α-lipoic acid in mild to moderate DSP is well tolerated and improves some neuropathic deficits and symptoms, but not nerve conduction (195). Additional long-term RCTs could further strengthen the rationale for the use of these agents in clinical practice. Safety profiles of α-lipoic acid are favorable during long-term treatment. An overview on the usual dosages of α-lipoic acid and benfothiamine, most frequent adverse events and scientific evidence can be found here (193,196,197,185).
Protein kinase C (PKC) activation is a critical step in the pathway to diabetic microvascular complications. It is activated by both hyperglycemia and disordered fatty-acid metabolism, resulting in increased production of vasoconstrictive, angiogenic, and chemotactic cytokines including transforming growth factor β (TGF-β), vascular endothelial growth factor (VEGF), endothelin (ET-1), and intercellular adhesion molecules (ICAMs). A multinational, randomized, phase-2, double blind, placebo-controlled trial with ruboxistaurin (a PKC-β inhibitor) failed to achieve the primary endpoints although significant changes were observed in a number of domains (198). Nevertheless, in a subgroup of patients with less severe DN (sural nerve action potential greater than 0.5 μV) at baseline and clinically significant symptoms, a statistically significant improvement in symptoms and vibratory detection thresholds was observed in the ruboxistaurin-treated groups as compared with placebo (199). A smaller, single center study showed improvement in symptom scores, endothelium dependent skin blood flow measurements, and quality of life scores in the ruboxistaurin treated group (200). These studies and the NATHAN studies have pointed out the change in the natural history of DPN with the advent of therapeutic lifestyle change, statins and ACE inhibitors, which have slowed the progression of DPN and drastically altered the requirements for placebo-controlled studies. Several studies (201,202) have demonstrated that patients with type 1 diabetes who retain some β-cell activity are considerably less prone to developing microvascular complications than those who are completely C-peptide deficient, and that C-peptide may have substantial anti-oxidant, cytoprotective, anti-anabolic, and anti-inflammatory effects. C-peptide administration for 6 months in type 1 diabetes has been shown to improve sensory nerve function (203).
GROWTH FACTORS
There is increasing evidence that there is a deficiency of nerve growth factor (NGF) in diabetes, as well as the dependent neuropeptides substance P (SP) and calcitonin gene-related peptide (CGRP) and that this contributes to the clinical perturbations in small-fiber function (204). Clinical trials with NGF have not been successful but are subject to certain caveats with regard to design; however, NGF still holds promise for sensory and autonomic neuropathies (205). The pathogenesis of DN includes loss of vasa nervorum, so it is likely that appropriate application of vascular endothelial growth factor (VEGF) would reverse the dysfunction. Introduction of VEGF gene into the muscle of DM animal models improved nerve function (206). However, VEGF gene studies with transfection of the gene into the muscle in humans failed to meet efficacy end points in painful DPN trials 207. Hepatocyte growth factor (208,209) (HGF) is another potent angiogenic cytokine under study for the treatment of painful neuropathy. INGAP peptide comprises the core active sequence of Islet Neogenesis Associated Protein (INGAP), a pancreatic cytokine that can induce new islet formation and restore euglycemia in diabetic rodents. Maysinger et al showed significant improvement in thermal hypoalgesia in diabetic mice after a 2-week treatment with INGAP peptide (210,211).
IMMUNE THERAPY
Several different autoantibodies in human sera have been reported that can react with epitopes in neuronal cells and have been associated with DN. Milicevic et al have reported a 12% incidence of a predominantly motor form of neuropathy in patients with diabetes associated with monosialoganglioside antibodies (anti GM1 antibodies) (63). Perhaps the clearest link between autoimmunity and neuropathy has been the demonstration of an 11-fold increased likelihood of CIDP, multiple motor polyneuropathy, vasculitis, and monoclonal gammopathies in diabetes (61). New data, however, support a predictive role of the presence of antineuronal antibodies on the later development of neuropathy, suggesting that these antibodies may not be innocent bystanders but neurotoxins (212). There may be selected cases, particularly those with autonomic neuropathy, evidence of antineuronal autoimmunity, and CIDP, that may benefit from intravenous immunoglobulin or large dose steroids (59).
Summary of Treatment of Diabetic Peripheral Neuropathy
In summary, the risk factors for DPN are well recognized and to-date only small-scale intervention studies targeting these risk factors that have used appropriate DPN biomarkers have been conducted. Nevertheless, these have provided preliminary evidence for the efficacy of multifactorial risk factor management in preventing the development and progression of DPN. Hence, early identifications of subjects with insipient/sub-clinical neuropathy using validated, yet novel non-invasive point of care devices will allow larger studies to determine if targeted intensified cardiometabolic risk factor control can prevent clinical DPN or halt disease progression. Unfortunately, despite several clinical trials, there has been relatively little progress in the development of disease modifying treatments despite some advances in the management of symptoms in painful DN, as described below.
PAINFUL DIABETIC PERIPHERAL NEUROPATHY
Pathogenesis
Peripheral neuropathic pain in diabetes is defined as “pain arising as a direct consequence of abnormalities in the peripheral somatosensory system” after exclusion of other causes (213). Nerve damage results in the release of inflammatory mediators which activate intracellular signal transduction pathways in the nociceptor terminal, prompting an increase in the production, transport, and membrane insertion of transducer channels and voltage-gated ion channels (214). Following nerve injury, different types of voltage-gated sodium and calcium channels are up-regulated at the site of the lesion and in the dorsal root ganglion membrane, promoting ectopic spontaneous activity along the primary afferent neuron and determining hyperexcitability associated with lowered activation threshold, hyper-reactivity to stimuli, and abnormal release of neurotransmitters such as substance P and glutamate (215, 216). As a consequence of this hyperactivity in primary afferent nociceptive neurons, important secondary changes may occur in the dorsal horn of the spinal cord and higher up in the central nervous system leading to neuron hyperexcitability. This phenomenon, called central sensitization, is a form of use-dependent synaptic plasticity, considered a major pathophysiological mechanism of neuropathic pain (217).
Diagnosis
Painful DPN is often underdiagnosed and under treated. Binns-Hall et al. trialed a ‘one-stop’ microvascular screening service, which tested a model for patients to receive combined eye, foot (DPN and painful-DPN), and renal screening (218). A new diagnosis of painful-DPN in this cohort was identified in 25% of participants using the validated screening tool for neuropathic pain, the Doleur Neuropathique en 4 Questions (DN4). Additionally, Daousi et al. found that in a community sample of 350 patients with diabetes 12.5% of patients with painful-DPN had not reported their symptoms to their treating physician (219). This study also found that 39.3% had never received treatment for their painful neuropathy. In the clinical environment, most cases of painful DPN can be diagnosed with a careful history to identify presence of typical painful neuropathic symptoms lasting ≥ 3 months and clinical examination to demonstrate the clinical signs of DPN. In these circumstances and other causes are excluded (see above), there is no need for further investigations.
A number of self-administered questionnaires have been developed, validated, translated, and subjected to cross-cultural adaptation both to diagnose and distinguish neuropathic as opposed to non-neuropathic pain (screening tools such as the Leeds Assessment of Neuropathic Symptoms and Signs (LANSS) Pain Scale (220), Douleur Neuropathique en 4 questions (DN4), Neuropathic Pain Questionnaire (NPS) (221), pain DETECT (89) and to assess pain quality and intensity such as the Short-Form McGill Pain Questionnaire (222), the Brief Pain Inventory (BPI) (223), and the Neuropathic Pain Symptom Inventory (NPSI) (224).
It is important to assess the intensity (severity) of neuropathic pain as it is helpful when assessing and monitoring response to therapy. The best approach is to use a simple 11-Point numerical rating scale (Likert scale) or a visual analogue scale. In clinical trials of neuropathic pain treatment a number of questionnaires are used to capture the complex, multidimensional impact of chronic pain. According to IMMPACT (Initiative on Methods, Measurement and Pain Assessment in Clinical Trials) the following assessments are performed to assess the efficacy and effectiveness of new treatments: 1. pain intensity measured on a 0 to 10 numerical rating scale (NRS); 2. physical functioning assessed by the Multidimensional Pain Inventory (MPI) and Brief Pain Inventory (BPI) Interferences scale; 3. emotional functioning, assessed by the Beck Depression Inventory (BPI) and Profile of Mood states; and 4. patient rating of overall improvement, assessed by the Patient Global Impression of Change (PGI-C) (225).
Quality of Life
Over time the persistence of extremely unpleasant painful symptoms can have a profound impact upon its sufferers’ lives. This often results in a poor quality of life (226), disruption of employment (227), and mood disturbance (13). This adds to the burden of suffering and increases the challenge of managing neuropathic effectively. This is further compounded when patients also suffer from other co-morbid conditions associated with diabetes. Painful-DPN is also an expensive condition, incurring high healthcare costs (228). Data from the US found that patients with DPN and painful-DPN have greater healthcare resource utilization and costs than those with diabetes alone (228). Patients with severe painful-DPN incurred five-fold higher annual direct medical costs (USD $30,755) than for patients with diabetes alone (USD $6632) (226).
Sensory Profiling
For many years, sensory profiling has been the mainstay for identifying a homogenous subgroup of neuropathic pain patients in clinical pain research. The basis of this approach is that painful symptoms reflect specific pathophysiological mechanisms, which are present to varying degrees in individual patients (229,230). Detailed sensory profiling using quantitative sensory testing (QST) can be used to subgroup patients into more homogenous cohorts (pain phenotypes), which could then be targeted with treatments known to act specifically on pathophysiological pathways underlying the phenotypes (231) (Figure 13). QST refers to a battery of standardized, psychophysical tests (e.g., thermal testing, pin prick, pressure algometry, and von Frey filaments) used to assess central and peripheral nervous system sensory function (232). In DPN, QST has been used for several decades mainly for diagnosing and quantifying the extent of small and large nerve fiber impairment in individuals predominantly with painless DPN. In the context of pain somatosensory phenotyping, a standardized QST protocol was developed by the German Research Network on Neuropathic Pain (DFNS), which includes 12 sensory testing parameters (i.e., cold and warm detection thresholds, paradoxical heat sensations, thermal sensory limen procedure, cold and heat pain thresholds, mechanical detection threshold, mechanical pain threshold, mechanical pain sensitivity, dynamic mechanical allodynia, wind-up ratio, vibration detection threshold, and pressure pain threshold) (232). The positive and negative results of individual patients are obtained by comparison against a normative QST reference dataset, comprised of age- and sex-stratified healthy individuals.

Figure 13.
Schematic representation of the generation of pain. (A) Normal: Central terminals of c-afferents project into the dorsal horn and make contact with secondary pain-signaling neurons. Mechanoreceptive Aβ afferents project without synaptic transmission into the dorsal columns (not shown) and also contact secondary afferent dorsal horn neurons. (B) C-fiber sensitization: Spontaneous activity in peripheral nociceptors (peripheral sensitization, black stars) induces changes in the central sensory processing, leading to spinal-cord hyperexcitability (central sensitization, gray star) that causes input from mechanoreceptive Aβ (light touch) and Aδ fibers (punctuate stimuli) to be perceived as pain (allodynia). (C) C-fiber loss: C-nociceptor degeneration and novel synaptic contacts of Aβ fibers with “free” central nociceptive neurons, causing dynamic mechanical allodynia. (D) Central disinhibition: Selective damage of cold-sensitive Aδ fibers that leads to central disinhibition, resulting in cold hyperalgesia. Sympat, sympathetic nerve
Two Distinct Pain Phenotypes – The Non-Irritable and Irritable Nociceptor
Application of the QST technique has shown that there are two distinct subgroups of patients who have particular patterns of sensory symptoms and signs: (a) a predominant differentiation with loss of sensory function (non-irritable nociceptor phenotype), and (b) a relatively preserved small fiber function associated with thermal/mechanical hypersensitivity (irritable nociceptor phenotype) (231). Using the DFNS protocol, the PiNS reported that the non-irritable nociceptor was the predominant phenotype in painful DPN, whilst only a minority of patients had the irritable nociceptor phenotype (6.3%) (233). Nevertheless, a small but significant proportion of patients (15%) did demonstrate signs of sensory gain with dynamic mechanical allodynia, often in combination with hyposensitivity across a range of small and large nerve fiber sensory assessments. The presence of allodynia would suggest that aberrant central processing of sensory inputs has an important role in these patients. Recent studies have demonstrated proof-of-concept for using sensory profiling to improve clinical trial efficiency by demonstrating that some treatments are more effective in patients with the irritable versus the non-irritable nociceptor phenotype (230-234). However, most of these studies examined patients with peripheral neuropathy of diverse causes.
Phenotype-Driven Therapeutic Experience in Painful DPN
Examples of studies that focused on painful DPN include an open label retrospective study using the DFNS protocol, which evaluated key phenotypic differences in sensory profiling associated with response to intravenous lidocaine in patients with severe, intractable painful DPN (235). Patients with the irritable nociceptor phenotype were more likely to respond to intravenous lidocaine, which inactivates sodium channels, compared to the non-irritable nociceptor phenotype (235). In fact, dynamic mechanical allodynia and pain summation to repetitive pinprick stimuli were the only evoked ‘gain of function’ QST parameters that informed treatment response. The presence of these sensory gain parameters suggests aberrant central processing with hyperexcitable neurons driven by abnormal sodium channel regulation, generating ectopic impulses and amplifying afferent sensory inputs. In another painful DPN study by Campbell et al. of topical clonidine, sensory profiling was performed using the capsaicin challenge test (236). The post-hoc analysis demonstrated a significant reduction in pain in the patient subgroup with increased spontaneous pain following cutaneous capsaicin administration, indicating the presence of functioning and sensitized nociceptors. Bouhassira et al. published post-hoc analysis data of treatment response based on sensory profiling using the Neuropathic Pain Symptom Inventory (NPSI) questionnaire from the Combination vs Monotherapy of pregabalin and duloxetine in Diabetic Neuropathy (COMBO-DN) study (237). This study examined the effect of high-dose duloxetine, a serotonin noradrenaline reuptake inhibitor, or pregabalin, a calcium channel blocker, as monotherapy versus combined pregabalin and duloxetine for painful DPN. The investigators showed that adding pregabalin (300 mg) to duloxetine (60 mg) improved the dimensions of ‘pressing pain’ and ‘evoked pain’ more significantly. On the other hand, increasing duloxetine from 60 mg to 120 mg daily improved the dimension ‘paresthesia/dysesthesia’ to a greater extent.
SENSORY PHENOTYPING TO PREDICT THERAPEUTIC RESPONSE
In a randomized, double-blind, placebo-controlled, and phenotype-stratified study of patients with painful DPN Demant et al. reported that oxcarbazepine was more efficacious for relief of peripheral neuropathic pain in patients with the irritable vs the nonirritable nociceptor phenotype (234). Based on this and other recent studies, current opinion with regard to neuropathic pain clinical trials recommends a detailed sensory profiling of participants at baseline; and even if there is no significant separation of a drug with placebo, a subgroup analysis can be performed to see if the drug was efficacious in a particular subgroup. If there is a clear signal that this was the case, a further, adequately powered, phenotype stratified trial would be designed.
Sensory profiling can also identify subgroups with altered endogenous pain modulation to predict treatment outcomes of drugs and other interventions that affect a given mechanism. Figure 14 describes the different nerve fibers affected and possible targeted treatments.
In a study of pain modulation in DPN, individuals were assessed using QST for conditioned pain modulation (CPM), a psychophysical paradigm in which central pain inhibition is measured via the phenomenon of ‘pain inhibiting pain,’ via the simultaneous administration of a conditioning painful stimulus at a distant body site. The pain in participants with abnormal CPM was more receptive to duloxetine, which is believed to increase descending inhibitory pain pathway activation, than individuals with normal pain modulation, although there was no comparison to placebo in this open-label study (238).

Figure 14.
Different mechanisms of pain and possible treatments. C fibers are modulated by sympathetic input with spontaneous firing of different neurotransmitters to the dorsal root ganglia, spinal cord and cerebral cortex. Sympathetic blockers (e.g., clonidine) and depletion of axonal substance P used by C fibers as their neurotransmitter (e.g., by capsaicin) may improve pain. In contrast Aδ fibers utilize Na+ channels for their conduction and agents that inhibit Na+ exchange such as antiepileptic drugs, tricyclic antidepressants, and insulin may ameliorate this form of pain. Anticonvulsants (carbamazepine, gabapentin, pregabalin, topiramate) potentiate activity of g-aminobutyric acid and inhibit Na+ and Ca2+ channels, N-methyl-D-aspartate receptors, and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors. Dextromethorphan blocks N-methyl-D-aspartate receptors in the spinal cord. Tricyclic antidepressants, selective serotonin reuptake inhibitors (e.g., fluoxetine), and serotonin and norepinephrine reuptake inhibitors inhibit serotonin and norepinephrine reuptake, enhancing their effect in endogenous pain-inhibitory systems in the brain. Tramadol is a central opioid analgesic. α2 antag, α 2 antagonists; 5HT, 5-hydroxytryptamine; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; DRG, dorsal root ganglia; GABA, g-aminobutyric acid; NMDA, N-methyl-D-aspartate; SNRIs, serotonin and norepinephrine reuptake inhibitors; SP, substance P; SSRIs, selective serotonin reuptake inhibitors; TCA, tricyclic antidepressants;
Taken together, these studies support the notion that mechanism-based approaches to pain management may be feasible in painful DPN. However, in an elegant mechanistic study, Haroutounian et al examined 14 patients with neuropathic pain of mixed etiology [unilateral foot pain from nerve injury (n=7) and distal polyneuropathy (n=7)] to determine the contribution of primary afferent input in maintaining peripheral neuropathic pain (239). Each patient underwent randomized ultrasound-guided peripheral nerve block with lidocaine versus intravenous lidocaine infusion. They found that peripheral afferent input was critical for maintaining neuropathic pain, but improvement in evoked hypersensitivity was not related to improvements in spontaneous pain intensity. This suggests that further studies are needed to rationalize sensory phenotyping in order to optimize clinical trial outcomes in painful DPN. Moreover, given the rarity of the irritable-nociceptor phenotype, as determined by QST, a single assessment modality may be unlikely to help stratify patients and combining with additional modalities may be necessary (e.g., brain imaging).
Brain Imaging in Painful Diabetic Peripheral Neuropathy
Recent advances in neuroimaging provide us with unique insights into the human central nervous system in chronic pain conditions (240). We now have a better understanding how the brain modulates nociceptive inputs to generate the pain experience, and how this is disrupted in patients with painful DPN. However, to date, brain imaging serves mainly as a research tool, with minimal direct application in clinical trials for pain or clinical practice. While mechanistic approaches that require carefully evaluating specific responses to guide therapy have significant appeal (e.g., cold, heat, von Frey etc.), in practice, these are time consuming and may be difficult to implement in busy clinical practices. Furthermore, these are psychophysical measures which rely on patient responses and may be subjective and biased. Sensory profiling methods also do not capture the complex and multidimensional pain experience, which affects emotional and cognitive processing in addition to sensory processing. For example, chronic pain patients often undergo neuropsychological changes, which include changes in emotion and motivation or changes in cognition (241). Chronic pain may also arise after the onset of depression, even in patients without a prior history of pain or depression. Collectively, these clinical insights suggest a better strategy for assessing and treating painful DPN, given it is a chronic disease of dynamic process (e.g., evolution of co-morbid phenotypes such as anxiety or depression), which is not easily reversed in most patients. It is important to determine specific targets that are relevant to pain across individuals, because modulating activation in these targets may provide evidence that a compound engages a target or attenuates nociceptive processing.
Structural and functional cortical plasticity is a fundamental property of the human central nervous system, which can adjust to nerve injury. However, it can have maladaptive consequences, possibly resulting in chronic pain. Studies using structural magnetic resonance (MR) neuroimaging have demonstrated a clear reduction in both spinal cord cross-sectional area (139) and primary somatosensory cortex (S1) gray matter volume in patients with DPN (141). These findings are supported by studies in other pain conditions, which have also reported dynamic structural and functional plasticity with profound effects on the brain in patients with neuropathic pain. More recently, it has been demonstrated how brain structural and functional changes are related to painful DPN clinical phenotypes (146). Patients with the painful insensate phenotype have a more pronounced reduction in S1 cortical thickness and a remapping of S1 sensory processing compared to painful DPN subjects with relatively preserved sensation (146). Furthermore, the extent to which S1 cortical structure and function is altered is related to the severity of neuropathy and the magnitude of self-reported pain. These data suggest a dynamic plasticity of the brain in DPN driven by the neuropathic process and may ultimately determine an individual’s clinical pain phenotypes.
Over the last decade, resting-state functional MR imaging (RS-fMRI) – a quick, and simple non-invasive technique – has become an increasingly appealing way to examine spontaneous brain activity in individuals, without relying on external stimulation tasks. During a typical RS-fMRI examination, the hemodynamic response to spontaneous neuronal activity (bold oxygen level dependent, BOLD) signal is acquired whilst subjects are instructed to simply rest in the MR scanner (242). The data acquired is used in brain mapping to evaluate regional interactions or functional connectivity, which occur in a resting state. Most studies use a machine learning approach to identify patterns of functional connectivity, which differentiates patients from controls. RS-fMRI experiments in painful DPN have reported greater thalamic-insula functional connectivity and decreased thalamic-somatosensory cortical functional connectivity in patients with the irritable versus non-irritable nociceptor phenotype (235). There was a significant positive correlation between thalamic-insula functional connectivity with self-reported pain scores (235). Conversely, there was a more significant reduction in thalamic-somatosensory cortical functional connectivity in those with more severe neuropathy. This demonstrates how RS-fMRI measures of functional connectivity relates to both the somatic and non-somatic assessments of painful DPN. In one study, using a machine learning approach to integrate anatomical and functional connectivity data achieved an accuracy of 92% and sensitivity of 90%, indicating good overall performance (235). Multimodal MR imaging combining structural and RS-fMRI has also been used to predict treatment response in painful DPN. Responders to intravenous lidocaine treatment have significantly greater S1 cortical volume and greater functional connectivity between the insular cortex and corticolimbic system compared to non-responders (235). The insular cortex plays a pivotal role in processing the emotion and cognitive dimensions of the chronic pain experience. The corticolimbic circuits have also long been implicated in reward, decision making, and fear learning. Hence, these findings suggest that this network may have a role in determining treatment response in painful DPN.
Using advanced multimodal MR neuroimaging, a number of studies have demonstrated alterations in pain processing brain regions, which relate to clinical pain phenotype, treatment response, and behavioral/psychological factors impacted by pain. Taken together, these assessments could serve as a possible Central Pain Signature for painful DPN. The challenge now is to apply this potential pain biomarker at an individual level in order to demonstrate clinical utility. To this end, applying machine learning (243) to leverage brain imaging features from a quick 6-minute RS-fMRI scan to classify individual patients into different clinical pain phenotypes is appealing. Future studies should externally validate and optimize current models in larger patient cohorts to examine if/how such models can be used as biomarkers in clinical trials of pain therapeutics. Although many of the findings described are consistent with neuroimaging studies in other chronic pain conditions, it is difficult to assess convergence of findings across a number of relatively small cohort studies employing different analytical methods to derive complex models involving a large number of distributed brain regions (244). These are important limitations that are being addressed with 1) a number of large scale multi-center studies in progress or in preparation (MAPP consortium (245) and Placebo Imaging consortium (246), and 2) several consensus statements by key stakeholders, promoting standardized approaches and reporting and transparent/sharable models.
General Principals of Managing Painful DPN
Managing painful symptoms in DPN may constitute a considerable treatment challenge. The efficacy of a single therapeutic agent is not the rule, and most patients require combination therapy to control the pain. The present ‘trial and error’ approach is to offer the available therapies in a stepwise fashion until an effective treatment is achieved (247,248). Effective pain treatment considers a favorable balance between pain relief and side effects without implying a maximum effect. The following general considerations in the pharmacotherapy of neuropathic pain require attention (249):
- 1.
The appropriate and effective drug has to be tried and identified in each patient by carefully titrating the dose based on efficacy and side effects.
- 2.
Lack of efficacy should be judged only after 2-4 weeks of treatment using an adequate dose.
- 3.
As the evidence from clinical trials suggests a ≥ 50% reduction in pain for any monotherapy, combination therapy is considered a ‘robust’ response. A reduction of pain of 30-49% may be considered a ‘clinically relevant’ response.
- 4.
Potential drug interactions have to be considered given the frequent use of polypharmacy in patients with diabetes.
For many patients, optimal management of chronic pain may require a multidisciplinary team approach with appropriate behavioral therapy, as well as input from a broad range of healthcare professionals. Here we highlight the common agents used to manage painful DPN and key papers to demonstrate the evidence base. The most recent guidelines for pharmacotherapy for neuropathic pain in general and specifically in painful DPN can be found elsewhere (16,250,251,252,253,254,67, 255,256).
ANTIDEPRESSANTS
Antidepressants are commonest agents used in the treatment of chronic neuropathic pain (217). The putative mechanisms of interrupting pain transmission by these agents include inhibition of norepinephrine and/or serotonin reuptake within the endogenous descending pain-inhibitory systems in the brain and spinal cord (257). Antagonism of N-Methyl-D-Aspartate receptors that mediate hyperalgesia and allodynia has also been proposed.
Tricyclic Antidepressants (TCAs)
Imipramine, amitriptyline, and clomipramine induce a balanced reuptake inhibition of both norepinephrine and serotonin, while desipramine is a relatively selective norepinephrine inhibitor. The most frequent adverse events of tricyclic antidepressants (TCAs) are anticholinergic symptoms including tiredness and dry mouth and may exacerbate cardiovascular and gastrointestinal autonomic neuropathy. The starting dose should be 25 mg (10 mg in frail patients) taken as a single night time dose one hour before sleep. The maximum dose is usually 150 mg per day and doses >100mg should be avoided in the elderly.
TCAs should be used with caution in patients with orthostatic hypotension and are contraindicated in patients with unstable angina, recent (<6 months) myocardial infarction, closed-angle glaucoma, heart failure, history of ventricular arrhythmias, significant conduction system disease, and long QT syndrome. Their use is limited by relatively high rates of adverse events and several contraindications.
Serotonin Noradrenaline Reuptake Inhibitors (SNRI)
The efficacy and safety of duloxetine has been evaluated in 7 RCTs establishing it as a mainstay treatment option in painful DPN. Several systematic reviews demonstrate a moderate strength of evidence for duloxetine reduces neuropathic pain to a clinically meaningful degree in patients with painful DPN (258,259,260). Patients with higher pain intensity tend to respond better than those with lower pain levels. The most frequent side effects of duloxetine (60/120 mg/day) include nausea (16.7/27.4%), somnolence (20.2/28.3%), dizziness (9.6/23%), constipation (4.9/10.6%), dry mouth (7.1/15%), and reduced appetite (2.6/12.4%). These adverse events are usually mild to moderate and transient. To minimize them the starting dose should be 30 mg/day for 4-5 days. In contrast to TCAs and some anticonvulsants, duloxetine does not cause weight gain, but a small increase in fasting blood glucose may occur (261).
Venlafaxine is another SNRI that has mixed action on catecholamine uptake. Compared to duloxetine, the strength of evidence for venlafaxine is lower and it could be considered an alternative if duloxetine is not tolerated. At lower doses, venlafaxine inhibits serotonin uptake and at higher doses it inhibits norepinephrine uptake (262). The extended release version of venlafaxine was found to be superior to placebo in painful DPN in non-depressed patients at doses of 150-225 mg daily, and when added to gabapentin there was improved pain, mood, and quality of life (263). In a 6-week trial comprised of 244 patients the analgesic response rates were 56%, 39%, and 34% in patients given 150-225 mg venlafaxine, 75 mg venlafaxine, and placebo, respectively. Because patients with depression were excluded, the effect of venlafaxine (150-225 mg) was attributed to an analgesic, rather than antidepressant, effect. The most common adverse events were tiredness and nausea (264); additionally, clinically important electrocardiogram changes were found in seven patients in the treatment arm.
ANTI-EPILEPTIC DRUGS
Calcium Channel Modulators (α2-δ ligands)
Gabapentin is an anticonvulsant structurally related to γ-aminobutyric acid (GABA), a neurotransmitter that plays a role in pain transmission and modulation. The exact mechanisms of action of this drug in neuropathic pain are not fully elucidated. Among others, they involve an interaction with the L-amino acid transporter system and high affinity binding to the α2-δ subunit of voltage-activated calcium channels. A Cochrane review reported 4 out of 10 patients with painful DPN achieved greater than 50% pain relief with gabapentin compared to placebo (2 out of 10). Pain was reduced by a third or more for 5 in 10 with gabapentin and 4 in 10 with placebo. Over half of those treated did not benefit from worthwhile pain relief but experienced adverse event (265).
In contrast to gabapentin, pregabalin is a more specific α2-δ ligand with a 6-fold higher binding affinity. It also has a more rapid onset with a dose-dependent linear pharmacokinetic profiled i.e., 600mg/day being more effective that 300mg/day (266). Hence, the administration (BD vs QDS) and dose titration of pregabalin in considerably easier compared to gabapentin. A recent Cochrane review reported moderate quality evidence for the efficacy of pregabalin in painful DPN compared to placebo (267). 3 or 4 in 10 people had pain reduced by half or more with pregabalin 300 mg or 600 mg daily, and 2 or 3 in 10 with placebo. Pain was reduced by a third or more for 5 or 6 in 10 people with pregabalin 300 mg or 600 mg daily, and 4 or 5 in 10 with placebo.
Common side-effects associated with the use of gabapentinoids include weight gain, edema, dizziness, and somnolence. They should be used with caution in patients with congestive cardiac failure (NYHA class III or IV) and renal impairment (dose reduction required). Pooled trial analysis of adverse events showed a higher risk of side-effects with increasing pregabalin dose but not older age (268). The misuse and abuse of gabapentinoids is a growing problem in the US and in Europe necessitating monitoring for signs of misuse/abuse and caution when used in at risk populations (269). Gabapentinoids may also increase the risk of respiratory depression, a serious concern for patients taking opioids or with underlying respiratory impairment (270,271,272).
TOPICAL CAPSAICIN
C-fibers utilize the neuropeptide substance P as their neurotransmitter, and depletion of axonal substance P (through the use of capsaicin) will often lead to amelioration of the pain. Prolonged application of capsaicin, a highly selective agonist of transient receptor potential vanilloid-1 (TRPV1), depletes stores of substance P, and possibly other neurotransmitters, from sensory nerve endings. This reduces or abolishes the transmission of painful stimuli from the peripheral nerve fibers to the higher centers (273). The 8% capsaicin patch (Qutenza) (274) is authorized for the treatment of peripheral neuropathic pain. In one RCT in painful DPN, a single application of 8% capsaicin patch applied for 30min provided modest pain relief for up to 3 months (275). Specialist trained staff are required for application which can be repeated every 2-3 months. A Cochrane review of low dose (0.025% and 0.075%) topical capsaicin cream was not able to provide any recommendations due to insufficient data (276).
LIDOCAINE
Lidocaine has unique analgesic properties. Although the exact mechanism by which intravenous lidocaine provides systemic analgesia is unknown, it is thought to have both peripheral and central mechanisms of action (277,278,279). It exhibits state-dependent binding where sodium channels that are rapidly and repeatedly activated and inactivated are more readily blocked (280). This state-dependence is thought to be very important in limiting the hyperexcitability of cells exhibiting abnormal activity. Thus, it is likely to have greater efficacy in patients with neuropathic pain (281,282) and has been used to relieve chronic pain for over 50 years (283). A Cochrane review of 30 RCT found that intravenous lidocaine (284), which is more effective than its oral analogue (mexilitine, NNT10-38) and gastrointestinal intolerance most common side effect and major factor limiting its use) (284,285) and is more effective than placebo in decreasing neuropathic pain. It was found to be generally well tolerated with little or no side effects (286). Hence, intravenous lidocaine is a recognized treatment option for patients with severe painful DPN (287), and is included in clinical guidelines (288).
Although 5% lidocaine patch is being used in patients with postherpetic neuralgia (289), there is insufficient evidence to recommend its use in those with painful DPN.
OPIOIDS
Tramadol and NMDA Receptor Antagonists
The most examined compounds in painful DPN are tramadol, oxycodone, and tapentadol. Tramadol is a centrally acting weak opioid and SNRI for use in treating moderate to severe pain. More severe pain requires administration of strong opioids such as oxycodone (µ-opioid agonist) or tapentadol (µ-opioid agonist and SNRI). There is limited data available on the efficacy of these agents from relatively small-scale studies. Recent Cochrane reviews graded the available evidence as mostly of low or very low quality and likely to overestimate the efficacy of tramadol and oxycodone in the treatment of painful DPN (290,291). Side effects typical of opioids were common including somnolence, headache, and nausea. There is an increased risk of serotonergic syndrome if tramadol and tapentadol are prescribed with other agents with serotonin reuptake inhibitor properties and thus best avoided. Nevertheless, there is role for these agents as 2nd or 3rd line analgesics for painful DPN in carefully selected patients unresponsive to standard treatments. Non-pharmacological and non-opioid analgesic treatments should be optimized and established and/or not tolerated/contraindicated before opioid treatment is considered (292). Regular monitoring/evaluation of efficacy is recommended particularly if treatment is longer than 3 months. Opioids are associated with less pain relief during longer trials possibly due to opioid tolerance or opioid induced hyperalgesia. Moreover, adverse outcomes such as dependence, overdose, depression, and impaired functional status were more common in patients with neuropathic pain (painful DPN 68%) receiving long-term (>90 days) vs short term (<90 days) of treatment (293). Hence, referral to specialist or centers with experience in opioid use is recommended to avoid risks.
PSYCHOLOGICAL SUPPORT
A psychological component to pain should not be underestimated. Hence, an explanation to the patient that even severe pain may remit, particularly in poorly controlled patients with acute painful neuropathy or in those painful symptoms precipitated by intensive insulin treatment. Thus, the empathetic approach addressing the concerns and anxieties of patients with neuropathic pain is essential for their successful management (294).
PHYSICAL MEASURES
The temperature of the painful neuropathic foot may be increased due to arterio-venous shunting. Cold water immersion may reduce shunt flow and relieve pain. Allodynia may be relieved by wearing silk pajamas or the use of a bed cradle. Patients who describe painful symptoms on walking as comparable to walking on pebbles may benefit from the use of comfortable footwear (255).
ACUPUNCTURE
A 10-week uncontrolled study with a follow-up period of 18-52 weeks in patients with diabetes showed significant pain relief after up to 6 courses of traditional Chinese acupuncture without any side effects (295). A single-blind placebo-controlled randomized trial of acupuncture in 45 subjects with painful DN recently reported an improvement in the outcome measures assessing pain in the acupuncture arm relative to sham treatment (296). However, Chen and colleagues warn that design flaws and lack of robust outcome measures of pain in acupuncture trials make meaningful conclusions difficult (297). Larger controlled studies are needed to confirm these early findings.
ELECTRICAL STIMULATION
Transcutaneous electrical nerve stimulation (TENS) influences neuronal afferent transmission and conduction velocity, increases the nociceptive flexion reflex threshold, and changes the somatosensory evoked potentials. In a 4-week study of TENS applied to the lower limbs, each for 30 minutes daily, pain relief was noted in 83% of the patients compared to 38% of a sham-treated group. In patients who only marginally responded to amitriptyline, pain reduction was significantly greater following TENS given for 12 weeks as compared with sham treatment. Thus, TENS may be used as an adjunctive modality combined with pharmacotherapy to augment pain relief (298).
Frequency-modulated electromagnetic nerve stimulation (FREMS) in 2 studies, including a recent double-blind randomized placebo controlled trial with 51 weeks of follow-up, proved to be a safe treatment for symptomatic diabetic neuropathy, with immediate but transient reduction in pain and no effect on nerve conduction velocities (299,300). Six out of eight trials analyzed in a recent review evaluating the use of electrical stimulation in painful DN found significant pain relief in patients treated with electrical stimulation compared with placebo or sham treatment (301).
Electrical spinal cord stimulation (SCS) was first reported in painful DPN in 1996 (302). With electrodes implanted between T9 and T11, 8 out of 10 patients reported greater than 50% pain relief. Most of these early devices utilized low-frequency stimulation (40-60Hz) with two RCTs demonstrating moderate utility (n=36 to 60) with 6-month to 24-month follow up (303,304,305) with responder attrition within 12 months (306). Modern iterations of SCS employ high-frequency stimulation (10kHz) provides pain relief without generating paresthesia (307,308,309,310). A recent RCT examine the use of 10kHz electrical SCS in patients with refractory painful DPN compared to conventional medical management in 216 randomized patients (311). 50% reduction in pain relief was observed in 5% in the control group compared to 79% in the electrical SCS group with 6 months follow up. The main limitation of this study was the lack of blinding and potential for placebo effects as an important confounding factor. Nevertheless, this is an interesting finding which should open a new area for further research. Overall complications of electrical SCS include wound infection and lead migration requiring reinsertion. Currently, therefore, this invasive treatment option should be reserved for patients who do not respond to analgesic combination pharmacotherapy.
SURGICAL DECOMPRESSION
Surgical decompression at the site of anatomic narrowing has been promoted as an alternative treatment for patients with symptomatic DPN. A systematic review of the literature revealed only Class IV studies concerning the utility of this therapeutic approach. Given the current evidence available, this treatment alternative should be considered unproven. Prospective randomized controlled trials with standard definitions and outcome measures are necessary to determine the value of this therapeutic intervention (312,313).
The odds ratios for efficacy of neuropathic pain medications are given in Figure 15. In addition, Table 5 shows the dosages of the different drugs and the commonly encountered side effects.
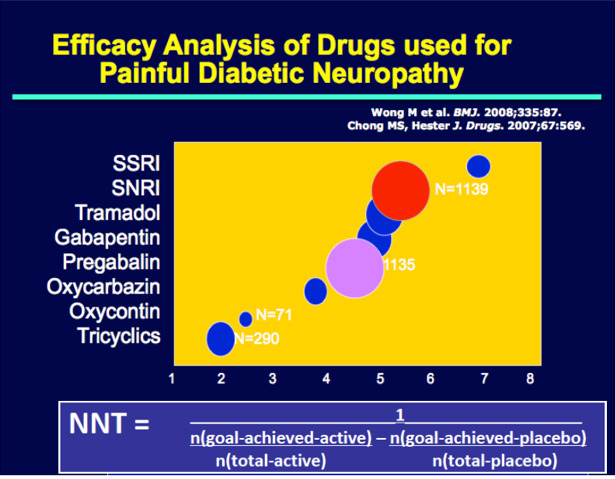
Figure 15.
Efficacy analysis of drugs used for the treatment of PDN
Table 5.
Treatments for Symptomatic Diabetic Polyneuropathy
Pain-Dosing and Side Effects
| Drug Class | Drug | Dose | Side Effects |
|---|---|---|---|
| Tricyclics | Amitriptyline | 5-75mg nocte | Somnolence, dizziness, dry mouth, tachycardia, |
| Nortriptyline | 50-150mg nocte | constipation, urinary retention, blurred vision | |
| Imipramine | 25-150mg nocte | Confusion | |
| SNRIs | Duloxetine | 30-60mg BD | Nausea, somnolence, dizziness, anorexia |
| Anticonvulsants | Gabapentin | 300-1200mg TDS | Somnolence, dizziness, Confusion, ataxia |
| Pregabalin | 50-300mg BD | Somnolence, confusion, edema, weight gain | |
| Carbamazepine/ Oxcarbazepine | Up to 200 QDS | Dizziness, somnolence, Nausea, leukopenia | |
| Opioids* | Tramadol | 50-100mg BD/QDS | Nausea, constipation, HA Somnolence |
| Oxycodone MR Tapentadol ER | 10-30mg BD 50mg BD up to 500mg/24hrs | Somnolence, nausea, constipation, HA Constipation, nausea, somnolence, dizziness | |
| Topical | Capsaicin | 0.075% QDS | Local irritation |
| Lidocaine | 0.04% QDS | Local irritation |
- *
Due to increased risk of adverse outcomes caution is advised if treatment is continued for >3 months.
Guidelines for Pharmacotherapy of Painful Neuropathy
Figure 16 is a pharmacotherapy algorithm that we propose for the management of painful neuropathy in diabetes. This presumes that the cause of the pain has been attributed to DPN and that all causes masquerading as DPN have been excluded. The identification of neuropathic pain as being focal or diffuse dictates the initial course of action. Focal neuropathic pain is best treated with splinting, steroid injections, and surgery to release entrapment. Diffuse neuropathies are treated with medical therapy and in a majority of cases, need combination therapy. Essential to the DPN evaluation is the identification of the patient’s comorbidities, potential adverse events, and drug interactions. When single agents fail, combinations of drugs with different mechanisms of action should be considered. Comorbidities that accompany pain include depression, anxiety, and sleep disturbances, all of which must be addressed for successful management of pain. Treatment of peripheral neuropathic pain conditions can benefit from further understanding of the impact of pain response and QOL, including activities of daily living (ADLs) and sleep. Patients often benefit from participation in pain management groups and psychological intervention to develop/gain better coping strategies and deal with harmful/disruptive pain-related behaviors. There is currently minimal evidence for the use of combination treatment for painful DPN – hence, most guidelines recommend switching to an alternative agent. There are also few head-to-head comparator trials of commonly used agent evaluating efficacy and safety between drugs. We await the outcome of the much-anticipated OPTION-DM study – head-to-head multicenter, RCT will inform clinicians of the most cost effective monotherapy (amitriptyline, pregabalin and duloxetine) followed by combination therapy for painful DPN (314).

Figure 16.
Algorithm for the Management of Symptomatic Diabetic Neuropathy. Non-pharmacological, topical or physical therapies can be useful at any time. SNRIs, serotonin and norepinephrine reuptake inhibitors; TCA, tricyclic antidepressants.
AUTONOMIC NEUROPATHY
Introduction
The autonomic nervous system (ANS) supplies all organs in the body and consists of an afferent and an efferent system, with long efferents in the vagus (cholinergic) and short postganglionic unmyelinated fibers in the sympathetic system (adrenergic). A third component is the neuropeptidergic system with its neurotransmitters substance P (SP), vasoactive intestinal polypeptide (VIP), and calcitonin gene related peptide (CGRP) amongst others. Diabetic autonomic neuropathy (DAN) is a serious and common complication of diabetes but remains among the least recognized and understood. Diabetic autonomic neuropathy (DAN) can cause dysfunction of every part of the body, and has a significant negative impact on survival and quality of life (315). The organ systems that most often exhibit prominent clinical autonomic signs and symptoms in diabetes include the pupils, sweat glands, genitourinary system, gastrointestinal tract, adrenal medullary system, and the cardiovascular system (Table 6). Clinical symptoms generally do not appear until long after the onset of diabetes. However, subclinical autonomic dysfunction can occur within a year of diagnosis in type 2 diabetes patients and within two years in type 1 diabetes patients (316).
Table 6.
Clinical Manifestations of Autonomic Neuropathy
| Cardiovascular Central: Tachycardia/ Bradycardia Systolic and diastolic dysfunction Decreased exercise tolerance Orthostasis, Orthostatic tachycardia and bradycardia syndrome Sleep apnea Anxiety/ depression Cardiac denervation syndrome Paradoxic supine or nocturnal hypertension Intraoperative and perioperative cardiovascular instability Peripheral: Decreased thermoregulation Decreased sweating Altered blood flow Impaired vasomotion Edema |
| Gastrointestinal Esophageal dysmotility Gastroparesis diabeticorum Diarrhea Constipation Fecal incontinence |
| Genitourinary Erectile dysfunction Retrograde ejaculation Neurogenic bladder and cystopathy Female sexual dysfunction (e.g., loss of vaginal lubrication) |
| Sudomotor Anhidrosis Hyperhidrosis Heat intolerance Gustatory sweating Dry skin |
| Metabolic Hypoglycemia unawareness Hypoglycemia unresponsiveness |
| Pupillary Pupillomotor function impairment (e.g., decreased diameter of dark-adapted pupil) Pseudo Argyll-Robertson pupil |
Microvascular flow is under the control of the ANS and is regulated by both the central and peripheral components of the ANS. Defective blood flow in the small capillary circulation is found with decreased responsiveness to mental arithmetic, cold pressor, hand grip, and heating (317). The defect is associated with a reduction in the amplitude of vasomotion (318) and resembles premature aging (277). There are differences in the glabrous and hairy skin (319) and is correctable with antioxidants (320). The clinical counterpart is a dry cold skin, loss of sweating, and development of fissures and cracks that are portals of entry for organisms leading to infectious ulcers and gangrenes. Silent myocardial infarction, respiratory failure, amputations, and sudden death are hazards for diabetes patients with cardiac autonomic neuropathy (321). Therefore, it is vitally important to make this diagnosis early so that appropriate intervention can be instituted (322).
Disturbances in the autonomic nervous system may be functional, e.g., gastroparesis with hyperglycemia and ketoacidosis, or organic wherein nerve fibers are actually lost. This creates inordinate difficulties in diagnosing, treating, and prognosticating as well as establishing true prevalence rates. Tests of autonomic function generally stimulate entire reflex pathways. Furthermore, autonomic control for each organ system is usually divided between opposing sympathetic and parasympathetic innervations, so that heart rate acceleration, for example, may reflect either decreased parasympathetic or increased sympathetic nervous system stimulation. Since many conditions affect the autonomic nervous system and autonomic neuropathy (AN) is not unique to diabetes, the diagnosis of DAN rests with establishing the diagnosis and excluding other causes (Table 7 and 8). The best studied diagnostic methods, for which there are large databases and evidence to support their use in clinical practice, relate to the evaluation of cardiovascular reflexes (Figure 17). In addition, the evaluation of orthostasis is fairly straightforward and is readily done in clinical practice (Figure 18), as is the establishment of the cause of gastrointestinal symptoms (Figure 19) and erectile dysfunction. The combination of cardiovascular autonomic tests with sudomotor function tests may allow a more accurate diagnosis of diabetic autonomic neuropathy (323). Tables 9 and 10 below present the diagnostic tests that would be applicable to the diagnosis of cardiovascular autonomic neuropathy. These tests can be used as a surrogate for the diagnosis of AN of any system since it is generally rare to find involvement (although it does occur) of any other division of the ANS in the absence of cardiovascular autonomic dysfunction. For example, if one entertains the possibility that the patient has erectile dysfunction due to AN, then prior to embarking upon a sophisticated and expensive evaluation of erectile status, a measure of heart rate and its variability in response to deep breathing would - if normal - exclude the likelihood that the erectile dysfunction is a consequence of disease of the autonomic nervous system. The cause thereof would have to be sought elsewhere. Similarly, it is extremely unusual to find gastroparesis secondary to AN in a patient with normal cardiovascular autonomic reflexes.
Table 7.
Differential Diagnosis of Diabetic Autonomic Neuropathy
| Clinical Manifestations | Differential Diagnosis |
|---|---|
| Cardiovascular Resting tachycardia, Exercise intolerance Orthostatic tachycardia and bradycardia syndromes Cardiac denervation, painless myocardial infarction Orthostatic hypotension Intraoperative and perioperative cardiovascular instability | Cardiovascular disorders Idiopathic orthostatic hypotension, multiple system atrophy with Parkinsonism, orthostatic tachycardia, hyperadrenergic hypotension Shy-Drager syndrome Panhypopituitarism Pheochromocytoma Hypovolemia Congestive heart disease Carcinoid syndrome |
| Gastrointestinal Esophageal dysfunction Gastroparesis diabeticorum Diarrhea Constipation Fecal incontinence | Gastrointestinal disorders Obstruction Bezoars Secretory diarrhea (endocrine tumors) Biliary disease Psychogenic vomiting Medications |
| Genitourinary Erectile dysfunction Retrograde ejaculation Cystopathy Neurogenic bladder | Genitourinary disorders Genital and pelvic surgery Atherosclerotic vascular disease Medications Alcohol abuse |
| Neurovascular Heat intolerance Gustatory sweating Dry skin Impaired skin blood flow | Other causes of neurovascular dysfunction Chaga's disease Amyloidosis Arsenic |
| Metabolic Hypoglycemia unawareness Hypoglycemia unresponsiveness Hypoglycemia associated autonomic failure | Metabolic disorders Other cause of hypoglycemia, intensive glycemic control and drugs that mask hypoglycemia |
| Pupillary Decreased diameter of dark- adapted pupil Argyll-Robertson type pupil | Pupillary disorders Syphilis |
Table 8.
Diagnosis and Management of Autonomic Nerve Dysfunction
| Symptoms | Assessment Modalities | Management |
|---|---|---|
| Resting tachycardia, exercise intolerance, early fatigue and weakness with exercise | HRV, respiratory HRV, MUGA thallium scan, 123I MIBG scan | Graded supervised exercise, beta blockers, ACE-inhibitors |
| Postural hypotension, dizziness, lightheadedness, weakness, fatigue, syncope, tachycardia/bradycardia | HRV, blood pressure measurement lying and standing | Mechanical measures, clonidine, midodrine, octreotide, erythropoietin, pyridostigmine |
| Hyperhidrosis | Sympathetic/parasympathetic balance | Clonidine, amitryptylline, trihexyphenidyl, propantheline, or scopolamine ,botox, Glycopyrrolate |
Table 9.
Diagnostic Tests of Cardiovascular Autonomic Neuropathy
| TEST | METHOD/ PARAMETERS |
|---|---|
| Resting heart rate Beat-to-beat heart rate Variation* | >100 beats/min is abnormal. With the patient at rest and supine (no overnight coffee or hypoglycemic episodes), breathing 6 breaths/min, heart rate monitored by EKG or ANSCORE device, a difference in heart rate of >15 beats/min is normal and <10 beats/min is abnormal, R-R inspiration/R-R expiration >1.17. All indices of HRV are age-dependent**. |
| Heart rate response to Standing* | During continuous EKG monitoring, the R-R interval is measured at beats 15 and 30 after standing. Normally, a tachycardia is followed by reflex bradycardia. The 30:15 ratio is normally >1.03. |
| Heart rate response to Valsalva maneuver* | The subject forcibly exhales into the mouthpiece of a manometer to 40 mmHg for 15 s during EKG monitoring. Healthy subjects develop tachycardia and peripheral vasoconstriction during strain and an overshoot bradycardia and rise in blood pressure with release. The ratio of longest R-R shortest R-R should be >1.2. |
| Spectral analysis of heart rate variation, very low frequency power (VLFP 0.003-0.04) and high frequency power (HFP 0.15-0.40 Hz) | Series of sequential R-R intervals into its various frequent components. It defines two fixed spectral regions for the low-frequency and high-frequency measure. |
| Systolic blood pressure response to standing | Systolic blood pressure is measured in the supine subject. The patient stands and the systolic blood pressure is measured after 2 min. Normal response is a fall of <10 mmHg, borderline is a fall of 10-29 mmHg, and abnormal is a fall of >30 mmHg with symptoms. |
| Diastolic blood pressure response to isometric exercise | The subject squeezes a handgrip dynamometer to establish a maximum. Grip is then squeezed at 30% maximum for 5 min. The normal response for diastolic blood pressure is a rise of >16 mmHg in the other arm. |
| EKG QT/QTc intervals Spectral analysis with respiratory frequency | The QTc (corrected QT interval on EKG) should be <440 ms. VLF peak (sympathetic dysfunction) LF peak (sympathetic dysfunction) HF peak (parasympathetic dysfunction) LH/HF ratio (sympathetic imbalance) |
| Neurovascular flow | Using noninvasive laser Doppler measures of peripheral sympathetic responses to nociception. |
- *
These can now be performed quickly (<15 min) in the practitioners' office, with a central reference laboratory providing quality control and normative values. LF, VLF, HF =low, very low and high frequency peaks on spectral analysis. These are now readily available in most cardiologist's practice.** Lowest normal value of E/I ratio: Age 20-24:1.17, 25-29:1.15, 30-34:1.13, 35-30:1.12, 40-44:1.10, 45-49:1.08, 50-54:1.07, 55-59:1.06, 60-64:1.04, 65-69:1.03, 70-75:1.02 .
Table 10.
Diagnostic Assessment of Cardiovascular Autonomic Function
| Parasympathetic | Sympathetic |
|---|---|
| Resting heart rate Beat to beat variation with deep breathing (E:I ratio) 30:15 heart rate ratio with standing Valsalva ratio Spectral analysis of heart rate variation , high frequency power (HFP 0.15-0.40 Hz) Spectral Analysis of HRV respiratory frequency | Resting heart rate Spectral analysis of heart rate variation, very low frequency power (VLFP 0.003-0.04) Orthostasis BP Hand grip BP Cold pressor response Sympathetic skin galvanic response (cholinergic) Sudorimetry (cholinergic) Cutaneous blood flow (peptidergic) |

Figure 17.
This is a sample power spectrum of the HRV signal from a subject breathing at an average rate of 7.5 breaths per minute (Fundamental Respiratory Frequency, FRF = 0.125 Hz). The method using HRV alone defines two fixed spectral regions for the low-frequency (LF) and high-frequency (HF) measure (dark gray and light gray, respectively). It is clear that the high-frequency (light gray) region includes very little area under the HRV spectral curve, suggesting very little parasympathetic activity. The great majority of the HRV spectral activity is under the low-frequency (dark gray) region suggesting primarily sympathetic activity. These representations are incorrect because the slow-breathing subject should have a large parasympathetic component reflective of the vagal activity. This parasympathetic component is represented correctly by the method using both HRV and respiratory activity which defines the red and blue regions of the spectrum in the graph. The blue region defined by the FRF represents purely parasympathetic activity whereas the remainder of the lower frequency regions (red region) represents purely sympathetic activity.

Figure 18.
Evaluation of postural dizziness in patients with diabetes

Figure 19.
Evaluation of a patient with suspected gastroparesis
The role of over-activation of the autonomic nervous system is illustrated in Figure 20 (324).
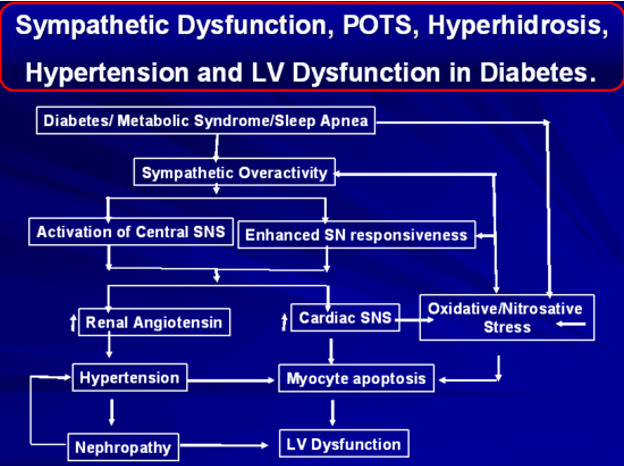
Figure 20.
Role of over-activation of autonomic nervous system
There are few data on the longitudinal trends in small fiber dysfunction. Much remains to be learned of the natural history of diabetic autonomic neuropathy. Karamitsos et al (325) reported that the progression of diabetic autonomic neuropathy is significant during the 2 years subsequent to its discovery.
The mortality for diabetic autonomic neuropathy has been estimated to be 44% within 2.5 years of diagnosing symptomatic autonomic neuropathy (29). In a meta-analysis, the Mantel-Haenszel estimates for the pooled prevalence rate risk for silent myocardial ischemia was 1.96, with 95% confidence interval of 1.53 to 2.51 (p<0.001; n = 1,468 total subjects). Thus, a consistent association between CAN and the presence of silent myocardial ischemia was shown (284) (Figure 21).

Figure 21.
Relative risks and 95% CIs for studies of cardiovascular neuropathy (CAN) and mortality. Pooled relative risk for 10 studies with CAN define by two or more measures: 3.45 (95% CI 2.66–4.47). Pooled relative risk for 4 studies with CAN defined by a single measure: 1.20 (1.02–1.41). REF: Maser, R. E., Mitchell, B. D., Vinik, A. I., and Freeman, R. Diabetes Care. 2003;26(6):1895-1901.
Prevention and Reversibility of Autonomic Neuropathy
It has now become clear that strict glycemic control (37) and a stepwise progressive management of hyperglycemia, lipids, and blood pressure as well as the use of antioxidants (326) and ACE inhibitors (327) reduce the odds ratio for autonomic neuropathy to 0.32 (328). It has also been shown that early mortality is a function of loss of beat-to-beat variability with MI. This can be reduced by 33% with acute administration of insulin (329). Kendall et al (330) reported that successful pancreas transplantation improves epinephrine response and normalizes hypoglycemia symptom recognition in patients with long standing diabetes and established autonomic neuropathy. Burger et al (331) showed that a reversible metabolic component of CAN exists in patients with early CAN.
Management of Autonomic Neuropathy
POSTURAL HYPOTENSION
The syndrome of postural hypotension is posture-related dizziness and syncope. Patients who have Type 2 diabetes mellitus and orthostatic hypotension are hypovolemic and have sympathoadrenal insufficiency; both factors contribute to the pathogenesis of orthostatic hypotension (332). Postural hypotension in the patient with diabetic autonomic neuropathy can present a difficult management problem. Elevating the blood pressure in the standing position must be balanced against preventing hypertension in the supine position.
Supportive Garments: Whenever possible, attempts should be made to increase venous return from the periphery using total body stockings. But leg compression alone is less effective, presumably reflecting the large capacity of the abdomen relative to the legs (333). Patients should be instructed to put them on while lying down and to not remove them until returning to the supine position.
Drug Therapy: Some patients with postural hypotension may benefit from treatment with 9-flurohydrocortisone. Unfortunately, symptoms do not improve until edema occurs, and there is a significant risk of developing congestive heart failure and hypertension. If fluorohydrocortisone does not work satisfactorily, various adrenergic agonists and antagonists may be used (Table 11). Enhancement of ganglionic transmission via the use of pyridostigmine (inhibitor of acetylcholinesterase) improved symptoms and orthostatic hypotension with only modest effects in supine BP for patients with POTS. Similarly, the use of β-adrenergic blockers may benefit the tachycardia, and anticholinergics, the orthostatic bradycardia. Pyridostigmine has also been shown to improve HRV in healthy young adults. If the adrenergic receptor status is known, then therapy can be guided to the appropriate agent. Metoclopramide may be helpful in patients with dopamine excess or increased sensitivity to dopaminergic stimulation. Patients with α2-adrenergic receptor excess may respond to the α2-antagonist yohimbine. Those few patients in whom ß-receptors are increased may be helped with propranolol. α2-adrenergic receptor deficiency can be treated with the α2-agonist clonidine, which in this setting may paradoxically increase blood pressure. One should start with small doses and gradually increase the dose. If the preceding measures fail, midodrine, an α1-adrenergic agonist, or dihydroergotamine in combination with caffeine may help. A particularly refractory form of postural hypotension occurs in some patients post-prandially and may respond to therapy with octreotide given subcutaneously in the mornings.
Table 11.
Pharmacologic Treatment of Autonomic Neuropathy
| Clinical status | Drug | Dosage | Side effects |
|---|---|---|---|
| Orthostatic hypotension | |||
| 9α flouro hydrocortisone, mineralocorticoid | 0.5-2 mg/day | Congestive heart failure, hypertension | |
| Clonidine, α2 adrenergic agonist | 0,1-0,5 mg, at bedtime | Orthostatic Hypotension, sedation, dry mouth, constipation, dizziness, bradycardia. | |
| Octreotide, somatostatin analogue | 0.1-0.5 mg/kg/day | Injection site pain, diarrhea | |
| Orthostatic tachycardia and bradycardia syndrome | |||
| Clonidine, α2 adrenergic agonist | 0.1-0.5 mg, at bedtime | Orthostatic Hypotension, sedation, dry mouth, constipation, dizziness, bradycardia. | |
| Octreotide, somatostatin analogue | 0.1-0.5 μg/kg/day | Injection site pain, diarrhea | |
| Gastroparesis diabeticorum | |||
| Domperidone, D2-receptor antagonist | 10-20 mg, 30-60 min before meal and bedtime | Galactorrhea | |
| Erythromycin, motilin receptor agonist | 250 mg, 30 minutes before meals | Abdominal cramps, nausea, diarrhea, rash | |
| Levosulphide, D2-receptor antagonist | 25 mg, 3 times/day | Galactorrhea | |
| Diabetic diarrhea | |||
| Metronidazole, broad spectrum antibiotics | 250 mg, 3 times/day, minimum 3 weeks | Anorexia, rash, GI upset, urine discoloration, dizziness, disulfiram like reaction. | |
| Clonidine, α2 adrenergic agonist | 0.1 mg, 2-3 times/day | Orthostatic Hypotension, sedation, dry mouth, constipation, dizziness, bradycardia. | |
| Cholestyramine, bile acid sequestrant | 4 g, 1-6 times/day | Constipation | |
| Loperamide, opiate-receptor agonist | 2 mg, four times/day | Toxic megacolon | |
| Octreotide, somatostatin analogue | 50 μg, 3 times/day | Aggravate nutrient malabsorption (at higher doses) | |
| Cystopathy | |||
| Bethanechol, acetylcholine receptor agonist | 10 mg, 4 times/day | Blurred vision, abdominal cramps, diarrhea, salivation, and hypotension. | |
| Doxazosin, α1 adrenergic antagonist | 1-2 mg, 2-3 times/day | Hypotension, headache, palpitation | |
| Exercise Intolerance | |||
| Graded supervised exercise | 20 minutes, 3 times/week | Foot injury, angina. | |
| Hyperhidrosis | |||
| Clonidine, α2 adrenergic agonist | 0.1-0.5 mg, at bedtime and divided doses above 0.2 mg | Orthostatic Hypotension, sedation, dry mouth, constipation, dizziness, bradycardia. | |
| Amitryptiline, Norepinephrine & serotonin reuptake inhibitor | 150 mg/ day | Tachycardia, palpitation | |
| Propantheline, Anti-muscarinic. | 15 mg/ day PO | Dry mouth, blurred vision | |
| Trihexyphenidyl, | 2-5 mg PO | Dry mouth, blurred vision, constipation, tachycardia, photosensitivity, arrhythmias. | |
| Botox, | |||
| Scopolamine, anti-cholinergic | 1.5 mg patch/ 3 days; 0.4 to 0.8mg PO | Dry mouth, blurred vision, constipation, drowsiness, and tachycardia. | |
| Glycopyrrolate, anti-cholinergic | 1-2 mg, 2-3 times daily. | Constipation, tachycardia, dry mouth. | |
| Erectile dysfunction | |||
| Sildenafil (Viagra), GMP type-5 phosphodiesterase inhibitor | 50 mg before sexual activity, only once per day | Hypotension and fatal cardiac event (with nitrate-containing drugs), headache, flushing, nasal congestion, dyspepsia, musculoskeletal pain, blurred vision | |
| Tadalafil (Cialis), GMP type-5 phosphodiesterase inhibitor | 10 mg PO before sexual activity only once per day. | Headache, flushing, dyspepsia, rhinitis, myalgia, back pain. | |
| Verdenafil (Levitra), GMP type-5 phosphodiesterase inhibitor | 10 mg PO, 60 minutes before sexual activity. | Hypotension, headache, dyspepsia, priapism. | |
SLEEP APNEA
During sleep, increased sympathetic drive is a result of repetitive episodes of hypoxia, hypercapnia, and obstructive apnea (OSA) acting through chemoreceptor reflexes. Increased sympathetic drive has been implicated in increased blood pressure variability with repetitive sympathetic activation and blood pressure surges impairing baroreflex and cardiovascular reflex functions (284). A direct relationship between the severity of OSA and the increase in blood pressure has been noted. Furthermore, the use of continuous positive airway pressure (CPAP) for the treatment of OSA has been shown to lower blood pressure and improve cardiovascular autonomic nerve fiber function for individuals with OSA. Withdrawal of CPAP for even a short period (i.e., 1 week) has been shown to result in a marked increase in sympathetic activity (284).
GASTROPATHY
Gastrointestinal motor disorders are frequent and widespread in patients with type 2 diabetes, regardless of symptoms (334) and there is a poor correlation between symptoms and objective evidence of a functional or organic defect. The first step in management of diabetic gastroparesis consists of multiple, small feedings; decreased fat intake as it tends to delay gastric emptying; maintenance of glycemic control (335,336); and a low-fiber diet to avoid bezoar formation. Metoclopramide may be used. Domperidone (337,338) has been shown to be effective in some patients, although probably no more so than metoclopramide. Erythromycin given as either a liquid or suppository also may be helpful. Erythromycin acts on the motilin receptor, "the sweeper of the gut," and shortens gastric emptying time (339). Several novel drugs, including the ghrelin (orexigenic hormone) and ghrelin receptor agonists, motilin agonist (mitemcinal), 5-HT4-receptor agonists and the muscarinic antagonist are being investigated for their prokinetic effects (340,341). If medications fail and severe gastroparesis persists, jejunostomy placement into normally functioning bowel may be needed. Different treatment modalities for gastroparesis include dietary modifications, prokinetic and antiemetic medications, measures to control pain and address psychological issues, and endoscopic or surgical options in selected instances (342).
For additional information see the Endotext chapter entitled “Gastrointestinal Disorders in Diabetes”.
ENTEROPATHY
Enteropathy involving the small bowel and colon can produce both chronic constipation and explosive diabetic diarrhea, making treatment of this complication difficult.
Antibiotics: Stasis of bowel contents with bacterial overgrowth may contribute to the diarrhea. Treatment with broad-spectrum antibiotics is the mainstay of therapy, including tetracycline or trimethoprim and sulfamethoxazole. Metronidazole appears to be the most effective and should be continued for at least 3 weeks.
Cholestyramine: Retention of bile may occur and can be highly irritating to the gut. Chelation of bile salts with cholestyramine 4g tid mixed with fluid may offer relief of symptoms.
Diphenoxylate plus atropine: Diphenoxylate plus atropine may help to control the diarrhea; however, toxic megacolon can occur, and extreme care should be used.
Diet: Patients with poor digestion may benefit from a gluten-free diet, while constipation may respond to a high-soluble-fiber diet supplemented with daily hydrophilic colloid. Beware of certain fibers in the neuropathic patient that can lead to bezoar formation because of bowel stasis in gastroparetic or constipated patients.
For additional information see the Endotext chapter entitled “Gastrointestinal Disorders in Diabetes”.
SEXUAL DYSFUNCTION
Erectile dysfunction (ED) occurs in 50-75% of men with diabetes, and it tends to occur at an earlier age than in the general population. The incidence of ED in men with diabetes aged 20-29 years is 9% and increases to 95% by age 70. It may be the presenting symptom of diabetes. More than 50% notice the onset of ED within 10 years of the diagnosis, but it may precede the other complications of diabetes. The etiology of ED in diabetes is multifactorial. Neuropathy, vascular disease, diabetes control, nutrition, endocrine disorders, psychogenic factors as well as drugs used in the treatment of diabetes and its complications play a role (343,344). The diagnosis of the cause of ED is made by a logical stepwise progression in all instances. An approach to therapy has been presented to which the reader is referred; Figure 22 below shows a flow chart modified from Vinik et. al., 1998 (302).

Figure 22.
Evaluation of patients with diabetes with erectile dysfunction
A thorough work-up for impotence will include: medical and sexual history; physical and psychological evaluations; blood tests for diabetes and levels of testosterone, prolactin, and thyroid hormones; tests for nocturnal erections; tests to assess penile, pelvic, and spinal nerve function; and a test to assess penile blood supply and blood pressure. The flow chart provided is intended as a guide to assist in defining the problem. The healthcare provider should initiate questions that will help distinguish the various forms of organic erectile dysfunction from those that are psychogenic in origin. Physical examination must include an evaluation of the autonomic nervous system, vascular supply, and the hypothalamic-pituitary-gonadal axis.
Autonomic neuropathy causing ED is almost always accompanied by loss of ankle jerks and absence or reduction of vibration sense over the large toes. More direct evidence of impairment of penile autonomic function can be obtained by (1) demonstrating normal perianal sensation, (2) assessing the tone of the anal sphincter during a rectal exam, and (3) ascertaining the presence of an anal wink when the area of the skin adjacent to the anus is stroked or contraction of the anus when the glans penis is squeezed, i.e., the bulbo-cavernosus reflex. These measurements are easily and quickly done at the bedside and reflect the integrity of sacral parasympathetic divisions.
Vascular disease is usually manifested by buttock claudication but may be due to stenosis of the internal pudendal artery. A penile/brachial index of <0.7 indicates diminished blood supply. A venous leak manifests as unresponsiveness to vasodilators and needs to be evaluated by penile Doppler sonography.
In order to distinguish psychogenic from organic erectile dysfunction, nocturnal penile tumescence (NPT) measurement can be done. Normal NPT defines psychogenic ED, and a negative response to vasodilators implies vascular insufficiency. Application of NPT is not so simple. It is much like having a sphygmomanometer cuff inflate over the penis many times during the night while one is trying to have a normal night's sleep and the REM sleep associated with erections. The individual may have to take home the device and become familiar with it over several nights before one has a reliable estimate of the failure of NPT.
Treatment of Erectile Dysfunction
A number of treatment modalities are available and each treatment has positive and negative effects; therefore, patients must be made aware of both aspects before a therapeutic decision is made. Before considering any form of treatment, every effort should be made to have the patient withdraw from alcohol and eliminate smoking. If possible, drugs that are known to cause erectile dysfunction should be removed. Additionally, metabolic control should be optimized.
Relaxation of the corpus cavernous smooth muscle cells is caused by NO and cGMP, and the ability to have and maintain an erection depends on NO and cGMP. The peripherally acting oral phosphodiesterase type 5 (PDE5) inhibitors block the action of PDE5, and cGMP accumulates, enhancing blood flow to the corpora cavernosum with sexual stimulation. This class of agents consists of sildenafil, vardenafil, and tadalafil. They have been evaluated in patients with diabetes with similar levels of efficacy of about 70%. A 50 mg tablet of sildenafil taken orally is the usual starting dose, 60 minutes before sexual activity. Lower doses should be considered in patients with renal failure and hepatic dysfunction. The duration of the drug effect is 4 hours. Generally, patients with diabetes require the maximum dose of each agent, sildenafil 100 mg, tadalafil 20 mg, and vardenafil 20 mg. Before prescribing a PDE5 inhibitor, it is important to exclude ischemic heart disease. These are absolutely contraindicated in patients being treated with nitroglycerine or other nitrate-containing drugs. Severe hypotension and fatal cardiac events can occur (345). Side-effects include headache, flushing, dyspepsia, and muscle pain (346). Direct injection of prostacyclin into the corpus cavernosum will induce satisfactory erections in a significant number of men. Also, surgical implantation of a penile prosthesis may be appropriate. The less expensive type of prosthesis is a semirigid, permanently erect type that may be embarrassing and uncomfortable for some patients. The inflatable type is three times more expensive and subject to mechanical failure, but it avoids the embarrassment caused by other devices.
Female Sexual Dysfunction
Women with diabetes mellitus may experience decreased sexual desire and more pain on sexual intercourse, and they are at risk of decreased sexual arousal, with inadequate lubrication (347). Diagnosis of female sexual dysfunction using vaginal plethysmography to measure lubrication and vaginal flushing has not been well established.
For additional information on this topic see the Endotext chapter entitled “Sexual Dysfunction in Diabetes”.
CYSTOPATHY
In diabetic autonomic neuropathy, the motor function of the bladder is unimpaired, but afferent fiber damage results in diminished bladder sensation. The urinary bladder can be enlarged to more than three times its normal size. Patients are seen with bladders filled to their umbilicus, yet they feel no discomfort. Loss of bladder sensation occurs with diminished voiding frequency, and the patient is no longer able to void completely. Consequently, dribbling and overflow incontinence are common complaints. A post-void residual of greater than 150cc is diagnostic of cystopathy. Cystopathy may put the patients at risk for urinary infections.
Treatment of Cystopathy
Patients with cystopathy should be instructed to palpate their bladder and, if they are unable to initiate micturition when their bladders are full, use Crede's maneuver (massage or pressure on the lower portion of abdomen just above the pubic bone) to start the flow of urine. The principal aim of the treatment should be to improve bladder emptying and to reduce the risk of urinary tract infection. Parasympathomimetics such as bethanechol are sometimes helpful, although frequently they do not help to fully empty the bladder. Extended sphincter relaxation can be achieved with an alpha-1-blocker, such as doxazosin. Self-catheterization can be particularly useful in this setting, with the risk of infection generally being low.
SWEATING DYSFUNCTION
Hyperhidrosis of the upper body, often related to eating (gustatory sweating), and anhidrosis of the lower body, are a characteristic feature of autonomic neuropathy. Gustatory sweating accompanies the ingestion of certain foods, particularly spicy foods, and cheeses. There is a suggestion that application of glycopyrrolate (an antimuscarinic compound) might benefit diabetes patients with gustatory sweating (348). Low-dose oral glycopyrrolate in the range of 1 mg to 2 mg once daily can be tolerated without problematic adverse effects to alleviate the symptoms of diabetic gustatory sweating. Although more long-term data are needed, the use of glycopyrrolate for diabetic gustatory sweating may be a viable option (349). Symptomatic relief can be obtained by avoiding the specific inciting food. Loss of lower body sweating can cause dry, brittle skin that cracks easily, predisposing one to ulcer formation that can lead to loss of the limb. Special attention must be paid to foot care.
METABOLIC DYSFUNCTION
Hypoglycemia Unawareness
Blood glucose concentration is normally maintained during starvation or increased insulin action by an asymptomatic parasympathetic response with bradycardia and mild hypotension, followed by a sympathetic response with glucagon and epinephrine secretion for short-term glucose counter regulation, and growth hormone and cortisol secretion for long-term regulation. The release of catecholamine alerts the patient to take the required measures to prevent coma due to low blood glucose. The absence of warning signs of impending neuroglycopenia is known as "hypoglycemic unawareness". The failure of glucose counter regulation can be confirmed by the absence of glucagon and epinephrine responses to hypoglycemia induced by a standard, controlled dose of insulin (350).
In patients with type 1 diabetes mellitus, the glucagon response is impaired with diabetes duration of 1-5 years; after 14-31 years of diabetes, the glucagon response is almost undetectable. Absence of the glucagon response is not present in those with autonomic neuropathy. However, a syndrome of hypoglycemic autonomic failure occurs with intensification of diabetes control and repeated episodes of hypoglycemia. The exact mechanism is not understood, but it does represent a real barrier to physiologic glycemic control. In the absence of severe autonomic dysfunction, hypoglycemia unawareness is at least in part reversible.
Patients with hypoglycemia unawareness and unresponsiveness pose a significant management problem for the physician. Although autonomic neuropathy may improve with intensive therapy and normalization of blood glucose, there is a risk to the patient, who may become hypoglycemic without being aware of it and who cannot mount a counterregulatory response. It is our recommendation that if a pump is used, boluses of smaller than calculated amounts should be used and, if intensive conventional therapy is used, long-acting insulin with very small boluses should be given. In general, normal glucose and HbA1 levels should not be goals in these patients to avoid the possibility of hypoglycemia. The use of continuous glucose monitoring with hypoglycemic alarms can be very helpful in warning patients of hypoglycemia and in preventing severe hypoglycemic reactions.
Further complicating management of some patients with diabetes is the development of a functional autonomic insufficiency associated with intensive insulin treatment, which resembles autonomic neuropathy in all relevant aspects. In these instances, it is prudent to relax therapy, as for the patient with bona fide autonomic neuropathy. If hypoglycemia occurs in these patients at a certain glucose level, it will take a lower glucose level to trigger the same symptoms in the next 24-48 hours. Avoidance of hypoglycemia for a few days will result in recovery of the adrenergic response.
For additional information on this topic see the Endotext chapter entitled “Hypoglycemia During Therapy of Diabetes”.
DIABETIC NEUROPATHIES: PROSPECTS FOR THE FUTURE
Management of DN encompasses a wide variety of therapies. Treatment must be individualized in a manner that addresses the particular manifestation and underlying pathogenesis of each patient's unique clinical presentation, without subjecting the patient to untoward medication effects. An increased understanding of the pathogenesis of DN will lead to more effective approaches to diagnose and treat this condition. Refinements and adoption of new approaches to measure quantitatively and diagnose DN early is crucial, so that appropriate therapies (risk factor modification or pathogenic) can be commenced before nerve damage is established. These tests must be validated and standardized to allow comparability between studies and a more meaningful interpretation of study results. Our ability to manage successfully the many different manifestations of DN depends ultimately on our success in uncovering the pathogenic processes underlying this disorder.
ACKNOWLEDGEMENTS
This chapter updates the original Endotext chapter on this topic written by Aaron Vinik, Carolina Casellini, and Marie-Laure Nevoret.
REFERENCES
- 1.
- Saeedi P., et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas. Diabetes research and clinical practice. 2019;157:107843. p. [PubMed: 31518657]
- 2.
- Williams R., et al. Global and regional estimates and projections of diabetes-related health expenditure: Results from the International Diabetes Federation Diabetes Atlas. Diabetes research and clinical practice. 2020;162:108072. p. [PubMed: 32061820]
- 3.
- Moxey P., et al. Lower extremity amputations—a review of global variability in incidence. Diabetic Medicine. 2011;28(10):1144–1153. [PubMed: 21388445]
- 4.
- Paisey R., et al. Diabetes‐related major lower limb amputation incidence is strongly related to diabetic foot service provision and improves with enhancement of services: peer review of the South‐West of England. Diabetic Medicine. 2018;35(1):53–62. [PMC free article: PMC5765400] [PubMed: 29023974]
- 5.
- Programme Budgeting. 2014 [cited 2019 08/10/2019]; Available from: https://bit
.ly/31WeHc5. - 6.
- Tesfaye S., et al. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes care. 2010;33(10):2285–2293. [PMC free article: PMC2945176] [PubMed: 20876709]
- 7.
- Ziegler D. Painful diabetic neuropathy: treatment and future aspects. Diabetes/metabolism research and reviews. 2008;24(S1):S52–S57. [PubMed: 18395890]
- 8.
- Boulton A.J., et al. Diabetic somatic neuropathies. Diabetes care. 2004;27(6):1458–1486. [PubMed: 15161806]
- 9.
- Vinik A.I., Ziegler D. Diabetic cardiovascular autonomic neuropathy. Circulation. 2007;115(3):387–397. [PubMed: 17242296]
- 10.
- Vinik, A.I., R.E. Maser, and D. Ziegler, Neuropathy: the crystal ball for cardiovascular disease? 2010, Am Diabetes Assoc. p. 1688-1690. [PMC free article: PMC2890382] [PubMed: 20587730]
- 11.
- Pop-Busui R., et al. Effects of cardiac autonomic dysfunction on mortality risk in the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Diabetes care. 2010;33(7):1578–1584. [PMC free article: PMC2890362] [PubMed: 20215456]
- 12.
- Abbott C.A., et al. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the UK. Diabetes care. 2011;34(10):2220–2224. [PMC free article: PMC3177727] [PubMed: 21852677]
- 13.
- Selvarajah D., et al. The contributors of emotional distress in painful diabetic neuropathy. Diabetes and Vascular Disease Research. 2014;11(4):218–225. [PubMed: 24821753]
- 14.
- Giordano J., Schatman M.E. A crisis in chronic pain care: an ethical analysis. Part three: toward an integrative, multi-disciplinary pain medicine built around the needs of the patient. Pain Physician. 2008;11(6):775–784. [PubMed: 19057625]
- 15.
- Thomas P. Classification, differential diagnosis, and staging of diabetic peripheral neuropathy. Diabetes. 1997;46 Supplement 2:S54–S57. [PubMed: 9285500]
- 16.
- Pop-Busui R., et al. Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes care. 2017;40(1):136–154. [PMC free article: PMC6977405] [PubMed: 27999003]
- 17.
- Ziegler D., et al. Somatic and autonomic nerve function during the first year after diagnosis of type 1 (insulin-dependent) diabetes. Diabetes Research (Edinburgh, Scotland). 1988;7(3):123–127. [PubMed: 3046819]
- 18.
- Papanas N., Ziegler D. Prediabetic neuropathy: does it exist? Current diabetes reports. 2012;12(4):376–383. [PubMed: 22562652]
- 19.
- The effect of intensive diabetes therapy on the development and progression of neuropathy. The Diabetes Control and Complications Trial Research Group. Ann Intern Med. 1995;122(8):561–8. [PubMed: 7887548]
- 20.
- Ziegler D., et al. The natural course of peripheral and autonomic neural function during the first two years after diagnosis of type 1 diabetes. Klinische Wochenschrift. 1988;66(21):1085–1092. [PubMed: 2853250]
- 21.
- Dyck P., et al. The Rochester Diabetic Neuropathy Study: design, criteria for types of neuropathy, selection bias, and reproducibility of neuropathic tests. Neurology. 1991;41(6):799–799. [PubMed: 2046920]
- 22.
- Casellini C.M., et al. A 6-month, randomized, double-masked, placebo-controlled study evaluating the effects of the protein kinase C-β inhibitor ruboxistaurin on skin microvascular blood flow and other measures of diabetic peripheral neuropathy. Diabetes care. 2007;30(4):896–902. [PubMed: 17392551]
- 23.
- Chalk, C., T.J. Benstead, and F. Moore, Aldose reductase inhibitors for the treatment of diabetic polyneuropathy. Cochrane database of systematic reviews, 2007(4). [PMC free article: PMC8406996] [PubMed: 17943821]
- 24.
- Ziegler D., et al. Epidemiology of polyneuropathy in diabetes and prediabetes. Handbook of clinical neurology. 2014;126:3–22. [PubMed: 25410210]
- 25.
- Hicks C.W., Selvin E. Epidemiology of peripheral neuropathy and lower extremity disease in diabetes. Current diabetes reports. 2019;19(10):1–8. [PMC free article: PMC6755905] [PubMed: 31456118]
- 26.
- Andersen S.T., et al. Risk factors for incident diabetic polyneuropathy in a cohort with screen-detected type 2 diabetes followed for 13 years: ADDITION-Denmark. Diabetes care. 2018;41(5):1068–1075. [PubMed: 29487078]
- 27.
- Mizokami-Stout K.R., et al. The contemporary prevalence of diabetic neuropathy in type 1 diabetes: findings from the T1D Exchange. Diabetes care. 2020;43(4):806–812. [PMC free article: PMC7085805] [PubMed: 32029635]
- 28.
- Partanen J., et al. Natural history of peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. New England Journal of Medicine. 1995;333(2):89–94. [PubMed: 7777034]
- 29.
- Martin C.L., et al. Neuropathy and related findings in the diabetes control and complications trial/epidemiology of diabetes interventions and complications study. Diabetes care. 2014;37(1):31–38. [PMC free article: PMC3868000] [PubMed: 24356595]
- 30.
- Dyck P.J., et al. The prevalence by staged severity of various types of diabetic neuropathy, retinopathy, and nephropathy in a population‐based cohort: the Rochester Diabetic Neuropathy Study. Neurology. 1993;43(4):817–817. [PubMed: 8469345]
- 31.
- Armstrong D.G., Lavery L.A., Harkless L.B. Validation of a diabetic wound classification system: the contribution of depth, infection, and ischemia to risk of amputation. Diabetes care. 1998;21(5):855–859. [PubMed: 9589255]
- 32.
- East, M. and N. Africa, IDF diabetes atlas. diabetes, 2017. 20: p. 79.
- 33.
- Hoffstad O., et al. Diabetes, lower-extremity amputation, and death. Diabetes Care. 2015;38(10):1852–1857. [PubMed: 26203063]
- 34.
- Diabetes, U., Twenty devastating amputations ever day. 2018, Retrieved on 12th March.
- 35.
- Cavanagh P., et al. Cost of treating diabetic foot ulcers in five different countries. Diabetes/metabolism research and reviews. 2012;28:107–111. [PubMed: 22271734]
- 36.
- Brownlee M., Hirsch I.B. Glycemic variability: a hemoglobin A1c–independent risk factor for diabetic complications. Jama. 2006;295(14):1707–1708. [PubMed: 16609094]
- 37.
- DCCT Research Group. B.N.D., Bethesda, MD 20892., Effect of intensive diabetes treatment on nerve conduction in the Diabetes Control and Complications Trial. Annals of Neurology. 1995;38(6):869–880. [PubMed: 8526459]
- 38.
- Callaghan, B.C., et al., Enhanced glucose control for preventing and treating diabetic neuropathy. Cochrane database of systematic reviews, 2012(6). [PMC free article: PMC4048127] [PubMed: 22696371]
- 39.
- Ismail-Beigi F., et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: an analysis of the ACCORD randomised trial. The Lancet. 2010;376(9739):419–430. [PMC free article: PMC4123233] [PubMed: 20594588]
- 40.
- Calles-Escandón J., et al. Effect of intensive compared with standard glycemia treatment strategies on mortality by baseline subgroup characteristics: the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial. Diabetes care. 2010;33(4):721–727. [PMC free article: PMC2845012] [PubMed: 20103550]
- 41.
- Tesfaye S., et al. Vascular risk factors and diabetic neuropathy. New England Journal of Medicine. 2005;352(4):341–350. [PubMed: 15673800]
- 42.
- Vincent A.M., et al. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nature Reviews Neurology. 2011;7(10):573–583. [PubMed: 21912405]
- 43.
- Boulton A.J., et al. Whither pathogenetic treatments for diabetic polyneuropathy? Diabetes/Metabolism Research and Reviews. 2013;29(5):327–333. [PubMed: 23381942]
- 44.
- Vinik A., et al. Diabetic neuropathies: clinical manifestations and current treatment options. Nature clinical practice Endocrinology & metabolism. 2006;2(5):269–281. [PubMed: 16932298]
- 45.
- Feldman E.L., et al. Diabetic neuropathy. Nature reviews Disease primers. 2019;5(1):1–18. [PubMed: 31197153]
- 46.
- Vinik A., et al. Focal entrapment neuropathies in diabetes. Diabetes care. 2004;27(7):1783–1788. [PubMed: 15220266]
- 47.
- Wilbourn, A., Diabetic entrapment and compression neuropathies. Diabetic neuropathy, 1999: p. 481-508.
- 48.
- Watanabe K., et al. Characteristics of cranial nerve palsies in diabetic patients. Diabetes research and clinical practice. 1990;10(1):19–27. [PubMed: 2249603]
- 49.
- Perkins B.A., Olaleye D., Bril V. Carpal tunnel syndrome in patients with diabetic polyneuropathy. Diabetes Care. 2002;25(3):565–569. [PubMed: 11874948]
- 50.
- Dawson D.M. Entrapment neuropathies of the upper extremities. New England Journal of Medicine. 1993;329(27):2013–2018. [PubMed: 8247077]
- 51.
- Rota E., Morelli N. Entrapment neuropathies in diabetes mellitus. World journal of diabetes. 2016;7(17):342–353. [PMC free article: PMC5027001] [PubMed: 27660694]
- 52.
- Llewelyn J., Thomas P., King R. Epineurial microvasculitis in proximal diabetic neuropathy. Journal of neurology. 1998;245(3):159–165. [PubMed: 9553846]
- 53.
- Dyck P.J.B., Windebank A.J. Diabetic and nondiabetic lumbosacral radiculoplexus neuropathies: new insights into pathophysiology and treatment. Muscle & nerve. 2002;25(4):477–491. [PubMed: 11932965]
- 54.
- Roberts A., James J., Dhatariya K. Management of hyperglycaemia and steroid (glucocorticoid) therapy: a guideline from the Joint British Diabetes Societies (JBDS) for Inpatient Care group. Diabet Med. 2018;35(8):1011–1017. [PubMed: 30152586]
- 55.
- Vinik, A.I., G.L. Pittenger, and L. Hopkins, Autoimmune mechanisms in the pathogenesis of diabetic neuropathy. 1998.
- 56.
- Steck A.J., Kappos L. Gangliosides and autoimmune neuropathies: classification and clinical aspects of autoimmune neuropathies. Journal of neurology, neurosurgery, and psychiatry. 1994;57 Suppl:26. [PMC free article: PMC1016719] [PubMed: 7964847]
- 57.
- Said G., et al. Nerve biopsy findings in different patterns of proximal diabetic neuropathy. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 1994;35(5):559–569. [PubMed: 8179302]
- 58.
- Sander, H.W. and S. Chokroverty. Diabetic amyotrophy: current concepts. in Seminars in neurology. 1996. © 1996 by Thieme Medical Publishers, Inc. [PubMed: 8987131]
- 59.
- Kendrel DA. D.C., LC Hopkins, Successful treatment of neuropathies in patients with diabetes mellitus. Arch Neurol. 1995;52:1053–1061. [PubMed: 7487556]
- 60.
- Britland S.T., et al. Acute and remitting painful diabetic polyneuropathy: a comparison of peripheral nerve fibre pathology. Pain. 1992;48(3):361–370. [PubMed: 1594258]
- 61.
- Sharma K.R., et al. Demyelinating neuropathy in diabetes mellitus. Archives of Neurology. 2002;59(5):758–765. [PubMed: 12020257]
- 62.
- Krendel D.A., Zacharias A., Younger D.S. Autoimmune diabetic neuropathy. Neurologic clinics. 1997;15(4):959–971. [PubMed: 9367975]
- 63.
- Milicevic Z., et al. Anti-ganglioside GM1 antibody and distal symmetric" diabetic polyneuropathy" with dominant motor features. Diabetologia. 1997;40(11):1364–1365. [PubMed: 9389432]
- 64.
- Ayyar D.R., Sharma K.R. Chronic inflammatory demyelinating polyradiculoneuropathy in diabetes mellitus. Current diabetes reports. 2004;4(6):409–412. [PubMed: 15539003]
- 65.
- BARADA. Proximal diabetic neuropathy-response to immunotherapy. Diabetes. 1999;48(5):SA148–SA148. A., et al. p.
- 66.
- Archer A., et al. The natural history of acute painful neuropathy in diabetes mellitus. Journal of Neurology, Neurosurgery & Psychiatry. 1983;46(6):491–499. [PMC free article: PMC1027437] [PubMed: 6875582]
- 67.
- NICE, Neuropathic pain in adults: pharmacological management in non-specialist settings. 2013. [PubMed: 31961628]
- 68.
- Tesfaye, S., et al., Arterio-venous shunting and proliferating new vessels in acute painful neuropathy of rapid glycaemic control (insulin neuritis). diabetologia, 1996. 39(3): p. 329-335. [PubMed: 8721779]
- 69.
- Gibbons C.H., Freeman R. Treatment-induced neuropathy of diabetes: an acute, iatrogenic complication of diabetes. Brain. 2015;138(1):43–52. [PMC free article: PMC4285188] [PubMed: 25392197]
- 70.
- Sinnreich M., Taylor B.V., Dyck P.J.B. Diabetic neuropathies: classification, clinical features, and pathophysiological basis. The neurologist. 2005;11(2):63–79. [PubMed: 15733329]
- 71.
- Oyibo S., et al. The relationship between blood glucose excursions and painful diabetic peripheral neuropathy: a pilot study. Diabetic Medicine. 2002;19(10):870–873. [PubMed: 12358878]
- 72.
- Ward J. The diabetic leg. Diabetologia. 1982;22(3):141–147. [PubMed: 7042425]
- 73.
- Katoulis E.C., et al. Gait abnormalities in diabetic neuropathy. Diabetes care. 1997;20(12):1904–1907. [PubMed: 9405916]
- 74.
- Cavanagh P.R., Ulbrecht J.S., Caputo G.M. New developments in the biomechanics of the diabetic foot. Diabetes/Metabolism Research and Reviews. 2000;16(S1):S6–S10. [PubMed: 11054880]
- 75.
- Reiber G.E., et al. Causal pathways for incident lower-extremity ulcers in patients with diabetes from two settings. Diabetes care. 1999;22(1):157–162. [PubMed: 10333919]
- 76.
- AN. A comparison of the Neuropen against standard quantitative sensory‐threshold measures for assessing peripheral nerve function. Diabetic Medicine. 2002;19(5):400–405. P., et al. p. [PubMed: 12027928]
- 77.
- Ziegler D., et al. Validation of a novel screening device (NeuroQuick) for quantitative assessment of small nerve fiber dysfunction as an early feature of diabetic polyneuropathy. Diabetes care. 2005;28(5):1169–1174. [PubMed: 15855584]
- 78.
- Viswanathan V., et al. Early recognition of diabetic neuropathy: evaluation of a simple outpatient procedure using thermal perception. Postgraduate medical journal. 2002;78(923):541–542. [PMC free article: PMC1742498] [PubMed: 12357015]
- 79.
- Martina I., et al. Measuring vibration threshold with a graduated tuning fork in normal aging and in patients with polyneuropathy. Journal of Neurology, Neurosurgery & Psychiatry. 1998;65(5):743–747. [PMC free article: PMC2170371] [PubMed: 9810949]
- 80.
- Rayman G., et al. The Ipswich Touch Test: a simple and novel method to identify inpatients with diabetes at risk of foot ulceration. Diabetes care. 2011;34(7):1517–1518. [PMC free article: PMC3120164] [PubMed: 21593300]
- 81.
- Association A.D., Professional Practice Committee. Standards of Medical Care in Diabetes—2022. Diabetes Care. 2021;45 Supplement_1:S3–S3. [PubMed: 33298410]
- 82.
- NICE Guidelines for the Management of Diabetes. Available from: https://www
.nice.org .uk/guidance/conditions-and-diseases /diabetes-and-other-endocrinal--nutritional-and-metabolic-conditions/diabetes. - 83.
- Feldman E.L., et al. A practical two-step quantitative clinical and electrophysiological assessment for the diagnosis and staging of diabetic neuropathy. Diabetes care. 1994;17(11):1281–1289. [PubMed: 7821168]
- 84.
- Bril V., Perkins B.A. Validation of the Toronto Clinical Scoring System for diabetic polyneuropathy. Diabetes care. 2002;25(11):2048–2052. [PubMed: 12401755]
- 85.
- Bril V., et al. Reliability and validity of the modified Toronto Clinical Neuropathy Score in diabetic sensorimotor polyneuropathy. Diabetic Medicine. 2009;26(3):240–246. [PMC free article: PMC2871179] [PubMed: 19317818]
- 86.
- Young M., et al. A multicentre study of the prevalence of diabetic peripheral neuropathy in the United Kingdom hospital clinic population. Diabetologia. 1993;36(2):150–154. [PubMed: 8458529]
- 87.
- Dyck P.J. Detection, characterization, and staging of polyneuropathy: assessed in diabetics. Muscle & Nerve. Official Journal of the American Association of Electrodiagnostic Medicine. 1988;11(1):21–32. [PubMed: 3277049]
- 88.
- Bastyr E.J. III, et al. Development and validity testing of the neuropathy total symptom score-6: questionnaire for the study of sensory symptoms of diabetic peripheral neuropathy. Clinical therapeutics. 2005;27(8):1278–1294. [PubMed: 16199253]
- 89.
- Freynhagen R., et al. Pain DETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Current medical research and opinion. 2006;22(10):1911–1920. [PubMed: 17022849]
- 90.
- Spallone V., et al. Validation of DN4 as a screening tool for neuropathic pain in painful diabetic polyneuropathy. Diabetic Medicine. 2012;29(5):578–585. [PubMed: 22023377]
- 91.
- Bouhassira D., et al. Development and validation of the neuropathic pain symptom inventory. Pain. 2004;108(3):248–257. [PubMed: 15030944]
- 92.
- Lauria G., et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Report of a joint task force of the European Fe‐deration of Neurological Societies and the Peripheral Nerve Society. European journal of neurology. 2010;17(7):903–e49. [PubMed: 20642627]
- 93.
- England J., et al. Practice Parameter: The Evaluation of Distal Symmetric Polyneuropathy: The Role of Autonomic Testing, Nerve Biopsy, and Skin Biopsy (An Evidence‐Based Review) Report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. PM&R. 2009;1(1):14–22. [PubMed: 19627868]
- 94.
- Papanas N., Ziegler D. New vistas in the diagnosis of diabetic polyneuropathy. Endocrine. 2014;47(3):690–698. [PubMed: 24839196]
- 95.
- Papanas N., et al. Sensitivity and specificity of a new indicator test (Neuropad) for the diagnosis of peripheral neuropathy in type 2 diabetes patients: a comparison with clinical examination and nerve conduction study. Journal of diabetes and its complications. 2007;21(6):353–358. [PubMed: 17967706]
- 96.
- Vinik, A.I., et al., Diabetic nerve conduction abnormalities in the primary care setting. Diabetes technology & therapeutics, 2006. 8(6): p. 654-662,Chatzikosma, G., et al., Evaluation of sural nerve automated nerve conduction study in the diagnosis of peripheral neuropathy in patients with type 2 diabetes mellitus. Archives of medical science: AMS, 2016. 12(2): p. 390. [PubMed: 17109597]
- 97.
- Lovblom L.E., et al. In vivo corneal confocal microscopy and prediction of future-incident neuropathy in type 1 diabetes: a preliminary longitudinal analysis. Canadian journal of diabetes. 2015;39(5):390–397. [PubMed: 25936902]
- 98.
- Alam U., et al. Diagnostic utility of corneal confocal microscopy and intra-epidermal nerve fibre density in diabetic neuropathy. PloS one. 2017;12(7):e0180175. p. [PMC free article: PMC5515394] [PubMed: 28719619]
- 99.
- Selvarajah D., et al. SUDOSCAN: a simple, rapid, and objective method with potential for screening for diabetic peripheral neuropathy. PloS one. 2015;10(10):e0138224. p. [PMC free article: PMC4601729] [PubMed: 26457582]
- 100.
- Mao F., et al. Sudoscan is an effective screening method for asymptomatic diabetic neuropathy in Chinese type 2 diabetes mellitus patients. Journal of diabetes investigation. 2017;8(3):363–368. [PMC free article: PMC5415453] [PubMed: 27607763]
- 101.
- Lee J.A., et al. Reliability and validity of a point-of-care sural nerve conduction device for identification of diabetic neuropathy. PloS one. 2014;9(1):e86515. p. [PMC free article: PMC3899274] [PubMed: 24466129]
- 102.
- Umapathi T., et al. Intraepidermal nerve fiber density as a marker of early diabetic neuropathy. Muscle & Nerve. Official Journal of the American Association of Electrodiagnostic Medicine. 2007;35(5):591–598. [PubMed: 17221881]
- 103.
- Quattrini C., et al. Surrogate markers of small fiber damage in human diabetic neuropathy. Diabetes. 2007;56(8):2148–2154. [PubMed: 17513704]
- 104.
- Malik R., et al. Small fibre neuropathy: role in the diagnosis of diabetic sensorimotor polyneuropathy. Diabetes/metabolism research and reviews. 2011;27(7):678–684. [PubMed: 21695760]
- 105.
- Papanas N., et al. Evaluation of a new indicator test for sudomotor function (Neuropad®) in the diagnosis of peripheral neuropathy in type 2 diabetic patients. Experimental and clinical endocrinology & diabetes. 2005;113(04):195–198. [PubMed: 15891953]
- 106.
- Ziegler D., et al. Neuropad as a diagnostic tool for diabetic autonomic and sensorimotor neuropathy. Diabetic medicine. 2009;26(7):686–692. Neuropad: evaluation of three cut‐off points of sudomotor dysfunction for early detection of polyneuropathy in recently diagnosed diabetes. Diabetic Medicine, 2011. 28(11): p. 1412-1415,Spallone, V., et al. p. [PubMed: 19573117]
- 107.
- Ponirakis G., et al. The diagnostic accuracy of Neuropad® for assessing large and small fibre diabetic neuropathy. Diabetic medicine. 2014;31(12):1673–1680. [PMC free article: PMC4236278] [PubMed: 24975286]
- 108.
- Papanas N., et al. Reproducibility of the new indicator test for sudomotor function (Neuropad®) in patients with type 2 diabetes mellitus. Experimental and clinical endocrinology & diabetes. 2005;113(10):577–581. [PubMed: 16320155]
- 109.
- Tavakoli M., et al. Corneal confocal microscopy: a novel noninvasive test to diagnose and stratify the severity of human diabetic neuropathy. Diabetes care. 2010;33(8):1792–1797. [PMC free article: PMC2909064] [PubMed: 20435796]
- 110.
- Zhivov A., et al. Real-time mapping of the subepithelial nerve plexus by in vivo confocal laser scanning microscopy. British journal of ophthalmology. 2010;94(9):1133–1135. [PubMed: 20813752]
- 111.
- Casanova-Molla J., et al. On the relationship between nociceptive evoked potentials and intraepidermal nerve fiber density in painful sensory polyneuropathies. Pain. 2011;152(2):410–418. [PubMed: 21185650]
- 112.
- Chao C.C., et al. Effects of aging on contact heat-evoked potentials: the physiological assessment of thermal perception. Muscle Nerve. 2007;36(1):30–8. [PubMed: 17503497]
- 113.
- Smith, A.G., et al., The diagnostic utility of Sudoscan for distal symmetric peripheral neuropathy. Journal of Diabetes and its Complications, 2014. 28(4): p. 511-516,Yajnik, C.S., et al., Quick and simple evaluation of sudomotor function for screening of diabetic neuropathy. International Scholarly Research Notices, 2012. 2012. [PMC free article: PMC4219320] [PubMed: 24661818]
- 114.
- Bourcier M.E., et al. Diabetic peripheral neuropathy: how reliable is a homemade 1-g monofilament for screening? A case-control study of sensitivity, specificity, and comparison with standardized sensory modalities. Journal of family practice. 2006;55(6):505–509. [PubMed: 16750065]
- 115.
- Kumar S., et al. Semmes-Weinstein monofilaments: a simple, effective and inexpensive screening device for identifying diabetic patients at risk of foot ulceration. Diabetes research and clinical practice. 1991;13(1-2):63–67. [PubMed: 1773715]
- 116.
- Armstrong D.G., et al. Choosing a practical screening instrument to identify patients at risk for diabetic foot ulceration. Archives of internal medicine. 1998;158(3):289–292. [PubMed: 9472210]
- 117.
- Vinik A.I., et al. Quantitative measurement of cutaneous perception in diabetic neuropathy. Muscle & Nerve. Official Journal of the American Association of Electrodiagnostic Medicine. 1995;18(6):574–584. [PubMed: 7753119]
- 118.
- Abbott C., et al. The North‐West Diabetes Foot Care Study: incidence of, and risk factors for, new diabetic foot ulceration in a community‐based patient cohort. Diabetic medicine. 2002;19(5):377–384. [PubMed: 12027925]
- 119.
- Dyck P.J., Melton L.J., O'Brien P.C. Approaches to improve epidemiological studies of diabetic neuropathy: insights from the Rochester Diabetic Neuropathy Study. Diabetes. 1997;46 Supplement 2:S5–S8. [PubMed: 9285491]
- 120.
- Herman W., et al. Use of the Michigan Neuropathy Screening Instrument as a measure of distal symmetrical peripheral neuropathy in Type 1 diabetes: results from the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications. Diabetic medicine. 2012;29(7):937–944. [PMC free article: PMC3641573] [PubMed: 22417277]
- 121.
- Dyck P.J., et al. “Unequivocally abnormal” vs “usual” signs and symptoms for proficient diagnosis of diabetic polyneuropathy: Cl vs N Phys Trial. Archives of neurology. 2012;69(12):1609–1614. [PMC free article: PMC3570730] [PubMed: 22986424]
- 122.
- Feng, Y., F.J. Schlösser, and B.E. Sumpio, The Semmes Weinstein monofilament examination as a screening tool for diabetic peripheral neuropathy. Journal of vascular surgery, 2009. 50(3): p. 675-682. e1. [PubMed: 19595541]
- 123.
- Richard J.L., et al. Screening patients at risk for diabetic foot ulceration: a comparison between measurement of vibration perception threshold and 10‐g monofilament test. International wound journal. 2014;11(2):147–151. [PMC free article: PMC7950904] [PubMed: 22892021]
- 124.
- Tan L.S. The clinical use of the 10 g monofilament and its limitations: a review. Diabetes research and clinical practice. 2010;90(1):1–7. [PubMed: 20655123]
- 125.
- Weisman A., et al. Identification and prediction of diabetic sensorimotor polyneuropathy using individual and simple combinations of nerve conduction study parameters. PLoS One. 2013;8(3):e58783. p. [PMC free article: PMC3606395] [PubMed: 23533591]
- 126.
- The Diabetic Retinopathy Barometer Report Global Findings. [cited 2019 28th December]; Available from: https://www
.idf.org/our-activities /advocacy-awareness /resources-and-tools /92:diabetic-retinopathy-barometer.html. - 127.
- Liew G., Michaelides M., Bunce C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ open. 2014;4(2):e004015. p. [PMC free article: PMC3927710] [PubMed: 24525390]
- 128.
- Marshall S. Diabetic nephropathy in type 1 diabetes: has the outlook improved since the 1980s? Diabetologia. 2012;55(9):2301–2306. [PubMed: 22696035]
- 129.
- Dyck P.J., et al. Patterns of quantitative sensation testing of hypoesthesia and hyperalgesia are predictive of diabetic polyneuropathy: a study of three cohorts. Nerve growth factor study group. Diabetes Care. 2000;23(4):510–517. [PubMed: 10857944]
- 130.
- Yarnitsky D., Sprecher E. Thermal testing: normative data and repeatability for various test algorithms. Journal of the neurological sciences. 1994;125(1):39–45. [PubMed: 7964887]
- 131.
- Shy M., et al. Quantitative sensory testing: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2003;60(6):898–904. [PubMed: 12654951]
- 132.
- Society P.N. Diabetic polyneuropathy in controlled clinical trials: consensus report of the Peripheral Nerve Society. Annals of Neurology. 1995;38(3):478–482. [PubMed: 7668839]
- 133.
- Gibbons C.H. Small fiber neuropathies. CONTINUUM: Lifelong Learning in Neurology. 2014;20(5):1398–1412. [PubMed: 25299289]
- 134.
- Pittenger G.L., et al. Intraepidermal nerve fibers are indicators of small-fiber neuropathy in both diabetic and nondiabetic patients. Diabetes care. 2004;27(8):1974–1979. [PubMed: 15277426]
- 135.
- Polydefkis M., et al. Skin biopsy as a tool to assess distal small fiber innervation in diabetic neuropathy. Diabetes technology & therapeutics. 2001;3(1):23–28. [PubMed: 11469706]
- 136.
- Smith A.G., et al. Lifestyle intervention for pre-diabetic neuropathy. Diabetes care. 2006;29(6):1294–1299. [PubMed: 16732011]
- 137.
- Vileikyte L., et al. The development and validation of a neuropathy-and foot ulcer-specific quality of life instrument. Diabetes care. 2003;26(9):2549–2555. [PubMed: 12941717]
- 138.
- Vinik E.J., et al. The development and validation of the Norfolk QOL-DN, a new measure of patients' perception of the effects of diabetes and diabetic neuropathy. Diabetes technology & therapeutics. 2005;7(3):497–508. [PubMed: 15929681]
- 139.
- Selvarajah D., et al. Early involvement of the spinal cord in diabetic peripheral neuropathy. Diabetes care. 2006;29(12):2664–2669. [PubMed: 17130202]
- 140.
- Eaton S.E., et al. Spinal-cord involvement in diabetic peripheral neuropathy. The Lancet. 2001;358(9275):35–36. [PubMed: 11454377]
- 141.
- Selvarajah D., et al. Magnetic resonance neuroimaging study of brain structural differences in diabetic peripheral neuropathy. Diabetes care. 2014;37(6):1681–1688. [PubMed: 24658391]
- 142.
- Selvarajah D., et al. Thalamic neuronal dysfunction and chronic sensorimotor distal symmetrical polyneuropathy in patients with type 1 diabetes mellitus. Diabetologia. 2008;51(11):2088–2092. [PubMed: 18773192]
- 143.
- Hansen T.M., et al. Brain spectroscopy reveals that N-acetylaspartate is associated to peripheral sensorimotor neuropathy in type 1 diabetes. Journal of Diabetes and its Complications. 2019;33(4):323–328. [PubMed: 30733057]
- 144.
- Selvarajah D., et al. Microvascular perfusion abnormalities of the Thalamus in painful but not painless diabetic polyneuropathy: a clue to the pathogenesis of pain in type 1 diabetes. Diabetes care. 2011;34(3):718–720. [PMC free article: PMC3041213] [PubMed: 21282344]
- 145.
- Cauda F., et al. Low-frequency BOLD fluctuations demonstrate altered thalamocortical connectivity in diabetic neuropathic pain. BMC neuroscience. 2009;10(1):1–14. [PMC free article: PMC2789078] [PubMed: 19941658]
- 146.
- Selvarajah D., et al. Structural and functional abnormalities of the primary somatosensory cortex in diabetic peripheral neuropathy: a multimodal MRI study. Diabetes. 2019;68(4):796–806. [PubMed: 30617218]
- 147.
- Reske-Nielsen E., Lundbæk K. Pathological changes in the central and peripheral nervous system of young long-term diabetics. Diabetologia. 1968;4(1):34–43. [PubMed: 4190608]
- 148.
- Reske-Nielsen E., et al. Pathological changes in the central and peripheral nervous system of young long-term diabetics. The terminal neuro-muscular apparatus. Diabetologia. 1970;6(2):98–103. [PubMed: 4192896]
- 149.
- Ziegler D., et al. Tibial nerve somatosensory evoked potentials at various stages of peripheral neuropathy in insulin dependent diabetic patients. Journal of Neurology, Neurosurgery & Psychiatry. 1993;56(1):58–64. [PMC free article: PMC1014767] [PubMed: 8381473]
- 150.
- Biessels G.-J., et al. Neurophysiological changes in the central and peripheral nervous system of streptozotocin-diabetic rats: course of development and effects of insulin treatment. Brain. 1999;122(4):757–768. [PubMed: 10219786]
- 151.
- Group L.A.R. Effects of a long-term lifestyle modification programme on peripheral neuropathy in overweight or obese adults with type 2 diabetes: the Look AHEAD study. Diabetologia. 2017;60(6):980. [PMC free article: PMC5423967] [PubMed: 28349174]
- 152.
- Control D., Group C.T.R. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. New England journal of medicine. 1993;329(14):977–986. [PubMed: 8366922]
- 153.
- Group U.P.D.S. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). The lancet. 1998;352(9131):837–853. [PubMed: 9742976]
- 154.
- Albers J.W., et al. Effect of prior intensive insulin treatment during the Diabetes Control and Complications Trial (DCCT) on peripheral neuropathy in type 1 diabetes during the Epidemiology of Diabetes Interventions and Complications (EDIC) Study. Diabetes care. 2010;33(5):1090–1096. [PMC free article: PMC2858182] [PubMed: 20150297]
- 155.
- Pop-Busui R., et al. Effects of prior intensive insulin therapy on cardiac autonomic nervous system function in type 1 diabetes mellitus: the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications study (DCCT/EDIC). Circulation. 2009;119(22):2886–2893. [PMC free article: PMC2757005] [PubMed: 19470886]
- 156.
- Group A.C. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. New England journal of medicine. 2008;358(24):2560–2572. [PubMed: 18539916]
- 157.
- Duckworth W., et al. Glucose control and vascular complications in veterans with type 2 diabetes. New England journal of medicine. 2009;360(2):129–139. [PubMed: 19092145]
- 158.
- Gæde P., et al. Effect of a multifactorial intervention on mortality in type 2 diabetes. New England Journal of Medicine. 2008;358(6):580–591. [PubMed: 18256393]
- 159.
- Vincent A.M., et al. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. Journal of the Peripheral Nervous System. 2009;14(4):257–267. [PMC free article: PMC4239691] [PubMed: 20021567]
- 160.
- Callaghan B.C., et al. Triglycerides and amputation risk in patients with diabetes: ten-year follow-up in the DISTANCE study. Diabetes care. 2011;34(3):635–640. [PMC free article: PMC3041196] [PubMed: 21285390]
- 161.
- Wiggin T.D., et al. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes. 2009;58(7):1634–1640. [PMC free article: PMC2699859] [PubMed: 19411614]
- 162.
- Knopp R.H., et al. Efficacy and safety of atorvastatin in the prevention of cardiovascular end points in subjects with type 2 diabetes: the Atorvastatin Study for Prevention of Coronary Heart Disease Endpoints in non-insulin-dependent diabetes mellitus (ASPEN). Diabetes care, 2006. 29(7): p. 1478-1485,Group, H.P.S.C., MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. The Lancet. 2003;361(9374):2005–2016. [PubMed: 16801565]
- 163.
- Colhoun H.M., et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. The Lancet. 2004;364(9435):685–696. [PubMed: 15325833]
- 164.
- Davis T., et al. Lipid-lowering therapy and peripheral sensory neuropathy in type 2 diabetes: the Fremantle Diabetes Study. Diabetologia. 2008;51(4):562–566. [PubMed: 18193189]
- 165.
- Zangiabadi N., et al. Atorvastatin treatment improves diabetic polyneuropathy electrophysiological changes in non-insulin dependent diabetic patients: a double blind, randomized clinical trial. Minerva Endocrinol. 2012;37(2):195–200. [PubMed: 22691892]
- 166.
- Hernández-Ojeda J., et al. Effect of rosuvastatin on diabetic polyneuropathy: a randomized, double-blind, placebo-controlled Phase IIa study. Diabetes, metabolic syndrome and obesity: targets and therapy. 2014;7:401. [PMC free article: PMC4159311] [PubMed: 25214797]
- 167.
- Ansquer J., et al. Fibrates and microvascular complications in diabetes-insight from the FIELD study. Current pharmaceutical design. 2009;15(5):537–552. [PubMed: 19199980]
- 168.
- Nielsen S.F., Nordestgaard B.G. Statin use before diabetes diagnosis and risk of microvascular disease: a nationwide nested matched study. The lancet Diabetes & endocrinology. 2014;2(11):894–900. [PubMed: 25217178]
- 169.
- Jarmuzewska E., Ghidoni A., Mangoni A.A. Hypertension and sensorimotor peripheral neuropathy in type 2 diabetes. European neurology. 2007;57(2):91–95. [PubMed: 17179711]
- 170.
- Estacio R.O., et al. Effect of blood pressure control on diabetic microvascular complications in patients with hypertension and type 2 diabetes. Diabetes care. 2000;23:B54. [PubMed: 10860192]
- 171.
- Forrest K.Y., et al. Hypertension as a risk factor for diabetic neuropathy: a prospective study. Diabetes. 1997;46(4):665–670. [PubMed: 9075809]
- 172.
- Malik R.A., et al. Effect of angiotensin-converting-enzyme (ACE) inhibitor trandolapril on human diabetic neuropathy: randomised double-blind controlled trial. The Lancet. 1998;352(9145):1978–1981. [PubMed: 9872248]
- 173.
- Ruggenenti P., et al. Effects of manidipine and delapril in hypertensive patients with type 2 diabetes mellitus: the delapril and manidipine for nephroprotection in diabetes (DEMAND) randomized clinical trial. Hypertension. 2011;58(5):776–783. [PubMed: 21931073]
- 174.
- Callaghan B.C., et al. Metabolic syndrome components are associated with symptomatic polyneuropathy independent of glycemic status. Diabetes care. 2016;39(5):801–807. [PMC free article: PMC4839175] [PubMed: 26965720]
- 175.
- Costa L., et al. Aggregation of features of the metabolic syndrome is associated with increased prevalence of chronic complications in type 2 diabetes. Diabetic Medicine. 2004;21(3):252–255. [PubMed: 15008835]
- 176.
- Ylitalo K.R., Sowers M., Heeringa S. Peripheral vascular disease and peripheral neuropathy in individuals with cardiometabolic clustering and obesity: National Health and Nutrition Examination Survey 2001–2004. Diabetes care. 2011;34(7):1642–1647. [PMC free article: PMC3120210] [PubMed: 21593304]
- 177.
- Callaghan B.C., et al. Association between metabolic syndrome components and polyneuropathy in an obese population. JAMA neurology. 2016;73(12):1468–1476. [PMC free article: PMC5217829] [PubMed: 27802497]
- 178.
- Kluding P.M., et al. The effect of exercise on neuropathic symptoms, nerve function, and cutaneous innervation in people with diabetic peripheral neuropathy. Journal of Diabetes and its Complications. 2012;26(5):424–429. [PMC free article: PMC3436981] [PubMed: 22717465]
- 179.
- Balducci S., et al. Exercise training can modify the natural history of diabetic peripheral neuropathy. Journal of diabetes and its complications. 2006;20(4):216–223. [PubMed: 16798472]
- 180.
- Singleton J.R., et al. Supervised exercise improves cutaneous reinnervation capacity in metabolic syndrome patients. Annals of neurology. 2015;77(1):146–153. [PMC free article: PMC4293306] [PubMed: 25388934]
- 181.
- Ziegler D., Sohr C.G., Nourooz-Zadeh J. Oxidative stress and antioxidant defense in relation to the severity of diabetic polyneuropathy and cardiovascular autonomic neuropathy. Diabetes care. 2004;27(9):2178–2183. [PubMed: 15333481]
- 182.
- Miyauchi Y., et al. Slowing of peripheral motor nerve conduction was ameliorated by aminoguanidine in streptozocin-induced diabetic rats. European journal of endocrinology. 1996;134(4):467–473. [PubMed: 8640299]
- 183.
- Haupt E., Ledermann H., Köpcke W. Benfotiamine in the treatment of diabetic. International journal of clinical pharmacology and therapeutics. 2005;43(2):71–77. [PubMed: 15726875]
- 184.
- Stracke H., Lindemann A., Federlin K. A benfotiamine-vitamin B combination in treatment of diabetic polyneuropathy. Experimental and clinical endocrinology & diabetes. 1996;104(04):311–316. [PubMed: 8886748]
- 185.
- Stracke H., et al. Benfotiamine in diabetic polyneuropathy (BENDIP): results of a randomised, double blind, placebo-controlled clinical study. Experimental and clinical endocrinology & diabetes. 2008;116(10):600–605. [PubMed: 18473286]
- 186.
- Greene D.A., et al. Effect of aldose reductase inhibition on nerve conduction and morphometry in diabetic neuropathy. Neurology. 1999;53(3):580–580. [PubMed: 10449124]
- 187.
- Hotta N., et al. Clinical efficacy of fidarestat, a novel aldose reductase inhibitor, for diabetic peripheral neuropathy: a 52-week multicenter placebo-controlled double-blind parallel group study. Diabetes care. 2001;24(10):1776–1782. [PubMed: 11574441]
- 188.
- Hotta N., et al. Long-term effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: a 3-y multicenter comparative study, ARI-Diabetes Complications Trial (ADCT). Diabetes. 2005;54:A213. [PubMed: 16801576]
- 189.
- Bril V., Buchanan R.A. Long-term effects of ranirestat (AS-3201) on peripheral nerve function in patients with diabetic sensorimotor polyneuropathy. Diabetes care. 2006;29(1):68–72. [PubMed: 16373898]
- 190.
- Keen H., et al. Treatment of diabetic neuropathy with γ-linolenic acid. Diabetes Care. 1993;16(1):8–15. [PubMed: 8380765]
- 191.
- Ametov A.S., et al. The sensory symptoms of diabetic polyneuropathy are improved with α-lipoic acid. Diabetes care. 2003;26(3):770–776. [PubMed: 12610036]
- 192.
- Reljanovic M., et al. Treatment of diabetic polyneuropathy with the antioxidant thioctic acid (α-lipoic acid): a two year multicenter randomized double-blind placebo-controlled trial (ALADIN II). Free radical research. 1999;31(3):171–179. [PubMed: 10499773]
- 193.
- Ziegler D., et al. Treatment of symptomatic diabetic polyneuropathy with the antioxidant α‐lipoic acid: a meta‐analysis. Diabetic Medicine. 2004;21(2):114–121. [PubMed: 14984445]
- 194.
- Ziegler D., et al. Oral treatment with α-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes care. 2006;29(11):2365–2370. [PubMed: 17065669]
- 195.
- Ziegler D., et al. Efficacy and safety of antioxidant treatment with α-lipoic acid over 4 years in diabetic polyneuropathy: the NATHAN 1 trial. Diabetes care. 2011;34(9):2054–2060. [PMC free article: PMC3161301] [PubMed: 21775755]
- 196.
- McIlduff C.E., Rutkove S.B. Critical appraisal of the use of alpha lipoic acid (thioctic acid) in the treatment of symptomatic diabetic polyneuropathy. Therapeutics and clinical risk management. 2011;7:377. [PMC free article: PMC3176171] [PubMed: 21941444]
- 197.
- Mijnhout, G.S., et al., Alpha lipoic acid for symptomatic peripheral neuropathy in patients with diabetes: a meta-analysis of randomized controlled trials. International Journal of Endocrinology, 2012. 2012. [PMC free article: PMC3272801] [PubMed: 22331979]
- 198.
- Vinik A.I., et al. Treatment of symptomatic diabetic peripheral neuropathy with the protein kinase C β-inhibitor ruboxistaurin mesylate during a 1-year, randomized, placebo-controlled, double-blind clinical trial. Clinical therapeutics. 2005;27(8):1164–1180. [PubMed: 16199243]
- 199.
- Vinik A.I., et al. Sural sensory action potential identifies diabetic peripheral neuropathy responders to therapy. Muscle & nerve. 2005;32(5):619–625. [PubMed: 16116628]
- 200.
- Boyd A., et al. Quality of life and objective measures of diabetic neuropathy in a prospective placebo-controlled trial of ruboxistaurin and topiramate. Journal of Diabetes Science and Technology. 2011;5(3):714–722. [PMC free article: PMC3192638] [PubMed: 21722587]
- 201.
- Forst T., et al. Molecular effects of C-Peptide in microvascular blood flow regulation. The review of diabetic studies: RDS. 2009;6(3):159. [PMC free article: PMC2827268] [PubMed: 20039005]
- 202.
- Wahren J., Kallas Å., Sima A.A. The clinical potential of C-peptide replacement in type 1 diabetes. Diabetes. 2012;61(4):761–772. [PMC free article: PMC3314360] [PubMed: 22442295]
- 203.
- Ekberg K., et al. C-Peptide Replacement Therapy and Sensory Nerve Function in Type 1 Diabetic Neuropathy. Diabetes Care. 2007;30(1):71–76. [PubMed: 17192336]
- 204.
- Pittenger G., Vinik A. Nerve growth factor and diabetic neuropathy. Experimental diabesity research. 2003;4(4):271–285. [PMC free article: PMC2478610] [PubMed: 14668049]
- 205.
- VINIK. A., Treatment of diabetic polyneuropathy (DPN) with recombinant human nerve growth factor (rhNGF). Diabetes. 1999;48(5):SA54–SA54.
- 206.
- Rivard A., et al. Rescue of diabetes-related impairment of angiogenesis by intramuscular gene therapy with adeno-VEGF. The American journal of pathology. 1999;154(2):355–363. [PMC free article: PMC1850015] [PubMed: 10027394]
- 207.
- Kessler J.A., et al. Gene therapy for diabetic peripheral neuropathy: A randomized, placebo‐controlled phase III study of VM202, a plasmid DNA encoding human hepatocyte growth factor. Clinical and Translational Science. 2021;14(3):1176–1184. [PMC free article: PMC8212761] [PubMed: 33465273]
- 208.
- Kessler J.A., et al. Double‐blind, placebo‐controlled study of HGF gene therapy in diabetic neuropathy. Annals of clinical and translational neurology. 2015;2(5):465–478. [PMC free article: PMC4435702] [PubMed: 26000320]
- 209.
- Ajroud-Driss S., et al. Phase 1/2 open-label dose-escalation study of plasmid DNA expressing two isoforms of hepatocyte growth factor in patients with painful diabetic peripheral neuropathy. Molecular Therapy. 2013;21(6):1279–1286. [PMC free article: PMC3677315] [PubMed: 23609019]
- 210.
- Tam J., Rosenberg L., Maysinger D. INGAP peptide improves nerve function and enhances regeneration in streptozotocin‐induced diabetic C57BL/6 mice. The FASEB journal. 2004;18(14):1767–1769. [PubMed: 15345684]
- 211.
- Granberg V., et al. Autoantibodies to autonomic nerves associated with cardiac and peripheral autonomic neuropathy. Diabetes care. 2005;28(8):1959–1964. [PubMed: 16043739]
- 212.
- Vinik A.I., Anandacoomaraswamy D., Ullal J. Antibodies to neuronal structures: innocent bystanders or neurotoxins? Diabetes care. 2005;28(8):2067–2072. [PubMed: 16043764]
- 213.
- Pain, I.A.f.t.S.o., Pain terms. Neuropathic Pain. IASP Taxonomy, 2017.
- 214.
- Colloca L., et al. Neuropathic pain. Nature reviews Disease primers. 2017;3(1):1–19. [PMC free article: PMC5371025] [PubMed: 28205574]
- 215.
- Bennett D.L., et al. The role of voltage-gated sodium channels in pain signaling. Physiological reviews. 2019;99(2):1079–1151. [PubMed: 30672368]
- 216.
- Saegusa H., et al. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N‐type Ca2+ channel. The EMBO journal. 2001;20(10):2349–2356. [PMC free article: PMC125247] [PubMed: 11350923]
- 217.
- Finnerup N.B. Nonnarcotic methods of pain management. New England Journal of Medicine. 2019;380(25):2440–2448. [PubMed: 31216399]
- 218.
- Binns‐Hall O., et al. One‐stop microvascular screening service: an effective model for the early detection of diabetic peripheral neuropathy and the high‐risk foot. Diabetic Medicine. 2018;35(7):887–894. [PMC free article: PMC6033008] [PubMed: 29608799]
- 219.
- Daousi C., et al. Chronic painful peripheral neuropathy in an urban community: a controlled comparison of people with and without diabetes. Diabetic medicine. 2004;21(9):976–982. [PubMed: 15317601]
- 220.
- Bennett M.I., et al. The S-LANSS score for identifying pain of predominantly neuropathic origin: validation for use in clinical and postal research. The Journal of Pain. 2005;6(3):149–158. [PubMed: 15772908]
- 221.
- Krause S.J., Backonja M.-M. Development of a neuropathic pain questionnaire. The Clinical journal of pain. 2003;19(5):306–314. [PubMed: 12966256]
- 222.
- Dworkin R.H., et al. Development and initial validation of an expanded and revised version of the Short-form McGill Pain Questionnaire (SF-MPQ-2). Pain. 2009;144(1-2):35–42. [PubMed: 19356853]
- 223.
- Daut R.L., Cleeland C.S., Flanery R.C. Development of the Wisconsin Brief Pain Questionnaire to assess pain in cancer and other diseases. Pain. 1983;17(2):197–210. [PubMed: 6646795]
- 224.
- Freeman R., et al. Sensory profiles of patients with neuropathic pain based on the neuropathic pain symptoms and signs. PAIN. 2014;155(2):367–376. [PubMed: 24472518]
- 225.
- Dworkin R.H., et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. The journal of pain. 2008;9(2):105–121. [PubMed: 18055266]
- 226.
- Davies M., et al. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes care. 2006;29(7):1518–1522. [PubMed: 16801572]
- 227.
- Tölle T., Xu X., Sadosky A.B. Painful diabetic neuropathy: a cross-sectional survey of health state impairment and treatment patterns. Journal of Diabetes and its Complications. 2006;20(1):26–33. [PubMed: 16389164]
- 228.
- Alleman C.J., et al. Humanistic and economic burden of painful diabetic peripheral neuropathy in Europe: a review of the literature. Diabetes research and clinical practice. 2015;109(2):215–225. [PubMed: 26008721]
- 229.
- Woolf, C.J., et al., Towards a mechanism-based classification of pain? 1998, LWW. p. 227-229. [PubMed: 9808347]
- 230.
- Edwards R.R., et al. Patient phenotyping in clinical trials of chronic pain treatments: IMMPACT recommendations. Pain. 2016;157(9):1851. [PMC free article: PMC5965275] [PubMed: 27152687]
- 231.
- Maier C., et al. Quantitative sensory testing in the German Research Network on Neuropathic Pain (DFNS): somatosensory abnormalities in 1236 patients with different neuropathic pain syndromes. Pain. 2010;150(3):439–450. [PubMed: 20627413]
- 232.
- Rolke R., et al. Quantitative sensory testing: a comprehensive protocol for clinical trials. European journal of pain. 2006;10(1):77–88. [PubMed: 16291301]
- 233.
- Themistocleous A.C., et al. The Pain in Neuropathy Study. (PiNS): a cross-sectional observational study determining the somatosensory phenotype of painful and painless diabetic neuropathy. Pain. 2016;157(5):1132. [PMC free article: PMC4834814] [PubMed: 27088890]
- 234.
- Demant D.T., et al. The effect of oxcarbazepine in peripheral neuropathic pain depends on pain phenotype: a randomised, double-blind, placebo-controlled phenotype-stratified study. PAIN. 2014;155(11):2263–2273. [PubMed: 25139589]
- 235.
- Wilkinson I.D., et al. Determinants of Treatment Response in Painful Diabetic Peripheral Neuropathy: A Combined Deep Sensory Phenotyping and Multimodal Brain MRI Study. Diabetes. 2020;69(8):1804–1814. [PubMed: 32471808]
- 236.
- Campbell C.M., et al. Randomized control trial of topical clonidine for treatment of painful diabetic neuropathy. PAIN. 2012;153(9):1815–1823. [PMC free article: PMC3413770] [PubMed: 22683276]
- 237.
- Bouhassira D., et al. Neuropathic pain phenotyping as a predictor of treatment response in painful diabetic neuropathy: data from the randomized, double-blind, COMBO-DN study. Pain. 2014;155(10):2171–2179. [PubMed: 25168665]
- 238.
- Yarnitsky D., et al. Conditioned pain modulation predicts duloxetine efficacy in painful diabetic neuropathy. Pain. 2012;153(6):1193–1198. [PubMed: 22480803]
- 239.
- Haroutounian S., et al. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. PAIN. 2014;155(7):1272–1279. [PubMed: 24704366]
- 240.
- Wager T.D., et al. An fMRI-based neurologic signature of physical pain. New England Journal of Medicine. 2013;368(15):1388–1397. [PMC free article: PMC3691100] [PubMed: 23574118]
- 241.
- Tracey I. “Seeing” how our drugs work brings translational added value. Anesthesiology. 2013;119(6):1247–1248. [PubMed: 24343284]
- 242.
- Buxton R. Interpreting oxygenation-based neuroimaging signals: the importance and the challenge of understanding brain oxygen metabolism. Frontiers in neuroenergetics. 2010;2:8. [PMC free article: PMC2899519] [PubMed: 20616882]
- 243.
- Pereira F., Mitchell T., Botvinick M. Machine learning classifiers and fMRI: a tutorial overview. Neuroimage. 2009;45(1):S199–S209. [PMC free article: PMC2892746] [PubMed: 19070668]
- 244.
- Wager T.D., et al. Evaluating the consistency and specificity of neuroimaging data using meta-analysis. Neuroimage. 2009;45(1):S210–S221. [PMC free article: PMC3318962] [PubMed: 19063980]
- 245.
- Alger J.R., et al. Multisite, multimodal neuroimaging of chronic urological pelvic pain: Methodology of the MAPP Research Network. NeuroImage: Clinical. 2016;12:65–77. [PMC free article: PMC4925887] [PubMed: 27408791]
- 246.
- Zunhammer M., et al. Placebo effects on the neurologic pain signature: a meta-analysis of individual participant functional magnetic resonance imaging data. JAMA neurology. 2018;75(11):1321–1330. [PMC free article: PMC6248115] [PubMed: 30073258]
- 247.
- Dworkin R.H., et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237–251. [PubMed: 17920770]
- 248.
- Finnerup N.B., Sindrup S.H., Jensen T.S. The evidence for pharmacological treatment of neuropathic pain. Pain. 2010;150(3):573–581. [PubMed: 20705215]
- 249.
- Ziegler D., Fonseca V. From guideline to patient: a review of recent recommendations for pharmacotherapy of painful diabetic neuropathy. Journal of Diabetes and its Complications. 2015;29(1):146–156. [PubMed: 25239450]
- 250.
- Bril, V., et al., Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Pm&r, 2011. 3(4): p. 345-352. e21. [PubMed: 21497321]
- 251.
- Bril V., et al. Neuropathy. Canadian journal of diabetes. 2018;42:S217–S221. [PubMed: 29650100]
- 252.
- Ziegler D., et al. Diabetic neuropathy. Experimental and Clinical Endocrinology & Diabetes. 2014;122(07):406–415. [PubMed: 25014092]
- 253.
- Finnerup N.B., et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. The Lancet Neurology. 2015;14(2):162–173. [PMC free article: PMC4493167] [PubMed: 25575710]
- 254.
- Moisset X., et al. Pharmacological and non-pharmacological treatments for neuropathic pain: systematic review and French recommendations. Revue neurologique. 2020;176(5):325–352. [PubMed: 32276788]
- 255.
- Sumitani M., et al. Executive summary of the clinical guidelines of pharmacotherapy for neuropathic pain: by the Japanese society of pain clinicians. Journal of anesthesia. 2018;32(3):463–478. [PMC free article: PMC5973958] [PubMed: 29737410]
- 256.
- Attal N., et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. European journal of neurology. 2010;17(9):1113–e88. [PubMed: 20402746]
- 257.
- Max M.B., et al. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. New England Journal of Medicine. 1992;326(19):1250–1256. [PubMed: 1560801]
- 258.
- Lunn, M.P., R.A. Hughes, and P.J. Wiffen, Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database of Systematic Reviews, 2014(1). [PMC free article: PMC10711341] [PubMed: 24385423]
- 259.
- Waldfogel J.M., et al. Pharmacotherapy for diabetic peripheral neuropathy pain and quality of life: a systematic review. Neurology. 2017;88(20):1958–1967. [PubMed: 28341643]
- 260.
- Dy, S.M., et al., Preventing complications and treating symptoms of diabetic peripheral neuropathy. 2017. [PubMed: 28749633]
- 261.
- Hardy T., et al. Does treatment with duloxetine for neuropathic pain impact glycemic control? Diabetes Care. 2007;30(1):21–26. [PubMed: 17192327]
- 262.
- Sansone R.A., Sansone L.A. Pain, pain, go away: antidepressants and pain management. Psychiatry (Edgmont). 2008;5(12):16. [PMC free article: PMC2729622] [PubMed: 19724772]
- 263.
- Simpson D.A. Gabapentin and venlafaxine for the treatment of painful diabetic neuropathy. Journal of clinical neuromuscular disease. 2001;3(2):53–62. [PubMed: 19078655]
- 264.
- Rowbotham M.C., et al. Venlafaxine extended release in the treatment of painful diabetic neuropathy: a double-blind, placebo-controlled study. Pain. 2004;110(3):697–706. [PubMed: 15288411]
- 265.
- Wiffen, P.J., et al., Gabapentin for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews, 2017(6). [PMC free article: PMC6452908] [PubMed: 28597471]
- 266.
- Freeman R., Durso-DeCruz E., Emir B. Efficacy, safety, and tolerability of pregabalin treatment for painful diabetic peripheral neuropathy: findings from seven randomized, controlled trials across a range of doses. Diabetes care. 2008;31(7):1448–1454. [PMC free article: PMC2453685] [PubMed: 18356405]
- 267.
- Derry, S., et al., Pregabalin for neuropathic pain in adults. Cochrane Database of Systematic Reviews, 2019(1). [PMC free article: PMC6353204] [PubMed: 30673120]
- 268.
- Semel D., et al. Evaluation of the safety and efficacy of pregabalin in older patients with neuropathic pain: results from a pooled analysis of 11 clinical studies. BMC family practice. 2010;11(1):1–12. [PMC free article: PMC2988717] [PubMed: 21054853]
- 269.
- Evoy K.E., et al. Abuse and misuse of pregabalin and gabapentin: a systematic review update. Drugs. 2021;81(1):125–156. [PubMed: 33215352]
- 270.
- Gabapentin and risk of Severe Respiratory Depression. Drug Ther Bull. 2018;53(1):3–4. [PubMed: 29288148]
- 271.
- Meisenberg B., et al. Implementation of solutions to reduce opioid-induced oversedation and respiratory depression. American Journal of Health-System Pharmacy. 2017;74(3):162–169. [PubMed: 27993764]
- 272.
- Eipe N., Penning J. Postoperative respiratory depression associated with pregabalin: A case series and a preoperative decision algorithm. Pain Research and Management. 2011;16(5):353–356. [PMC free article: PMC3206785] [PubMed: 22059207]
- 273.
- Rains C., Bryson H.M. Topical capsaicin. Drugs & aging. 1995;7(4):317–328. [PubMed: 8535059]
- 274.
- Backonja M., et al. NGX-4010, a high-concentration capsaicin patch, for the treatment of postherpetic neuralgia: a randomised, double-blind study. The Lancet Neurology. 2008;7(12):1106–1112. [PubMed: 18977178]
- 275.
- Bonezzi C., et al. Capsaicin 8% dermal patch in clinical practice: an expert opinion. Expert opinion on pharmacotherapy. 2020;21(11):1377–1387. [PubMed: 32511032]
- 276.
- Derry, S. and R.A. Moore, Topical capsaicin (low concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews, 2012(9). [PMC free article: PMC6540838] [PubMed: 22972149]
- 277.
- Omana-Zapata I., et al. QX-314 inhibits ectopic nerve activity associated with neuropathic pain. Brain research. 1997;771(2):228–237. [PubMed: 9401743]
- 278.
- Devor M., Wall P.D., Catalan N. Systemic lidocaine silences ectopic neuroma and DRG discharge without blocking nerve conduction. Pain. 1992;48(2):261–268. [PubMed: 1589245]
- 279.
- Abdi S., Lee D.H., Chung J.M. The anti-allodynic effects of amitriptyline, gabapentin, and lidocaine in a rat model of neuropathic pain. Anesthesia & Analgesia. 1998;87(6):1360–1366. [PubMed: 9842827]
- 280.
- Sheets P.L., Jarecki B.W., Cummins T.R. Lidocaine reduces the transition to slow inactivation in Nav1. 7 voltage‐gated sodium channels. British journal of pharmacology. 2011;164(2b):719–730. [PMC free article: PMC3188891] [PubMed: 21232038]
- 281.
- Jasmin L., et al. The cold plate as a test of nociceptive behaviors: description and application to the study of chronic neuropathic and inflammatory pain models. Pain. 1998;75(2-3):367–382. [PubMed: 9583773]
- 282.
- Chaplan S.R., et al. Prolonged alleviation of tactile allodynia by intravenous lidocaine in neuropathic rats. The Journal of the American Society of Anesthesiologists. 1995;83(4):775–785. [PubMed: 7574057]
- 283.
- BARTLETT. E.E. and O. HUTASERANI, Xylocaine for the relief of postoperative pain. Anesthesia & Analgesia. 1961;40(3):296–304. [PubMed: 14448503]
- 284.
- Challapalli, V., et al., Systemic administration of local anesthetic agents to relieve neuropathic pain. Cochrane Database of Systematic Reviews, 2005(4). [PMC free article: PMC6483498] [PubMed: 16235318]
- 285.
- McGavin C., Gupta S., McHardy G. Treatment of chronic painful diabetic neuropathy with intravenous lidocaine inusion. British Medical Journal. 1986:173. [PMC free article: PMC1339043] [PubMed: 3080123]
- 286.
- Kastrup J., et al. Intravenous lidocaine infusion—a new treatment of chronic painful diabetic neuropathy? Pain. 1987;28(1):69–75. [PubMed: 3822496]
- 287.
- Viola V., Newnham H.H., Simpson R.W. Treatment of intractable painful diabetic neuropathy with intravenous lignocaine. Journal of Diabetes and its Complications. 2006;20(1):34–39. [PubMed: 16389165]
- 288.
- Tesfaye S., et al. Painful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assessment and management. Diabetes/metabolism research and reviews. 2011;27(7):629–638. [PubMed: 21695762]
- 289.
- Finnerup N.B., Sindrup S.H., Jensen T.S. Management of painful neuropathies. Handbook of Clinical Neurology. 2013;115:279–290. [PubMed: 23931787]
- 290.
- Duehmke, R.M., et al., Tramadol for neuropathic pain in adults. Cochrane Database of Systematic Reviews, 2017(6). [PMC free article: PMC6481580] [PubMed: 28616956]
- 291.
- Gaskell, H., et al., Oxycodone for neuropathic pain in adults. Cochrane Database of Systematic Reviews, 2016(7). [PMC free article: PMC6457997] [PubMed: 27465317]
- 292.
- Häuser W., et al. European* clinical practice recommendations on opioids for chronic noncancer pain–Part 1: Role of opioids in the management of chronic noncancer pain. European Journal of Pain. 2021;25(5):949–968. [PMC free article: PMC8248186] [PubMed: 33655607]
- 293.
- Hoffman E.M., et al. Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA neurology. 2017;74(7):773–779. [PMC free article: PMC5710534] [PubMed: 28531306]
- 294.
- Tesfaye, S., Painful diabetic neuropathy. Aetiology and nonpharmacological treatment. Clinical management of diabetic neuropathy. Humana Press, Totowa, New jersey, 1998.
- 295.
- Abuaisha B., Costanzi J., Boulton A. Acupuncture for the treatment of chronic painful peripheral diabetic neuropathy: a long-term study. Diabetes research and clinical practice. 1998;39(2):115–121. [PubMed: 9597381]
- 296.
- Garrow A.P., et al. Role of acupuncture in the management of diabetic painful neuropathy (DPN): a pilot RCT. Acupuncture in Medicine. 2014;32(3):242–249. [PubMed: 24657491]
- 297.
- Chen W., et al. Manual acupuncture for treatment of diabetic peripheral neuropathy: a systematic review of randomized controlled trials. PloS one. 2013;8(9):e73764. p. [PMC free article: PMC3771980] [PubMed: 24069229]
- 298.
- Kumar D., et al. Diabetic peripheral neuropathy: effectiveness of electrotherapy and amitriptyline for symptomatic relief. Diabetes Care. 1998;21(8):1322–1325. [PubMed: 9702441]
- 299.
- Bosi E., et al. Effectiveness of frequency-modulated electromagnetic neural stimulation in the treatment of painful diabetic neuropathy. Diabetologia. 2005;48(5):817–823. [PubMed: 15834546]
- 300.
- Bosi E., et al. Frequency-modulated electromagnetic neural stimulation (FREMS) as a treatment for symptomatic diabetic neuropathy: results from a double-blind, randomised, multicentre, long-term, placebo-controlled clinical trial. Diabetologia. 2013;56(3):467–475. [PMC free article: PMC3563945] [PubMed: 23238789]
- 301.
- Thakral G., et al. Electrical stimulation as an adjunctive treatment of painful and sensory diabetic neuropathy. Journal of diabetes science and technology. 2013;7(5):1202–1209. [PMC free article: PMC3876364] [PubMed: 24124947]
- 302.
- Tesfaye S., et al. Electrical spinal-cord stimulation for painful diabetic peripheral neuropathy. The Lancet. 1996;348(9043):1698–1701. [PubMed: 8973433]
- 303.
- Kumar K., Toth C., Nath R.K. Spinal cord stimulation for chronic pain in peripheral neuropathy. Surgical neurology. 1996;46(4):363–369. [PubMed: 8876718]
- 304.
- de Vos C.C., et al. Spinal cord stimulation in patients with painful diabetic neuropathy: a multicentre randomized clinical trial. PAIN. 2014;155(11):2426–2431. [PubMed: 25180016]
- 305.
- van Beek M., et al. Sustained treatment effect of spinal cord stimulation in painful diabetic peripheral neuropathy: 24-month follow-up of a prospective two-center randomized controlled trial. Diabetes Care. 2015;38(9):e132–e134. [PubMed: 26116722]
- 306.
- Slangen R., et al. Spinal cord stimulation and pain relief in painful diabetic peripheral neuropathy: a prospective two-center randomized controlled trial. Diabetes Care. 2014;37(11):3016–3024. [PubMed: 25216508]
- 307.
- van Beek M., et al. Severity of neuropathy is associated with long-term spinal cord stimulation outcome in painful diabetic peripheral neuropathy: five-year follow-up of a prospective two-center clinical trial. Diabetes Care. 2018;41(1):32–38. [PubMed: 29109298]
- 308.
- Sills S. Treatment of painful polyneuropathies of diabetic and other origins with 10 kHz SCS: a case series. Postgraduate medicine. 2020;132(4):352–357. [PubMed: 32073352]
- 309.
- Galan V., et al. 10-kHz spinal cord stimulation treatment for painful diabetic neuropathy: results from post-hoc analysis of the SENZA-PPN study. Pain Management. 2020;10(5):291–300. [PubMed: 32779967]
- 310.
- Bradley K., Redwood City C. Paresthesia-independence: an assessment of technical factors related to 10 kHz paresthesia-free spinal cord stimulation. Pain Physician. 2017;20:331–341. [PubMed: 28535555]
- 311.
- Petersen E.A., et al. Effect of high-frequency (10-kHz) spinal cord stimulation in patients with painful diabetic neuropathy: a randomized clinical trial. JAMA neurology. 2021 [PMC free article: PMC8022268] [PubMed: 33818600]
- 312.
- Chaudhry V., et al. Practice Advisory: utility of surgical decompression for treatment of diabetic neuropathy: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2006;66(12):1805–1808. [PubMed: 16801641]
- 313.
- Chaudhry, V., J. Russell, and A. Belzberg, Decompressive surgery of lower limbs for symmetrical diabetic peripheral neuropathy. Cochrane Database of Systematic Reviews, 2008(3). [PMC free article: PMC8990523] [PubMed: 18646138]
- 314.
- Selvarajah D., et al. Multicentre, double-blind, crossover trial to identify the Optimal Pathway for TreatIng neurOpathic paiN in Diabetes Mellitus (OPTION-DM): study protocol for a randomised controlled trial. Trials. 2018;19(1):1–12. [PMC free article: PMC6196556] [PubMed: 30348206]
- 315.
- Vinik A.I., Erbas T. Diabetic autonomic neuropathy. Handbook of clinical neurology. 2013;117:279–294. [PubMed: 24095132]
- 316.
- Pfeifer M.A., et al. Autonomic neural dysfunction in recently diagnosed diabetic subjects. Diabetes care. 1984;7(5):447–453. [PubMed: 6499637]
- 317.
- Stansberry K.B., et al. Impairment of peripheral blood flow responses in diabetes resembles an enhanced aging effect. Diabetes care. 1997;20(11):1711–1716. [PubMed: 9353614]
- 318.
- Stansberry K.B., et al. Impaired peripheral vasomotion in diabetes. Diabetes care. 1996;19(7):715–721. [PubMed: 8799625]
- 319.
- Stansberry K.B., et al. Primary nociceptive afferents mediate the blood flow dysfunction in non-glabrous (hairy) skin of type 2 diabetes: a new model for the pathogenesis of microvascular dysfunction. Diabetes Care. 1999;22(9):1549–1554. [PubMed: 10480524]
- 320.
- Haak E.S., et al. The effect of α-lipoic acid on the neurovascular reflex arc in patients with diabetic neuropathy assessed by capillary microscopy. Microvascular Research. 1999;58(1):28–34. [PubMed: 10388600]
- 321.
- Ziegler D. Diabetic cardiovascular autonomic neuropathy: prognosis, diagnosis and treatment. Diabetes/metabolism reviews. 1994;10(4):339–383. [PubMed: 7796704]
- 322.
- Valensi P. Diabetic autonomic neuropathy: what are the risks? Diabetes & metabolism. 1998;24:66–72. [PubMed: 9881235]
- 323.
- Vinik A.I., et al. Neurovascular function and sudorimetry in health and disease. Current diabetes reports. 2013;13(4):517–532. [PubMed: 23681491]
- 324.
- Vinik A.I., Maser R., Nakave A. Diabetic cardiovascular autonomic nerve dysfunction. US Endocr Dis. 2007;2:2–9.
- 325.
- Karamitsos D., et al. The natural history of recently diagnosed autonomic neuropathy over a period of 2 years. Diabetes research and clinical practice. 1998;42(1):55–63. [PubMed: 9884034]
- 326.
- Ziegler D., Gries F.A. α-Lipoic acid in the treatment of diabetic peripheral and cardiac autonomic neuropathy. Diabetes. 1997;46 Supplement 2:S62–S66. [PubMed: 9285502]
- 327.
- Athyros V., et al. Long-term effect of converting enzyme inhibition on circadian sympathetic and parasympathetic modulation in patients with diabetic autonomic neuropathy. Acta cardiologica. 1998;53(4):201–209. [PubMed: 9842405]
- 328.
- Gæde P., et al. Intensified multifactorial intervention in patients with type 2 diabetes mellitus and microalbuminuria: the Steno type 2 randomised study. The Lancet. 1999;353(9153):617–622. [PubMed: 10030326]
- 329.
- Malmberg K., et al. Glycometabolic state at admission: important risk marker of mortality in conventionally treated patients with diabetes mellitus and acute myocardial infarction: long-term results from the Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI) study. Circulation. 1999;99(20):2626–2632. [PubMed: 10338454]
- 330.
- Kendall D.M., et al. Pancreas transplantation restores epinephrine response and symptom recognition during hypoglycemia in patients with long-standing type I diabetes and autonomic neuropathy. Diabetes. 1997;46(2):249–257. [PubMed: 9000702]
- 331.
- Burger A.J., et al. Effect of glycemic control on heart rate variability in type I diabetic patients with cardiac autonomic neuropathy. The American journal of cardiology. 1999;84(6):687–691. [PubMed: 10498140]
- 332.
- Laederach-Hofmann K., Weidmann P., Ferrari P. Hypovolemia contributes to the pathogenesis of orthostatic hypotension in patients with diabetes mellitus. The American journal of medicine. 1999;106(1):50–58. [PubMed: 10320117]
- 333.
- Denq J.-C., et al. Efficacy of compression of different capacitance beds in the amelioration of orthostatic hypotension. Clinical Autonomic Research. 1997;7(6):321–326. [PubMed: 9430805]
- 334.
- Annese V., et al. Gastrointestinal motor dysfunction, symptoms, and neuropathy in noninsulin-dependent (type 2) diabetes mellitus. Journal of clinical gastroenterology. 1999;29(2):171–177. [PubMed: 10478880]
- 335.
- Melga P., et al. Chronic administration of levosulpiride and glycemic control in IDDM patients with gastroparesis. Diabetes Care. 1997;20(1):55–58. [PubMed: 9028694]
- 336.
- Stacher G., et al. Cisapride versus placebo for 8 weeks on glycemic control and gastric emptying in insulin-dependent diabetes: a double blind cross-over trial. The Journal of Clinical Endocrinology & Metabolism. 1999;84(7):2357–2362. [PubMed: 10404803]
- 337.
- Barone J.A. Domperidone: a peripherally acting dopamine2-receptor antagonist. Annals of Pharmacotherapy. 1999;33(4):429–440. [PubMed: 10332535]
- 338.
- Silvers D., et al. Domperidone in the management of symptoms of diabetic gastroparesis: efficacy, tolerability, and quality-of-life outcomes in a multicenter controlled trial. Clinical therapeutics. 1998;20(3):438–453. [PubMed: 9663360]
- 339.
- Erbas T., et al. Comparison of metoclopramide and erythromycin in the treatment of diabetic gastroparesis. Diabetes Care. 1993;16(11):1511–1514. [PubMed: 8299441]
- 340.
- Camilleri M., Acosta A. Emerging treatments in Neurogastroenterology: relamorelin: a novel gastrocolokinetic synthetic ghrelin agonist. Neurogastroenterology & Motility. 2015;27(3):324–332. [PMC free article: PMC4424792] [PubMed: 25545036]
- 341.
- Alam U., Asghar O., Malik R.A. Diabetic gastroparesis: Therapeutic options. Diabetes Therapy. 2010;1(1):32–43. [PMC free article: PMC3118275] [PubMed: 22127672]
- 342.
- Abell T., et al. Treatment of gastroparesis: a multidisciplinary clinical review: The American Motility Society Task Force on Gastroparesis (members in alphabetical order). Neurogastroenterology & Motility. 2006;18(4):263–283. [PubMed: 16553582]
- 343.
- Vinik A., Richardson D. Erectile dysfunction in diabetes: pills for penile failure. Clinical Diabetes. 1998;16(3):108–109.
- 344.
- Vinik A., Richardson D. Erectile dysfunction in diabetes. Clinical Diabetes. 1996;14(5):111–121.
- 345.
- Rendell M.S., et al. Sildenafil for treatment of erectile dysfunction in men with diabetes: a randomized controlled trial. Jama. 1999;281(5):421–426. [PubMed: 9952201]
- 346.
- Goldstein I., et al. Vardenafil, a new phosphodiesterase type 5 inhibitor, in the treatment of erectile dysfunction in men with diabetes: a multicenter double-blind placebo-controlled fixed-dose study. Diabetes care. 2003;26(3):777–783. [PubMed: 12610037]
- 347.
- Enzlin P., et al. Diabetes mellitus and female sexuality: a review of 25 years’ research. Diabetic Medicine. 1998;15(10):809–815. [PubMed: 9796879]
- 348.
- Shaw J., et al. A randomised controlled trial of topical glycopyrrolate, the first specific treatment for diabetic gustatory sweating. Diabetologia. 1997;40(3):299–301. [PubMed: 9084967]
- 349.
- Edick C.M. Oral glycopyrrolate for the treatment of diabetic gustatory sweating. Annals of Pharmacotherapy. 2005;39(10):1760–1760. [PubMed: 16159997]
- 350.
- Meyer C., et al. IMPROVED GLUCOSE COUNTERREGULATION AND AUTONOMIC SYMPTOMS AFTER INTRAPORTAL ISLET TRANSPLANTS ALONE IN PATIENTS WITH LONG-STANDING TYPE I DIABETES MELLITUS1. Transplantation. 1998;66(2):233–240. [PubMed: 9701271]
- ABSTRACT
- INTRODUCTION
- SCOPE OF THE PROBLEM
- CLASSIFICATION OF DIABETIC NEUROPATHIES
- NATURAL HISTORY OF DIABETIC NEUROPATHIES (DN)
- MODIFIABLE RISK FACTORS FOR DPN INCIDENCE AND PROGRESSION
- PATHOGENESIS OF DIABETIC NEUROPATHIES
- CLINICAL PRESENTATION
- DIAGNOSIS OF DIABETIC NEUROPATHIES
- TREATMENT OF DIABETIC POLYNEUROPATHIES
- PAINFUL DIABETIC PERIPHERAL NEUROPATHY
- AUTONOMIC NEUROPATHY
- DIABETIC NEUROPATHIES: PROSPECTS FOR THE FUTURE
- ACKNOWLEDGEMENTS
- REFERENCES
- Review Painful and non-painful diabetic neuropathy, diagnostic challenges and implications for future management.[Brain. 2021]Review Painful and non-painful diabetic neuropathy, diagnostic challenges and implications for future management.Jensen TS, Karlsson P, Gylfadottir SS, Andersen ST, Bennett DL, Tankisi H, Finnerup NB, Terkelsen AJ, Khan K, Themistocleous AC, et al. Brain. 2021 Jul 28; 144(6):1632-1645.
- Review Diabetic Peripheral Neuropathy: Epidemiology, Diagnosis, and Pharmacotherapy.[Clin Ther. 2018]Review Diabetic Peripheral Neuropathy: Epidemiology, Diagnosis, and Pharmacotherapy.Iqbal Z, Azmi S, Yadav R, Ferdousi M, Kumar M, Cuthbertson DJ, Lim J, Malik RA, Alam U. Clin Ther. 2018 Jun; 40(6):828-849. Epub 2018 Apr 30.
- Review Diabetic autonomic neuropathy.[Diabetes Care. 2003]Review Diabetic autonomic neuropathy.Vinik AI, Maser RE, Mitchell BD, Freeman R. Diabetes Care. 2003 May; 26(5):1553-79.
- Review Diabetic peripheral neuropathy essentials: a narrative review.[Ann Palliat Med. 2023]Review Diabetic peripheral neuropathy essentials: a narrative review.Chang MC, Yang S. Ann Palliat Med. 2023 Mar; 12(2):390-398. Epub 2023 Feb 8.
- Review Pharmacotherapy of Painful Diabetic Neuropathy: A Clinical Update.[Sisli Etfal Hastan Tip Bul. 2022]Review Pharmacotherapy of Painful Diabetic Neuropathy: A Clinical Update.James CF, Tripathi S, Karampatou K, Gladston DV, Pappachan JM. Sisli Etfal Hastan Tip Bul. 2022; 56(1):1-20. Epub 2022 Mar 28.
- Diabetic Neuropathies - EndotextDiabetic Neuropathies - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...