

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Pituitary adenomas comprise approximately 10-20% of intracranial tumors. Non-functioning pituitary adenomas (NFPAs) are benign adenohypophyseal tumors not associated with clinical evidence of hormonal hypersecretion. NFPAs comprise different histological subtypes, classified according to their immunostaining to different adenohypophyseal hormones and transcription factors. The silent gonadotroph adenoma is the most common subtype, followed by corticotroph, PIT1 (POU1F1) gene lineage, and null cell tumors. Patients with NFPAs usually come to medical attention as a result of “mass effects” symptoms such as headaches, visual disorders, and/or cranial nerve dysfunction caused by lesions large enough to damage surrounding structures. Hypopituitarism, caused by the compression of the normal anterior pituitary, and hyperprolactinemia due to pituitary stalk deviation can also be present. Some cases may be diagnosed incidentally through imaging studies performed for other purposes. Patients with NFPAs should undergo hormonal, clinical, and laboratory evaluation to rule out hyper and hypopituitarism. Assessment of prolactin and IGF-1 levels have been recommended in all patients whereas screening for cortisol excess is suggested in the presence of clinical symptoms. Impairment of pituitary function should be assessed by baseline hormonal measurements and/or stimulatory tests, if needed. Patients in whom the tumor abuts the optic chiasm should be submitted to visual field perimetry. Surgical resection is the primary treatment for symptomatic patients with NFPAs, i.e., those with neuro-ophthalmologic complaints and/or tumors affecting the optic pathway. Visual deficits and, less commonly, hormone deficiencies may improve following surgical treatment although new hormone deficiencies may also occasionally develop after a surgical approach. For patients with residual NFPAs following transsphenoidal surgery, a therapeutic attempt using cabergoline can be made according to clinical judgment in individual cases. Radiotherapy in the postoperative period is not consensual and is generally reserved for cases of tumors not completely resected by surgery, those cases that present progressive tumor growth during follow-up, or for patients who, at diagnosis, already have tumors with aggressive features. Highly aggressive tumors need special care during follow-up, including temozolomide with or without radiotherapy complementation or new potential emerging treatments. For patients with asymptomatic NFPAs a “watch and wait” option is reasonable. Follow up is individualized and should consider tumor size, prior treatments, and clinical symptoms. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Pituitary adenomas are common, predominantly indolent neoplasms comprising approximately 10-20% of intracranial tumors (1,2). Non-functioning pituitary adenomas (NFPAs) are benign neoplasms that originate from the adenohypophyseal cells and are not associated with clinical evidence of hormonal hypersecretion (3). They comprise a large and heterogeneous group, representing a sizeable proportion (22% to 54% in different series) of all pituitary adenomas (4-7). Malignant transformation in a non-functioning pituitary adenoma (pituitary carcinoma) is extremely rare and is characterized by craniospinal or systemic dissemination (8).
NFPAs can be further classified according to their pituitary hormone and transcription factor profile, as defined by the 2017 World Health Organization (WHO) classification for endocrine tumors (9). Tumors that express one or more anterior pituitary hormones or their transcription factors with immunohistochemistry (IHC) but do not secrete hormones at a clinically relevant level can be referred to as silent pituitary adenomas (SPAs) (10,11). As a consequence, the definition of “null cell adenoma” is now limited to an exceptionally rare primary adenohypophyseal tumor that shows immunonegativity for all adenohypophyseal hormones as well as for cell-type specific transcription factors (12) - Figure 1.

Figure 1
- Classification of non-functioning pituitary adenomas rely upon clinical features and histopathological data. Reprinted with permission from Drummond et al., 2019
The term “totally silent” has been proposed to be used when a patient with an NFPA presents basal and stimulated serum concentrations of the correspondent hormones within the normal range and there are no clinical signs or symptoms that can be attributed to hormone excess (13). The term “clinically silent” may be used when NFPAs secrete hormonal products that cause an elevation of the serum concentration but do not result in clinical signs or symptoms of hormonal hypersecretion (13). Some cases are referred to as “whispering” adenomas with borderline, mild, often overlooked, clinical symptoms and signs (14,15).
EPIDEMIOLOGY
The prevalence of NFPAs is variable and is often based upon autopsy or magnetic resonance imaging (MRI) series. Data from Europe, North and South America have estimated that the prevalence of clinically relevant NFPAs is 7–41.3 cases per 100,000 of population (16). This is likely an underestimate of the true prevalence, as many NFPAs go undiagnosed until they are large enough to cause mass effect or are accidentally discovered. Data are discordant about gender predominance and the peak occurrence is from the fourth to the eighth decade. A recent population-based study from South Korea showed an annual incidence of 3.5 cases/ 100,000 population for NFPAs, which is noticeably higher than the annual incidences previously reported in other countries, such as Sweden, Finland, and Argentina (17). These differences may be related to genetic and environmental factors in Asian populations or inter-study heterogeneity and may also reflect the good local access to diagnostic evaluations using magnetic resonance imaging and computed tomography.
CLINICAL PRESENTATION
The absence of clinical manifestations of hormonal hypersecretion usually results in significant diagnostic delay and therefore NFPAs may not be diagnosed until they cause mass effects to surrounding structures (3), causing symptoms such as headaches, visual disorders, and/or cranial nerve dysfunction. Other manifestations are hormone deficiencies or hyperprolactinemia due to pituitary stalk deviation and, less frequently, pituitary apoplexy (18,19). Additionally, some cases may be diagnosed incidentally through imaging studies performed for other purposes, the so-called pituitary incidentaloma.
Neurologic Manifestations
VISUAL IMPAIRMENT
Impaired vision, caused by suprasellar extension of the adenoma that compresses the optic chiasm, is the most common neuro-ophthalmological symptom (19). Different types of visual defects depend on the degree and site of optic nerve compression. Both eyes are usually affected, although a significant proportion of patients may have unilateral or altitudinal problems in 33 and 16% of the cases, respectively (20). Diplopia, induced by oculomotor nerve compression resulting from parasellar expansion of the adenoma may occur, and the fourth, fifth and sixth cranial nerves may also be occasionally involved when there is parasellar expansion (16). Nevertheless, the typical visual field defect associated with pituitary tumors is bitemporal hemianopia, reported in approximately 40% of the patients.
HEADACHE
Headaches, the second most common neurologic symptom, occur in 19-75% of patients with pituitary tumors, regardless of size (21). In a retrospective case series of incidentally-discovered NFPAs, headache was present in approximately 20% of the cases (22). In a recent prospective series, 138 out of 269 patients with NFPAs complained of tumor-related symptoms and, among them, 23% presented with headaches (23). Although it is not always clear whether the presenting headache is related to the tumor, proposed mechanisms for headache include increased intrasellar pressure, stretching of dural membrane pain receptors, and activation of trigeminal pain pathways (16,24). Cerebrospinal fluid (CSF) rhinorrhea, associated or not with headache, can occur in cases where the tumor causes erosion of the sellar floor and extends inferiorly to the sphenoid sinus.
PITUITARY APOPLEXY
Pituitary apoplexy (sudden hemorrhage into a pituitary adenoma) is rare. According to a retrospective case series of 485 Mexican patients with NFPAs, pituitary apoplexy was the initial presentation in 8% of the cases (25). It causes acute onset of a severe headache associated with visual disturbances and can occur in all types of pituitary tumors, although some series suggest pituitary apoplexy might be more common in NFPAs than other adenomas subtypes (26). It has been suggested that the combination of the high metabolism of pituitary adenomas combined with their special blood supply would make them more prone to vascular events (27). Pituitary apoplexy may occur without an identified risk factor, but it has also been reported as being related to pregnancy, use of anticoagulants, surgical procedures, as well as in association with dynamic tests, such as thyrotropin-releasing hormone (TRH), gonadotropin-releasing hormone (GnRH), and insulin-tolerance stimulation tests (28).
The actual long-term risk of apoplexy in NFPAs has not been clearly defined (29). In a Japanese prospective cohort study of 42 asymptomatic patients with NFPAs (mean initial tumor size of 18.3 mm) followed-up for approximately four years, pituitary apoplexy was reported in 9.5% of the cases (30). A systematic review and meta-analysis evaluating the outcomes of patients with NFPAs and pituitary incidentalomas who were treated conservatively and followed-up for mean period of 3.9 years showed that the development of pituitary apoplexy was rare (0.2/100 patients-year), although a trend for a greater incidence of pituitary apoplexy was seen in macroadenomas compared with microadenomas, with a reported incidence of apoplexy of 1.1% per year (31). These findings are in line with two recent retrospective studies including a Korean series of 197 patients with NFPAs without mass effect symptoms or pituitary apoplexy at baseline who were followed-up for a median of 37 months (32) and an Italian series of 296 patients with incidentally discovered NFPAs (macroadenomas in 49% of the cases) who we followed-up for approximately three years (33). Five out of 169 patients with macroadenomas developed pituitary apoplexy with an overall incidence of 0.83 per 100 patients-years in the Korean series while no cases of pituitary apoplexy were reported in the Italian series.
Endocrine Manifestations
HORMONAL DEFICIENCIES
Most patients with nonfunctioning pituitary macroadenomas present with deficiency of at least one pituitary hormone resulting from the compression of the normal anterior pituitary and/or pituitary stalk, preventing the stimulation of pituitary cells by hypothalamic factors. Hypogonadism can result from either a direct compressive effect on gonadotropic cells or by stalk compression-induced hyperprolactinemia that inhibits the pulsatile secretion of gonadotropin releasing hormone via interfering with hypothalamic kisspeptin-secreting cells (34). This “disconnection hyperprolactinemia” usually <2000mIU/L (95 ng/mL) (35) is characterized by compression of the pituitary stalk, which prevents the arrival of dopamine to the anterior pituitary, the main inhibitor of prolactin (stalk effect). GH and gonadotroph axes are most commonly affected, followed by adrenal insufficiency and central hypothyroidism (16,23,32).
HORMONAL EXCESS
Gonadotroph adenomas are usually considered to be "nonfunctioning" although they can secrete intact gonadotropins, as they do not generally result in a clinical syndrome. Occasionally, gonadotroph adenomas secrete primarily FSH but also LH in quantities high enough to raise serum gonadotropin levels, which in turn, may lead to the development of some specific symptoms, such as ovarian hyperstimulation in young women (36-38) or, more rarely, precocious puberty or testicular enlargement in men. In addition, low serum LH:FSH ratios (usually < 1.0) have been described in clinically-secreting gonadotroph adenomas (39). Measurement of α-subunit may also contribute to a pre-operative diagnosis in clinically silent but biochemically-secreting NFPAs, as it may be the sole biochemical marker of the gonadotroph subtype in a number of cases. In addition, circulating FSH, LH and α-subunit levels can help the post-operative surveillance of these patients (39).
HISTOPATHOLOGICAL CLASSIFICATION
The 2017 WHO classification for endocrine tumors (9) defines pituitary adenomas, including NFPAs, according to their pituitary hormone and transcription factor profile. It is noteworthy that in this classification system, IHC forms the basis of the new categorization, in which an adenohypophyseal cell lineage designation of the pituitary adenoma has replaced the concept of a “hormone-producing pituitary adenoma” (40) -Table 1.
Table 1
– Classification of Silent Pituitary Adenomas According to Adenohypophyseal Hormones and Transcription Factors.
| Cell Lineage | Hormone Staining | Transcription Factors and Other Co-Factors |
|---|---|---|
| Somatotroph adenoma Sparsely granulated Densely granulated | GH, α-subunit Weak and patchy Diffuse and strong | PIT-1 |
| Lactotroph adenomas Sparsely granulated Densely granulated Acidophil stem-cell adenoma | PRL Perinuclear Diffuse PRL Focal and variable PRL, GH | PIT-1, ERα |
| Thyrotroph adenomas | TSHβ, α-subunit | PIT-1, GATA2 |
| Corticotroph adenomas Densely granulated (type I) Sparsely granulated (type II) Crooke-cell | ACTH Diffuse and strong ACTH Weak and patchy ACTH Periphery (ring-like) | TPIT |
| Gonadotroph adenomas | FSHβ, LHβ, α-subunit | SF1, GATA2, ERα |
| Null cell adenomas | None | None |
| Plurihormonal PIT-1-positive adenomas Adenomas with unusual IHC combinations | GH, PRL, TSHβ±, α-subunit Various combinations | PIT-1 |
Adapted from Mete et al, 2017.
Indeed, the prevalence of the different histological subtypes of NFPAs is dependent on the extent of the IHC profile. According to a large retrospective case series (11), the gonadotroph adenoma is the most common subtype, followed by corticotroph, PIT1 (GH/ Prolactin/TSH) lineage and null cell - Figure 2.
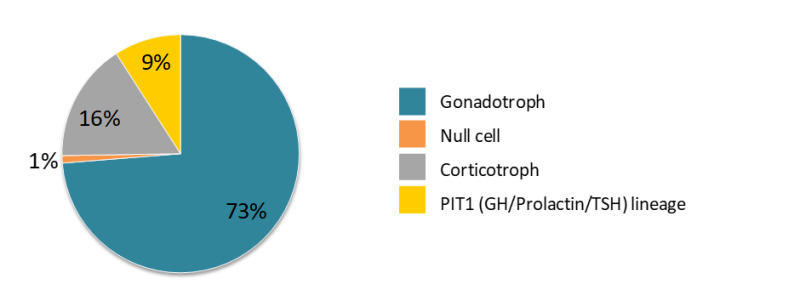
Figure 2
- Prevalence of silent pituitary adenoma subtypes according to immunochemistry for anterior pituitary hormones and transcription factors. Adapted from Nishioka et al., 2015
Pituitary hormones gene expression analysis using real-time quantitative PCR (RT-qPCR) has been shown to complement the IHC classification of pituitary tumors, according to a large observational, cross-sectional study evaluating 268 patients (41). Addition of RT-qPCR analysis reduced the proportion of null-cell subtype in this series from 36% to 19%. Lower specific adenohypophyseal hormone gene expression was observed in some SPA variants when compared with their secreting counterparts, including somatotroph, lactotroph, mixed somatotroph-lactotroph and plurihormonal adenomas, which could contribute to the absence of endocrine manifestations.
The European Pituitary Pathology Group has recently proposed a standardized report for the diagnosis of pituitary adenomas which includes a multi-step approach, comprising the summary of clinical and neuroimaging features, IHC for hormones and transcription factors, assessment of proliferation and, when indicated, the use of markers predictive of treatment response (42). This proposition highlights the role of the pituitary pathologist as part of a multidisciplinary pituitary team helping the clinician define the most appropriate post-operative strategy for each patient, including appropriate follow-up and early recognition and treatment of potentially aggressive tumors (43).
Subtypes of NFPAs
NULL CELL ADENOMAS AND SILENT GONADOTROPH ADENOMAS
Null cell adenomas and silent gonadotroph adenomas (SGAs) were previously misunderstood to be the same type of tumor. The addition of immunostaining for steroidogenic factor 1 (SF1) as a tool in the diagnosis of pituitary lesions has shown that many LH/FSH immunonegative adenomas are in fact SGAs (44), which comprise almost 80% of resected NFPAs. The distinction between SGAs and null cell adenomas is of clinical relevance because true null cell adenomas are rare and likely to be more invasive and aggressive than SGAs (45). In addition, these rare cases of ‘null cell adenomas’ may require a broad panel of immunohistochemistry in order to exclude a sellar metastasis of a neuroendocrine tumor (NET) from another organ. Several markers, including TTF1, serotonin, ATRX, DAXX and CDX2 may be useful in the differential diagnosis between an NFPA and a metastatic NET in the sellar region (46). Data from a retrospective case series of 516 patients with NFPAs have shown that from the 23% of the tumors initially classified as null cell adenomas by using only classical pituitary hormone IHC, only 5% of them remained as true null cell type. Indeed, immunostaining using lineage-specific markers, namely, PIT-1 (coded by the POU class 1 homeobox 1 (POU1F1) gene), SF1, TPIT (coded by the T-Box transcription factor 19 (TBX19) gene) and estrogen receptor-α (ERα) provided tumor reclassification in 95% of cases (11).
SILENT CORTICOTROPH ADENOMAS
Silent corticotroph adenomas (SCAs) are characterized by the absence of clinical features of Cushing syndrome, along with normal circadian cortisol secretion (totally silent) or elevated ACTH (clinically silent) (5,47-49). They currently account for approximately 15% of NFPAs, an underestimated proportion, since IHC for the transcription factor TPIT, a marker of corticotroph differentiation which regulates the proopiomelanocortin (POMC) lineage giving origin to the corticotrophs, is still not widely available (50). For instance, in a recent retrospective series, inclusion of IHC for TPIT allowed identification of eight SCAs out of 18 (44%) pituitary tumors previously classified as null cell adenomas (51).
SCAs often present themselves as macroadenomas associated with mass-related symptoms. In comparison with SGAs, SCAs show female preponderance, are more frequently giant adenomas, and are more often associated with marked cavernous sinus invasion (49). Importantly, the presence of multiple microcysts in T2-weighted pituitary MRI sequences in a NFPA has a high specificity (> 90%) for the corticotroph subtype. The first study showed that multiple microcysts were present in 76% (13/17) of SCAs as opposed to 5% (3/60) of SGAs (52) while a more recent study showed comparable results, as multiple microcysts were present in 58% (7/12) SCAs but in only 9.5% of SGAs (53). Histologically, SCAs can be further divided into type 1 (densely granulated), type 2 (sparsely granulated) and Crooke-cell adenoma. Type 1 SCAs show strong ACTH immunoreactivity whereas type 2 SCAs resemble the rare chromophobe corticotroph adenoma and show weak and focal ACTH immunoreactivity. Type 2 SCAs seem to be more common and are likely to display higher expression of migration and proliferation factors compared with type 1 SCAs (5,11,40). Clinically silent Crooke-cell adenoma is a rare yet highly aggressive subtype as it carries significant risk of morbidity (54). Since SCAs are nonfunctional, an important point to note is the absence of Crooke’s hyalinization in the normal surrounding pituitary gland, as there is no exposure to high circulating glucocorticoid levels to promote hyaline deposits of cytokeratin filaments in the cytoplasm of normal corticotrophs (49).
The transformation of a silent corticotroph tumor into Cushing disease has been described, although the mechanism involved in this phenomenon is not yet well understood (55,56). It has been proposed that the clinical manifestations of Cushing disease are dependent on the processing of the pro-hormone POMC in corticotrophs. The pro-hormone convertase 1/3 (PC1/3) is involved in the post-translational processing of POMC into mature and biologically active ACTH. SCAs show a decrease in PC1/3 expression associated with a down-regulation of PC1/3 genes compared with corticotroph adenomas associated with Cushing disease (47). An attractive and more likely hypothesis involves epigenetic mechanisms: DNA hypermethylation of regulatory regions could lead to reduced expression of POMC transcripts impairing the production of secreted ACTH. Differences in methylation status were observed when comparing ACTH-secreting pituitary tumors and SCAs: the second promoter was highly methylated in SCAs, partially demethylated in normal pituitary tissue and highly demethylated in pituitary and ectopic ACTH-secreting tumors (57).
SILENT SOMATOTROPH ADENOMAS
Silent somatotroph adenomas are PIT1 and GH-immunoreactive tumors without clinical and biological signs of acromegaly. They represent approximately 2-4% of all pituitary adenomas in surgical case series (58). Patients with silent somatotroph adenomas usually present with normal pre-operative GH and IGF-1 levels but there have been few reports of "clinically silent" cases, with non-suppressible serum GH and elevated IGF-1 levels (59).
Similar to secreting somatotroph adenomas, silent somatotroph adenomas are classified into densely granulated and sparsely granulated types, based on the presence and pattern of the low molecular weight cytokeratin staining. More than 50% of silent somatotroph adenoma cases constitute mixed GH–PRL adenomas, a proportion twofold higher than somatotroph adenomas causing acromegaly (60). Unlike clinically functioning somatotroph tumors, the silent ones are more frequently sparsely granulated with more aggressive behavior and a lower response to somatostatin analog therapy (61). Furthermore, silent somatotroph adenomas are more frequent in females, present at a younger age, are larger, more invasive, and recur earlier and more frequently than their secreting counterparts (62).
SILENT THYROTROPH ADENOMAS
Silent thyrotroph adenomas usually present TSHβ, α-subunit, and PIT-1 expression on IHC in a variable manner (40). They are more frequent when compared with their functioning counterparts and seem to behave similarly regarding treatment outcomes and recurrence rates (63). In a recent retrospective series of 20 patients with silent thyrotroph adenomas who underwent transsphenoidal surgery, 95% were macroadenomas and 85% showed extrasellar growth. Gross total tumor resection was achieved in 45% while major tumor progression or recurrence occurred in 10% of the patients over a median follow-up period of 18.5 months (64). These results show that, although silent thyrotroph adenomas tend to be large and invasive tumors, good overall outcomes with low complication rates can be achieved.
SILENT LACTOTROPH ADENOMAS
Clinically silent lactotroph adenomas are rare. The positive prolactin staining by IHC with no clinical signs of hyperprolactinemia is usually encountered concomitantly with GH positive staining (silent mixed somatotroph-lactotroph adenoma) (61). They can also express ERα on immunostaining (9).
PLURIHORMONAL ADENOMAS
According to the WHO 2017 classification, plurihormonal adenomas can be classified on the basis of their transcription factor expression into two groups: 1) “PIT1-positive adenomas” (previously known as silent subtype 3 pituitary adenoma); and 2) plurihormonal with more than one transcription factor, termed “plurihormonal adenomas with unusual immunohistochemical combinations” (PAWUC) (9,40). In a recent large retrospective series comprising 665 patients who underwent endoscopic endonasal transsphenoidal surgery, plurihormonal adenomas were identified in 4% of the cases (18 patients had PAWUC and 9 patients were diagnosed with PIT-1 positive adenomas); 24 (89%) were macroadenomas (including 6 giant adenomas) and cavernous sinus invasion was found in 12 patients (44%) (65).
PIT1-positive plurihormonal adenomas are a distinct entity, with reportedly aggressive behavior. A single-center retrospective case series reported a prevalence of 0.9% for ‘silent adenomas type 3’ among resected pituitary tumors over a period of 13 years. All tumors were macroadenomas, aggressive, invasive, with a high rate of persistent/recurrent disease (66). Interestingly, these tumors may present clinical symptoms of hormone excess, such as acromegaly, hyperthyroidism or marked hyperprolactinemia (67) and the diagnosis of a PIT-1 positive plurihormonal adenoma should be kept in mind in case of large and invasive NFPA tumors in young patients, particularly those showing TSH and often minor reactivities for PRL and GH as well as cytologic atypia in conjunction with an elevated Ki-67 labelling index (66).
PAWUC are also characterized by aggressive behavior and higher rates of cavernous sinus invasion. A recent single-center retrospective series comparing clinical characteristics, outcome parameters and rate of invasiveness of 22 PAWUC to 51 SGAs showed that patients with PAWUC were younger, were more likely to present with intraoperatively signs of tumor invasion and less likely to achieve gross-total resection than patients with SGAs, suggesting a more aggressive behavior of NFPAs if additional transcription factors other than SF1, GATA2/3 and ER α are expressed within the tumor cells (68).
EVALUATION
According to current practice guidelines on incidentally discovered sellar masses, all patients, including those without symptoms, should undergo hormonal, clinical, and laboratory evaluation for hyper and hypopituitarism (69). The extent of the evaluation is still debatable and has commonly relied on clinical experience - assessment of prolactin and IGF-1 levels have been recommended, whereas screening for cortisol excess (i.e., overnight 1mg dexamethasone and/or late-evening salivary cortisol and/or urinary-free cortisol) is suggested in the presence of clinical symptoms, while measurement of ACTH levels is not routinely recommended. (69).
Patients with macroadenoma, especially those >3 cm and with normal or slightly elevated prolactin, must have their serum prolactin diluted in order to exclude the "hook effect" (70,71). This effect occurs when excessively high levels of this hormone interfere with the formation of the complex antibody-antigen-antibody sandwich, and a prolactinoma might be mistaken for a NFPA (72).
It is also suggested to check for pituitary hormone deficiencies in patients with non-functioning tumors, irrespective of symptoms. Macroadenomas can cause impairment of pituitary function by the involvement of the normal gland or pituitary stalk compression, and the risk of hypopituitarism is directly related to tumor volume. Microadenomas eventually lead to pituitary dysfunction, particularly if larger than 5mm (32,69). A large retrospective cohort of 218 patients with NFPAs found at least one pituitary deficiency at diagnosis in 33.3% of microadenomas (73) although other studies have failed to identify hormonal deficiencies in microadenomas. An interesting and cost-effective approach for microincidentalomas could be no hormonal evaluation for cystic and solid lesions smaller than 5 mm. For solid microadenomas (between 6-9 mm), it is suggested a simple investigation with measurements of PRL and IGF1 (74).
Concerning macroadenomas, current guidelines suggest that serum cortisol levels at 8-9 AM should be done as the first-line test for diagnosing central adrenal insufficiency (75). Despite well recognized limitations of most commercial cortisol immunoassays (76), baseline cortisol levels <3 µg/dL (83 nmol/L) are suggestive of central adrenal insufficiency, while baseline cortisol levels >15 µg/dL (414 nmol/L) likely exclude it, and levels between 3 and 15 µg/dL require a corticotropin/insulin stimulation test. Central hypothyroidism is assessed by measuring serum TSH and free T4 (fT4), and fT4 level below the laboratory reference range in conjunction with a low, normal, or mildly elevated TSH in the setting of pituitary disease usually confirms the diagnosis (75). Diagnosing GH deficiency (GHD) is relatively straightforward in patients with IGF1 concentration lower than the gender- and age-specific lower limit of normal and structural pituitary disease. Low IGF1 in patients with non-functioning tumors who have at least three pituitary hormone deficiencies is also suggestive of GHD (29). Nevertheless, some adults with suspected GHD may have normal IGF1, in such cases GH stimulation testing may be useful. Central hypogonadism in males, manifests with low serum testosterone, low or inappropriately normal FSH/LH and features of testosterone deficiency. For females, pre-menopausal women with oligomenorrhea or amenorrhea should be screened with serum E2, FSH, and LH. In postmenopausal women, if the patient is not in hormonal therapy, the absence of high serum FSH and LH is sufficient for a diagnosis of central hypogonadism (75).
Sellar MRI with gadolinium is the best imaging study for suspected adenomas because it provides images of high resolution of the mass as well as its relationship with surrounding structures. Pituitary adenomas usually appear hypo or isointense compared to normal pituitary tissue in T1 images on MRI. In addition, while the normal pituitary tissue enhances earlier with contrast, pituitary adenomas commonly present with delayed contrast uptake (77). Patients whose tumor abuts the optic chiasm should have visual field perimetry, preferably by the Goldmann method, to assess for visual deficits (69).
Functional (FDG- and/or SSTR- positron emission tomography (PET) imaging studies should only be considered in aggressive pituitary adenomas/carcinomas in the setting of site-specific symptoms (neck/back pain or neurological complaints), and/or where laboratory measures are discordant with known visible extent of disease (78). Recently, 37 patients diagnosed with NFPAs enrolled in a clinical trial underwent 68Ga-DOTATATE PET/computed tomography (CT) of the head. 68Ga-DOTATATE uptake was positive in 34/37 patients (92%), demonstrating in vivo SSTR expression in NFPAs and ultimately showing superior sensitivity of PET imaging when compared with 111In-DTPA-octreotide scintigraphy (79).
The pre-operative differential diagnosis of NFPAs includes several additional primary non-hormonal-secreting lesions (80). This distinction is challenging in many cases because the clinical presentations and radiological aspects may be similar between adenomas and other pituitary lesions. The presence of diabetes insipidus (DI) is common in tumors of non-pituitary origin and indicates that the sellar mass is most likely not a pituitary adenoma (81). An increase in α subunit of the glycoprotein hormones (elevated in 30% of NFPA patients) can help in the differential diagnosis although normal values do not rule them out (82).
TREATMENT
In spite of current knowledge of definite NFPAs pathologic subtypes, initial treatment strategies are similar regardless of the subset. A small number of studies have reported treatment outcome results taking into consideration adenohypophyseal hormone immuno-profile (82-84) and, for the future, precise pathologic characterization utilizing adenohypophyseal hormones and transcription factors may potentially aid in predicting the response to specific adjunctive therapies.
Surgery
Surgical resection is the primary treatment for symptomatic patients with NFPAs (85), i.e., those with neuro-ophthalmologic complaints and/or tumors affecting the optic pathway. Surgery is also urgently indicated for patients with apoplexy who develop neuro-ophthalmologic complaints. In some experts’ opinion, tumors larger than 2cm should also be considered for surgery due to their propensity for growth (86). Treatment for hypopituitarism, primarily central adrenal insufficiency, and central hypothyroidism, should be commenced prior to surgical resection. Mortality rate is low (<1%) and reported surgical complication rates are acceptable. Postoperative complications such as CSF leakage, fistula, meningitis, vascular injury, persistent diabetes insipidus (DI), or new visual field defect occurred in ≤ 5% of patients, as reported by a systematic review and metanalysis (87). Accomplishment of total or near-total resection can be challenging and varies in different series, ranging from 20% to 80% (88,89). According to a recent large retrospective series evaluating 254 patients who underwent endoscopic endonasal surgery for a macroadenoma (including 72 patients with NFPAs), cavernous sinus invasion as assessed by the modified Knosp classification (90) effectively predicted surgical outcomes. Gross total resection was negatively correlated with tumor grade and success rates of gross-total resection were between 30% and 56% for tumors extending beyond the internal carotid artery and into the cavernous sinus compartments (grades 3A and 3B) (91).
A new “shape grading system” (Figure 3) has been recently proposed for predicting outcomes in patients with NFPAs operated by transsphenoidal surgery. In a retrospective single center study, 191 NFPAs were assessed according to different radiological growth patterns: spherical (Shape I), oval (Shape II), dumbbell (Shape III), mushroom (Shape IV), and polylobulated (Shape V) (92). Gross total resection was achieved in 53% of the patients, with decreasing likelihood of accomplishment in higher shape grades - shape I (82%), shape II (74%), shape III (24%), shape IV (17%) and shape 5 (0%). Furthermore, shape grades predicted resection rate better than Knosp grades. During a mean follow-up of 59 months, tumor recurrence or regrowth was observed in 6% and 12% of the patients, respectively. Higher shape grades also correlated with higher risk of tumor recurrence/regrowth as well as the need for reoperation and/or radiotherapy. Of note, SCAs more often grew as shape IV or V, while silent somatotroph adenomas were more often detected in the shape V group. Thus, the shape grading system seems to be a complimentary tool to better predict NFPA surgical outcomes, however, these findings need validation in other cohorts.

Figure 3
- The Shape grading system. Abbreviations: OC, optic chiasm; PS, pituitary stalk. From Berkaman et al Acta Neuropathologica 2021 (92)
Visual field deficits and, less commonly, hormone deficiencies may improve following surgical treatment although new hormone deficiencies may also occasionally develop after a surgical approach (87,93). The incidence of DI after endoscopic transsphenoidal surgery to remove NFPAs was recently investigated in 168 patients. Seventy-seven (45.8%) patients experienced postoperative DI and 10 (6.0%) patients suffered from permanent DI. A large cephalocaudal tumor diameter (cut off value-of 2.7 cm) was predictive of postoperative DI in such patients (94). Moreover, younger age and the absence or intrasellar location of the bright spot (as opposed to suprasellar location) on preoperative T1-weighted MRI were further factors predicting postoperative DI in a Japanese series of 333 patients with NFPAs undergoing transsphenoidal surgery (95).
Pituitary function should be reassessed one to three months after surgery and treatment of hypopituitarism introduced according to hormone deficiencies. Whether or not GH deficiency should be replaced requires a thoughtful and individualized evaluation of risks and benefits (96,97). Current data supports the absence of any stimulation of remnant or induction of recurrence by growth hormone replacement in patients with NFPAs solely treated by surgical removal (98). In a retrospective series, tumor regrowth occurred in 38/107 (36%) of non-GH treated subjects and in 8/23 (35%) of GH-treated subjects, followed up for a mean period of 6.8 years. The Cox regression analysis showed that after adjusting for sex and age at tumor diagnosis, cavernous sinus invasion at diagnosis, and type of tumor removal, GH treatment was not a significant independent predictor of recurrence.
A sellar MRI should be obtained three to six months after surgery to assess the extent of tumor resection. Also, as a significant number of patients with NFPAs may develop tumor re-growth, long-term imaging surveillance is recommended. In a retrospective analysis of 155 patients with NFPAs treated solely by surgery and followed-up for a mean period of six years (twenty-nine were followed up for more than 10 years), re-growth was reported in 34.8% of the cases, with 20% of relapse/re-growth occurring after 10 years (88). Likewise, in patients with NFPAs who present with classical apoplexy, tumor re-growth rate is not negligible. In a retrospective series of thirty-two patients with NFPAs who underwent surgery for pituitary apoplexy, tumor re-growth was reported in 11% of the cases at just over five years (99). Therefore, it is advisable that patients with NFPAs, particularly those with post-operative tumor remnants, be closely monitored following surgery, and follow-up surveillance needs to certainly be continued for more than 10 years.
Radiation Therapy
Radiation therapy (RT) has been shown to be effective as an adjunct to surgical resection in cases of post-operative residual tumor or recurrence. However, it carries a major long-term risk of hypopituitarism (85,100,101). Stereotactic techniques such as stereotactic radiosurgery or fractionated stereotactic radiotherapy have been developed with the purpose of delivering more localized irradiation and reducing long-term side-effects. Both techniques provide excellent tumor control in patients with NFPAs, ranging from 85% to 95% at five to ten years (102). However, at the present time, there is no consensus concerning the systematic use of RT in the postoperative period for patients with incompletely resected NFPAs (29) and whether an earlier approach would be preferable over conservative management. In general, RT is reserved for cases with large tumor remnants and for those cases that present progressive tumor growth during follow-up (85). Adjuvant RT may also be considered for patients who, at diagnosis, already present aggressive tumors, such as those invading parasellar structures or with extensive positive immunostaining for Ki-67, a proliferative index significantly associated with recurrence in NFPAs (9,103). Furthermore, NFPA subtype may be a relevant factor in the expected response to radiotherapy. A retrospective multicenter study demonstrated that overall tumor control rate after radiosurgery was lower in SCAs compared to other NFPA subtypes (104) suggesting that, in SCAs, an elevated margin dose may be considered in order to achieve a better chance of tumor control. In line with these findings, a recent and large retrospective surgical series showed a significantly lower progression-free survival in patients with SCAs compared to other NFPAs (24.5 vs 51.1 months) (105). Among the SCA cohort, progression was noted despite the use of adjuvant radiosurgery in one third of the patients. Notwithstanding, a recent systematic review and meta-analysis showed no evidence supporting higher recurrence rate after primary treatment of SCAs compared to other NFPAs (106), however the evaluated study samples included only a small number of patients who had been offered adjuvant radiotherapy.
Asymptomatic Tumors
The best treatment strategy for an asymptomatic NFPA is not yet defined since there are few natural history studies in the literature (85). Hypopituitarism is not, per se, an indication for surgical resection but requires treatment and follow-up, when present. Approximately half of these tumors will increase in size during long-term follow-up and nearly 20% of patients will require surgical intervention (30,107). Unfortunately, until now, no independent clinical predictors for an increase in tumor volume have been identified. A conservative approach in selected patients without visual field defects has been suggested (107) while an early intervention may be justified in younger patients with large lesions (30). It is recommended that microadenomas be followed up annually with MRI imaging to detect tumor enlargement, for three years. Subsequent repeat imaging can be done less frequently. For tumors <5mm, no imaging surveillance is suggested, according to a recently published guideline (108). Surgery is indicated only when significant tumor enlargement is demonstrated or if the tumor abuts the optic chiasm. Regarding asymptomatic macroadenomas, if surgery is not performed, then visual fields should be tested and an MRI scan performed at six monthly intervals initially, then annually for three years and less frequently afterwards (86). Recent evidence of gadolinium-based contrast agents (GBCAs) deposition in human brain has raised long-term safety concerns (109), possibly impacting current updates to management. Clinicians should offer reassurance to patients regarding the judicious use of GBCAs with preference to the use of safer macrocyclic agents.
Medical Therapy
Primary treatment of NFPAs with medical therapy is not currently recommended (15,85). Small series have demonstrated significant tumor volume stabilization under medical treatment in non-operated patients with NFPAs due to contraindications or refusal (110), whereas other studies have revealed tumor volume progression in the long term (111). The main medical agents that have been evaluated in NFPAs are dopamine agonists (DA) and somatostatin analogues (SAs), mainly in patients with residual tumor after transsphenoidal surgery. Figure 4 shows a suggested algorithm for the management of patients with NFPAs.

Figure 4
- Suggested algorithm for the management of patients with NFPAs. CBA: cabergoline; NFPA: non-functioning pituitary adenoma, MRI: magnetic resonance imaging; PRRT: peptide receptor radionuclide therapy RT: radiotherapy, *potential option, less evidenced-based approach; SSTR: somatostatin receptor
DOPAMINE AGONISTS
Dopamine receptor type 2 (D2R) expression has been demonstrated in patients with NFPAs. In a small series of 18 patients with hormone-negative NFPAs, two thirds of them (12/18) expressed D2R by real-time polymerase chain reaction. Patients who presented residual tumor (9/18) were treated with cabergoline up to 3mg per week. After 12 months of treatment, tumor shrinkage was observed in 56% of the patients and tumor reduction was significantly greater in those expressing D2R (112). A historical cohort analysis on the adjunctive role of DA in adult patients with GH and ACTH negative NFPAs showed favorable results (83). The treatment group consisted of patients who were either initiated on DA upon post-surgical residual tumor detection or when tumor growth was subsequently detected on follow-up, while the control group received no medication after surgical treatment. Tumor control was significantly superior in patients who were treated upon detection of post-operative residual tumor, compared to those who were treated after presenting tumor progression or the control group, 87% vs. 58% vs. 47%, respectively. The requirement for additional treatment (surgery and/or radiotherapy) during follow-up was significantly decreased from 47% to 16% with adjunctive DA therapy. In this series, there were no correlations between clinical response to DA and D2R expression, the isoform type or their expression levels.
A randomized open-label clinical trial was recently conducted aiming to compare cabergoline with non-intervention in 116 Brazilian patients with residual NFPA after transsphenoidal surgery followed-up for over two years. NFPAs were classified solely based on IHC for anterior pituitary hormones and included 59% hormone-negative adenomas and 34% silent gonadotroph adenomas. Silent corticotroph adenomas were excluded from this study. By the end of the study, residual tumor shrinkage was significantly more frequently observed in the medical-therapy group compared to patients in the control group (28.8% vs. 10.5%). Tumor response was not associated with D2R expression (113). Thus, although cabergoline has not been a consensual treatment for patients with NFPAs, a therapeutic attempt can be made according to clinical judgment in individual cases; particularly in patients with large extrasellar remnants after surgery and a high probability of tumor progression (114). On the other hand, most studies published so far included only patients with hormone-negative or silent gonadotroph adenomas, such that this proposition should not be extended to silent corticotroph adenomas or the other rarer silent tumors. It is also important to bear in mind the natural history of untreated NFPAs, which may show a spontaneous decrease in tumor volume in up to 30% of them (115).
Cabergoline doses are variable, but usually started at 0.5 mg weekly, increasing 0.5 mg each week until a maximum dose of 3.0 mg/week is reached. For these patients, tumor shrinkage is not the major target although it can occur in 38% of them; otherwise, prevention of tumor growth is the main treatment goal. Tumor progression can be assessed by a sellar MRI every six months for the first two years, and annually thereafter. Once stability is achieved without significant side effects, the drug can be maintained indefinitely, taking into account individual cost-benefits (82,116).
SOMATOSTATIN RECEPTOR LIGANDS
The finding of somatostatin receptors (SSTRs) expression by NFPAs has raised the possibility that the use of somatostatin receptor ligands (SRLs) could be an effective treatment strategy (117,118). The SRL octreotide which binds with high affinity to SSTR2 was not effective in controlling tumor size or improving visual field in a small group of patients with NFPAs (119). A case-control study evaluated the results of long-acting octreotide in patients harboring post-surgical NFPA residues and it demonstrated tumor remnant stabilization in 81% (21/26) of patients in the treated group compared to 47% (6/13) of patients in the control group after a mean follow-up of 37 months (120). However, neither visual field nor pituitary function changed in any of the groups. This cohort of 39 NFPAs consisted of a heterogeneous group as IHC revealed positivity for pituitary hormones in 28 cases (20 of those showing positivity for FSH and/or LH) whereas the remaining 11 cases were negative for adenohypophyseal hormones. SSTR5 was the predominant SSTR expressed (84%), followed by SSTR3 (61%), while SSTR2 was expressed in 46% of the cases.
The expression of SSTRs and zinc finger protein 1 (ZAC1), a protein regulating apoptosis and cell cycle arrest, were assessed in a group of NFPAs (SGAs and hormone-negative adenomas), active somatotroph adenomas, and normal pituitary. SSTR2 and ZAC1 expression was reduced whereas SSTR3 expression was increased in SGAs compared to active somatotroph adenomas and normal pituitary (121). Likewise, other studies have suggested that SSTR3 is the predominant SSTR expressed in most NFPAs, both by IHC studies (118,122) and mRNA levels (11,122). However, a few other studies have demonstrated higher SSTR2 expression than SSTR3 or SSTR5 expression in SGAs and hormone-negative adenomas (103,123). Regarding corticotroph pituitary tumors, SSTR5 expression was significantly less frequently found in SCAs as compared to their secreting counterparts (2/23 vs. 24/39) (124). Furthermore, amongst the 23 SCAs, somatic ubiquitin-specific protease 8 (USP8) mutation was detected in only two tumors, in line with previous reports showing a significantly lower prevalence of USP8 mutations in SCAs compared to functioning corticotroph tumors (125,126).
There is an ongoing multicenter, randomized, double-blind, placebo-controlled trial (GALANT study) evaluating the effectiveness of first generation SRLs in patients with NFPAs and suprasellar extension, either surgery-naive or with a postoperative remnant. Forty-four patients with positive results on 68Ga-DOTATATE PET are being randomized to treatment with either the SRL lanreotide or placebo (127).
Pasireotide is a universal SRL with action on SSTR1, SSTR2, SSTR3 and STR5 subtypes and, therefore, seems more attractive than first-generation SRLs as an alternative medical treatment for NFPAs. A head-to-head comparison of octreotide and pasireotide in MENX-affected rat harboring a mutation I the gene encoding p27, an in vivo model of NFPAs, was recently performed. Pasireotide showed superior anti-tumor effect vs. octreotide, especially in females, which also showed higher SSTR3 expression (128). There are two phase 2 clinical trials evaluating the safety and efficacy of pasireotide for the treatment of NFPAs. Both trials have been recently completed but the results are not yet available. Passion 1 (NCT01283542) is an open-label single arm study evaluating tumor volume response to pasireotide in naive patients with NFPAs >1cm. Another phase 2 clinical trial (NCT01620138) is currently comparing the response of patients with NFPAs and surgical remnants to cabergoline vs. pasireotide. Notwithstanding, until further data are available, the use of SSRLs is not currently recommended for the treatment of patients with NFPAs.
Treatment of NFPAs with dopastatin, i.e., chimeric compound with dopamine and somatostatin agonist activity, has also been under investigation. BIM-23A760, a compound with potent agonist activity both at D2R and SSTR2 effectively inhibited cell proliferation in primary cultures of NFPAs (129). Interestingly, TBR-760 (formerly BIM-23A760) was recently tested in a mouse model of aggressive NFPA and resulted in nearly complete inhibition of tumor growth (130). A one year, randomized, double-blind, placebo-controlled phase 2 study of TBR-760 in adult patients with residual NFPAs > 1cm after transsphenoidal surgery (NCT04335357) is being conducted and is expected to be completed by 2023.
TEMOZOLOMIDE
Temozolomide (TMZ) was the first alkylating chemotherapeutic drug to show significant response rates in aggressive pituitary tumors (131). Notwithstanding, TMZ seems to be less effective in NFPAs compared to their secreting counterparts as 45% of 110 clinically functioning pituitary tumors showed regression on first-line TMZ while only 17% of 47 NFPAs did (132).
Responsiveness to TMZ is probably dependent on the immuno-expression of O (6)-methylguanine DNA methyltransferase (MGMT), a DNA repair protein that acts by removing the alkyl group and therefore is associated with TMZ resistance (133). Low immuno-expression of MGMT by pituitary tumors has been associated with higher response rates to TMZ (131). MGMT immuno-expression was assessed in a group of 45 NFPAs and the degree of expression was correlated with tumor aggressiveness (133). Low MGMT expression was observed in 50% of the aggressive NFPAs compared to 24% of the non-aggressive NFPAs. These findings suggest that aggressive NFPAs with low MGMT expression could be potential candidates for treatment with TMZ.
Regarding dosing regimens and indications, current guidelines suggest using TMZ (150-200 mg/m2daily for 5/28 days) as an alternative treatment for patients with aggressive NFPAs presenting tumor progression despite radiotherapy and other therapeutic measures, and for exceptional cases of pituitary carcinomas (29,132). Furthermore, in the case of rapid tumor growth in patients who have not previously reached maximal doses of radiotherapy, the use of the STUPP protocol (six weeks of concomitant fractionated radiotherapy and TMZ 75 mg/m2 daily, followed by TMZ alone 150-200 mg/m2 daily for 5/28 days) has been suggested (132). Studies on long-term administration of TMZ are scarce but clinical observations indicate that TMZ should be continued for as long as it is effective and well tolerated. Some authors suggest continuing TMZ at standard dosage for two years and later reducing to half-dose (134). Patients receiving TMZ chemotherapy are at risk for hematologic toxicity, occurring in approximately 15-20% of the cases. The most common non-hematologic side effects of TMZ are nausea, anorexia, and fatigue (135).
PEPTIDE RECEPTOR RADIONUCLIDE THERAPY
In vivo SSTR expression by NFPAs has been previously demonstrated both by positive uptake on somatostatin receptor scintigraphy (120) and on 68Gallium DOTATATE PET/ CT (79) providing a rationale for the administration of PRRT in these patients. So far, PRRT has been studied in few patients with aggressive pituitary tumors (clinically functioning and NFPAs) with different response patterns (136,137). Among the six patients with nonfunctioning pituitary adenoma or carcinoma, three patients (not previously treated with TMZ) showed stable disease (138-140), two patients had progressive disease (138,141) and one patient died in the following months while information on tumor volume was lacking (142).
QUALITY OF LIFE AND LONG-TERM MORBIDITY AND MORTALITY
A substantial number of patients with NFPAs suffer from morbidities related to the tumor itself, as well as to the treatments offered. Standardized mortality ratios in these patients seem to be higher than that of the general population with deaths associated mainly with circulatory, respiratory, and infectious causes (143). Until now, there was no consensus on predictive factors of mortality but those most consistently described are older age at diagnosis (144,145) and high doses of glucocorticoid replacement therapy (146). A retrospective series of 546 patients operated on for a macro NFPA between 1963 and 2011 and followed up for a median period of 8 years reported a standardized mortality ratio of 3.6 (95% CI, 2.9–4.5) (144). After adjustment for factors proven to be significant in univariate analysis - radiotherapy, tumor regrowth, and untreated growth hormone deficiency - age at diagnosis was an independent predictor of mortality, with shorter survival observed in older patients.
Following NFPA treatment, patient-reported health-related quality of life (HR-QoL) substantially improves. Nevertheless, there are conflicting findings about HR-QoL normalization, which may also be related to the lack of a disease-specific HR-QoL questionnaire for NFPAs (147). Some studies have described a persistent decreased HR-QoL compared to healthy controls and reference data (148,149), while others have not (150). In the latest study, HR-QoL was evaluated in 193 consecutive patients with NFPAs followed up in a tertiary endocrine referral center (150). The overall health-related quality of life and perception of subjective health in patients with NFPAs was not compromised although specific groups, such as females, patients with tumor recurrences, and with visual defects, were shown to be affected in various dimensions. Altered sleep-wake rhythmicity has also been described in a cohort of 69 patients with NFPA in long term remission after transsphenoidal surgery on stable replacement treatment for hypopituitarism (151). NFPA patients reported severely impaired QoL, sleep quality, and increased daytime sleepiness. Preoperative visual field defects were associated with sleep-wake rhythm fragmentation and vasopressin deficiency was associated with decreased sleep efficiency, independent of age, hypopituitarism, or radiotherapy.
More recently, the use of the Wilson–Cleary model, a conceptual biopsychosocial model of HR-QoL, suggested that elements at each stage of this model could be contributing to the impairment in HR-QoL observed in patients with a NFPA (147). The authors concluded that currently available biomedical treatments, i.e., surgery, radiotherapy, and hormone replacement therapy, are clearly not sufficient for achievement of good HR-QoL in patients with a NFPA, and further improvement should be supported by a pituitary specific care trajectory, targeting not only biological and physiological variables, but also psychosocial care.
PROGNOSTIC FACTORS AND FUTURE PERSPECTIVES
There has been a search to identify reliable factors related to aggressiveness and the risk of recurrence in NFPAs. A single-center retrospective study which evaluated 108 surgically-resected NFPAs followed for up to 15 years (152) showed that 22% of the patients required further treatment, either second surgery or radiotherapy. Factors determining recurrence were the presence of residual tumor, tumor growth rate (>80 mm3/year), and suprasellar extension (152). According to another retrospective case series evaluating patients with NFPAs who presented tumor regrowth after primary treatment, the NFPA subtype, categorized by anterior pituitary hormone immunostaining, was not a predictive factor for the requirement of secondary treatment or tumor regrowth. Significant risk factors were female gender and treatment approach (monitoring vs. interventions); secondary progression was significantly higher in those patients who were followed conservatively (63%) as compared to those who received surgery (36%), radiotherapy (13%), and surgery/adjuvant radiotherapy (13%) (84). In patients with SGAs, ERα seems to be a prognostic factor for re-intervention (reoperation or radiation) in males - the combination of the absence of ERα expression and young age served as good predictive markers of aggressiveness (122).
The role of cellular markers, associated with cell proliferation and apoptosis, in predicting the recurrence of NFPAs has also been investigated (153). Proliferative indexes such as a high Ki-67 index, assessed by IHC, were significantly associated with a tumor size greater than 3 cm, as well as with tumor recurrence (103). Evaluation of tumor proliferation by using Ki-67 IHC is widely available and is recommended as part of the assessment of NFPAs (40). According to a large retrospective series evaluating 601 patients with surgically resected pituitary tumors, including approximately 30% NFPAs, the optimal cut-off points of the Ki-67 proliferation index that predicted recurrence was 2.5% with 84.6% sensitivity and 47.4% specificity (154). Moreover, a recent retrospective analysis of 120 patients operated on for an NFPA showed that invasive and proliferative tumors, i.e., grade 2b tumors. according to the clinicopathological classification of Trouillas et al. (155) showed an overall likelihood of recurrence that was approximately 9 times greater than those of grade 1a, i.e., non-invasive and non-proliferative tumors (156). Further risk factors associated with an increased risk of recurrence in this cohort were a younger age and the presence of residual tumor.
Minichromosome maintenance 7 (MCM7), a cell-cycle regulator protein, has been recently proposed as a marker of tumor progression in NFPAs. In a cohort study of 97 surgically treated NFPAs, the probability for reintervention within 6 years for patients harboring residual tumors with high MCM7 expression was 93% (157). Ki-67 expression >3%, age ≤55 years and mitotic index≥1, but not tumor subtype, were also associated with reintervention. Attempts to develop clinical nomograms to predict post-operative recurrence of NFPAs have recently been demonstrated based on age, tumor size, cavernous sinus invasion, sphenoid sinus invasion, and surgery extension. The nomogram model proposed by Lyu et al. (158) showed an area under the ROC curve of 0.953 and correlation analyses indicated that sphenoid sinus invasion, cavernous sinus invasion and tumor size could promote the recurrence of NFPA while advanced age and gross total resection could effectively inhibit it.
Genetic and epigenetic mechanisms underlying the development and aggressiveness of NFPAs have not been fully elucidated (159). The mutational landscape of a cohort of pituitary tumors including 37 NFPAs was recently published (160). Aside from demonstrating gonadotroph signatures in seven out of eight SCAs, retrieving the question whether a subset of SCAs arise from a specific pituitary lineage distinct from other corticotroph (161), this study showed that most NFPAs displayed no functional somatic variant and no chromosome alterations. Further characterization of these tumors with techniques such as whole genome sequencing coupled to chromatin structure analysis may disclose mutations in non-coding regions affecting chromatin opening and/or the binding of specific transcription factors. allowing for the development of novel therapeutic strategies.
An adverse pituitary adenoma phenotype is defined not only by the intrinsic activity of tumor cells but also by the infiltrated immune cells in the tumor microenvironment (162). The study of the immune profile of pituitary tumors for predicting immunotherapy responsiveness is an evolving field. Wang et al (163) have recently proposed classification of these tumors on three clusters based on tumor-infiltrating immune cells and the expression of immune checkpoint molecules This study cohort included 57 “unspecified“ NFPAs since they were not pathologically classified with pituitary transcription factors. The majority of them exhibited a “hot” immune microenvironment and were predicted to exhibit higher immunotherapy responsiveness.
CONCLUSION
NFPAs are frequent in endocrine practice. Clinically they range from being completely asymptomatic (incidental findings on head MRI or computed tomography scans) to causing significant hypothalamic/pituitary dysfunction and visual symptoms due to mass effect., For microadenomas and asymptomatic/relatively small macroadenomas (1–2 cm), a “watch and wait” option is reasonable. If tumor growth, development of visual field defects, or progressive pituitary dysfunction is detected during follow up, then surgery is indicated. It is now recommended that NFPAs are classified according to their pituitary hormone and transcription factor profiles along with proliferation markers such as Ki-67. This refined stratification may potentially aid in predicting disease course and in selecting adjunctive therapies. Radiotherapy is an effective treatment, although usually reserved for those patients with aggressive tumors and significant tumor remnant after pituitary surgery as considerable side effects may occur. Medical treatment with dopamine agonists stands as an alternative in selected cases, despite the lack of placebo-controlled trials. Highly aggressive tumors need special care during follow-up, including treatment with TMZ with or without RT complementation or new potential emerging treatments. Close collaboration of a multidisciplinary pituitary team is crucial to better serve this challenging group of patients.
REFERENCES
- 1.
- Daly AF, Rixhon M, Adam C, Dempegioti A, Tichomirowa MA, Beckers A. High prevalence of pituitary adenomas: a cross-sectional study in the province of Liege, Belgium. J Clin Endocrinol Metab. 2006;91(12):4769–4775. [PubMed: 16968795]
- 2.
- Ho K, Fleseriu M, Kaiser U, Salvatori R, Brue T, Lopes MB, Kunz P, Molitch M, Camper SA, Gadelha M, Syro LV, Laws E, Reincke M, Nishioka H, Grossman A, Barkan A, Casanueva F, Wass J, Mamelak A, Katznelson L, van der Lely AJ, Radovick S, Bidlingmaier M, Boguszewski M, Bollerslev J, Hoffman AR, Oyesiku N, Raverot G, Ben-Shlomo A, Fowkes R, Shimon I, Fukuoka H, Pereira AM, Greenman Y, Heaney AP, Gurnell M, Johannsson G, Osamura RY, Buchfelder M, Zatelli MC, Korbonits M, Chanson P, Biermasz N, Clemmons DR, Karavitaki N, Bronstein MD, Trainer P, Melmed S. Pituitary Neoplasm Nomenclature Workshop: Does Adenoma Stand the Test of Time? J Endocr Soc. 2021;5(3):bvaa205. [PMC free article: PMC7874572] [PubMed: 33604494]
- 3.
- Fleseriu M, Karavitaki N. Non-functioning pituitary adenomas, not all the same and certainly not boring! Pituitary. 2018;21(2):109–110.
- 4.
- Fernandez A, Karavitaki N, Wass JA. Prevalence of pituitary adenomas: a community-based, cross-sectional study in Banbury (Oxfordshire, UK). Clin Endocrinol (Oxf). 2010;72(3):377–382. [PubMed: 19650784]
- 5.
- Horvath E, Kovacs K, Killinger DW, Smyth HS, Platts ME, Singer W. Silent corticotropic adenomas of the human pituitary gland: a histologic, immunocytologic, and ultrastructural study. Am J Pathol. 1980;98(3):617–638. [PMC free article: PMC1903510] [PubMed: 6244736]
- 6.
- Aflorei ED, Korbonits M. Epidemiology and etiopathogenesis of pituitary adenomas. J Neurooncol. 2014;117(3):379–394. [PubMed: 24481996]
- 7.
- Tjornstrand A, Gunnarsson K, Evert M, Holmberg E, Ragnarsson O, Rosen T, Filipsson Nystrom H. The incidence rate of pituitary adenomas in western Sweden for the period 2001-2011. Eur J Endocrinol. 2014;171(4):519–526. [PubMed: 25084775]
- 8.
- Lenders N, McCormack A. Malignant transformation in non-functioning pituitary adenomas (pituitary carcinoma). Pituitary. 2018;21(2):217–229. [PubMed: 29299820]
- 9.
- Lloyd R, Osamura, R., Klöppel, G. & Rosai, J (eds). World Health Organization Classification of Tumours of Endocrine Organs. Vol 10. 4th ed: IARC Publication; 2017.
- 10.
- Saeger W, Ludecke DK, Buchfelder M, Fahlbusch R, Quabbe HJ, Petersenn S. Pathohistological classification of pituitary tumors: 10 years of experience with the German Pituitary Tumor Registry. Eur J Endocrinol. 2007;156(2):203–216. [PubMed: 17287410]
- 11.
- Nishioka H, Inoshita N, Mete O, Asa SL, Hayashi K, Takeshita A, Fukuhara N, Yamaguchi-Okada M, Takeuchi Y, Yamada S. The Complementary Role of Transcription Factors in the Accurate Diagnosis of Clinically Nonfunctioning Pituitary Adenomas. Endocr Pathol. 2015;26(4):349–355. [PubMed: 26481628]
- 12.
- Lopes MBS. Classification, Pathobiology, Molecular Markers, and Intraoperative Pathology. In: Laws JE, Cohen-Gadol A., Schwartz T., Sheehan J., ed. Transsphenoidal Surgery: Springer, Cham; 2017:113-143.
- 13.
- Mayson SE, Snyder PJ. Silent (clinically nonfunctioning) pituitary adenomas. J Neurooncol. 2014;117(3):429–436. [PubMed: 24676675]
- 14.
- Grossman AB. The 2004 World Health Organization classification of pituitary tumors: is it clinically helpful? Acta Neuropathol. 2006;111(1):76–77. [PubMed: 16328520]
- 15.
- Drummond J, Roncaroli F, Grossman AB, Korbonits M. Clinical and Pathological Aspects of Silent Pituitary Adenomas. J Clin Endocrinol Metab. 2019;104(7):2473–2489. [PMC free article: PMC6517166] [PubMed: 30020466]
- 16.
- Ntali G, Wass JA. Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas. Pituitary. 2018;21(2):111–118. [PubMed: 29368293]
- 17.
- Oh JS, Kim HJ, Hann HJ, Kang TU, Kim DS, Kang MJ, Lee JY, Shim JJ, Lee MR, Ahn HS. Incidence, mortality, and cardiovascular diseases in pituitary adenoma in Korea: a nationwide population-based study. Pituitary. 2021;24(1):38–47. [PubMed: 32949324]
- 18.
- Chen L, White WL, Spetzler RF, Xu B. A prospective study of nonfunctioning pituitary adenomas: presentation, management, and clinical outcome. J Neurooncol. 2011;102(1):129–138. [PubMed: 20730474]
- 19.
- Ferrante E, Ferraroni M, Castrignano T, Menicatti L, Anagni M, Reimondo G, Del Monte P, Bernasconi D, Loli P, Faustini-Fustini M, Borretta G, Terzolo M, Losa M, Morabito A, Spada A, Beck-Peccoz P, Lania AG. Non-functioning pituitary adenoma database: a useful resource to improve the clinical management of pituitary tumors. Eur J Endocrinol. 2006;155(6):823–829. [PubMed: 17132751]
- 20.
- Ogra S, Nichols AD, Stylli S, Kaye AH, Savino PJ, Danesh-Meyer HV. Visual acuity and pattern of visual field loss at presentation in pituitary adenoma. J Clin Neurosci. 2014;21(5):735–740. [PubMed: 24656736]
- 21.
- Rizzoli P, Iuliano S, Weizenbaum E, Laws E. Headache in Patients With Pituitary Lesions: A Longitudinal Cohort Study. Neurosurgery. 2016;78(3):316–323. [PubMed: 26485333]
- 22.
- Losa M, Donofrio CA, Barzaghi R, Mortini P. Presentation and surgical results of incidentally discovered nonfunctioning pituitary adenomas: evidence for a better outcome independently of other patients' characteristics. Eur J Endocrinol. 2013;169(6):735–742. [PubMed: 23999643]
- 23.
- Freda PU, Bruce JN, Khandji AG, Jin Z, Hickman RA, Frey E, Reyes-Vidal C, Otten M, Wardlaw SL, Post KD. Presenting Features in 269 Patients With Clinically Nonfunctioning Pituitary Adenomas Enrolled in a Prospective Study. J Endocr Soc. 2020;4(4):bvaa021. [PMC free article: PMC7101088] [PubMed: 32258955]
- 24.
- Greenman Y, Melmed S. Diagnosis and management of nonfunctioning pituitary tumors. Annu Rev Med. 1996;47:95–106. [PubMed: 8712806]
- 25.
- Vargas G, Gonzalez B, Ramirez C, Ferreira A, Espinosa E, Mendoza V, Guinto G, Lopez-Felix B, Zepeda E, Mercado M. Clinical characteristics and treatment outcome of 485 patients with nonfunctioning pituitary macroadenomas. Int J Endocrinol. 2015;2015:756069. [PMC free article: PMC4337176] [PubMed: 25737722]
- 26.
- Briet C, Salenave S, Chanson P. Pituitary apoplexy. Endocrinol Metab Clin North Am. 2015;44(1):199–209. [PubMed: 25732655]
- 27.
- Oldfield EH, Merrill MJ. Apoplexy of pituitary adenomas: the perfect storm. J Neurosurg. 2015;122(6):1444–1449. [PubMed: 25859802]
- 28.
- Wildemberg LE, Glezer A, Bronstein MD, Gadelha MR. Apoplexy in nonfunctioning pituitary adenomas. Pituitary. 2018;21(2):138–144. [PubMed: 29383476]
- 29.
- Vieira LN, Boguszewski CL, Araujo LA, Bronstein MD, Miranda PA, Musolino NR, Naves LA, Vilar L, Ribeiro-Oliveira AJ, Gadelha MR. A review on the diagnosis and treatment of patients with clinically nonfunctioning pituitary adenoma by the Neuroendocrinology Department of the Brazilian Society of Endocrinology and Metabolism. Archives of endocrinology and metabolism. 2016;60(4):374–390. [PMC free article: PMC10118716] [PubMed: 27533614]
- 30.
- Arita K, Tominaga A, Sugiyama K, Eguchi K, Iida K, Sumida M, Migita K, Kurisu K. Natural course of incidentally found nonfunctioning pituitary adenoma, with special reference to pituitary apoplexy during follow-up examination. J Neurosurg. 2006;104(6):884–891. [PubMed: 16776331]
- 31.
- Fernandez-Balsells MM, Murad MH, Barwise A, Gallegos-Orozco JF, Paul A, Lane MA, Lampropulos JF, Natividad I, Perestelo-Perez L, Ponce de Leon-Lovaton PG, Erwin PJ, Carey J, Montori VM. Natural history of nonfunctioning pituitary adenomas and incidentalomas: a systematic review and metaanalysis. J Clin Endocrinol Metab. 2011;96(4):905–912. [PubMed: 21474687]
- 32.
- Kim JH, Dho YS, Kim YH, Lee JH, Lee JH, Hong AR, Shin CS. Developing an optimal follow-up strategy based on the natural history of nonfunctioning pituitary adenomas. J Neurosurg. 2018;131(2):500–506. [PubMed: 30215565]
- 33.
- Tresoldi AS, Carosi G, Betella N, Del Sindaco G, Indirli R, Ferrante E, Sala E, Giavoli C, Morenghi E, Locatelli M, Milani D, Mazziotti G, Spada A, Arosio M, Mantovani G, Lania AGA. Clinically Nonfunctioning Pituitary Incidentalomas: Characteristics and Natural History. Neuroendocrinology. 2020;110(7-8):595–603. [PubMed: 31525736]
- 34.
- Sonigo C, Bouilly J, Carre N, Tolle V, Caraty A, Tello J, Simony-Conesa FJ, Millar R, Young J, Binart N. Hyperprolactinemia-induced ovarian acyclicity is reversed by kisspeptin administration. J Clin Invest. 2012;122(10):3791–3795. [PMC free article: PMC3461919] [PubMed: 23006326]
- 35.
- Karavitaki N, Thanabalasingham G, Shore HC, Trifanescu R, Ansorge O, Meston N, Turner HE, Wass JA. Do the limits of serum prolactin in disconnection hyperprolactinaemia need re-definition? A study of 226 patients with histologically verified non-functioning pituitary macroadenoma. Clin Endocrinol (Oxf). 2006;65(4):524–529. [PubMed: 16984247]
- 36.
- Gryngarten MG, Braslavsky D, Ballerini MG, Ledesma J, Ropelato MG, Escobar ME. Spontaneous ovarian hyperstimulation syndrome caused by a follicle-stimulating hormone-secreting pituitary macroadenoma in an early pubertal girl. Horm Res Paediatr. 2010;73(4):293–298. [PubMed: 20215777]
- 37.
- Graillon T, Castinetti F, Chabert-Orsini V, Morange I, Cuny T, Albarel F, Brue T, Dufour H. Functioning gonadotroph adenoma with severe ovarian hyperstimulation syndrome: A new emergency in pituitary adenoma surgery? Surgical considerations and literature review. Ann Endocrinol (Paris). 2019;80(2):122–127. [PubMed: 30825998]
- 38.
- Broughton C, Mears J, Williams A, Lonnen K. A clinically functioning gonadotroph adenoma presenting with abdominal pain, ovarian hyperstimulation and fibromatosis. Endocrinol Diabetes Metab Case Rep. 2018;2018. [PMC free article: PMC6300858] [PubMed: 30532999]
- 39.
- Takeda M, Otsuka F, Suzuki J, Kishida M, Ogura T, Tamiya T, Makino H. Involvement of activin/BMP system in development of human pituitary gonadotropinomas and nonfunctioning adenomas. Biochem Biophys Res Commun. 2003;306(4):812–818. [PubMed: 12821114]
- 40.
- Mete O, Lopes MB. Overview of the 2017 WHO Classification of Pituitary Tumors. Endocr Pathol. 2017;28(3):228–243. [PubMed: 28766057]
- 41.
- Torregrosa-Quesada ME, García-Martínez A, Sánchez-Barbie A, Silva-Ortega S, Cámara R, Fajardo C, Lamas C, Aranda I, Pico A. The silent variants of pituitary tumors: demographic, radiological and molecular characteristics. J Endocrinol Invest. 2021;44(8):1637–1648. [PubMed: 33476035]
- 42.
- Villa C, Vasiljevic A, Jaffrain-Rea ML, Ansorge O, Asioli S, Barresi V, Chinezu L, Gardiman MP, Lania A, Lapshina AM, Poliani L, Reiniger L, Righi A, Saeger W, Soukup J, Theodoropoulou M, Uccella S, Trouillas J, Roncaroli F. A standardised diagnostic approach to pituitary neuroendocrine tumours (PitNETs): a European Pituitary Pathology Group (EPPG) proposal. Virchows Arch. 2019;475(6):687–692. [PubMed: 31578606]
- 43.
- Trouillas J, Jaffrain-Rea ML, Vasiljevic A, Raverot G, Roncaroli F, Villa C. How to Classify the Pituitary Neuroendocrine Tumors (PitNET)s in 2020. Cancers (Basel). 2020;12(2) [PMC free article: PMC7072139] [PubMed: 32098443]
- 44.
- Gomez-Hernandez K, Ezzat S, Asa SL, Mete O. Clinical Implications of Accurate Subtyping of Pituitary Adenomas: Perspectives from the Treating Physician. Turk Patoloji Derg. 2015;31 Suppl 1:4–17. [PubMed: 26177314]
- 45.
- Balogun JA, Monsalves E, Juraschka K, Parvez K, Kucharczyk W, Mete O, Gentili F, Zadeh G. Null cell adenomas of the pituitary gland: an institutional review of their clinical imaging and behavioral characteristics. Endocr Pathol. 2015;26(1):63–70. [PubMed: 25403448]
- 46.
- Manojlovic-Gacic E, Bollerslev J, Casar-Borota O. Invited Review: Pathology of pituitary neuroendocrine tumours: present status, modern diagnostic approach, controversies and future perspectives from a neuropathological and clinical standpoint. Neuropathol Appl Neurobiol. 2020;46(2):89–110. [PubMed: 31112312]
- 47.
- Raverot G, Wierinckx A, Jouanneau E, Auger C, Borson-Chazot F, Lachuer J, Pugeat M, Trouillas J. Clinical, hormonal and molecular characterization of pituitary ACTH adenomas without (silent corticotroph adenomas) and with Cushing's disease. Eur J Endocrinol. 2010;163(1):35–43. [PubMed: 20385723]
- 48.
- Righi A, Faustini-Fustini M, Morandi L, Monti V, Asioli S, Mazzatenta D, Bacci A, Foschini MP. The changing faces of corticotroph cell adenomas: the role of prohormone convertase 1/3. Endocrine. 2017;56(2):286–297. [PubMed: 27491554]
- 49.
- Ben-Shlomo A, Cooper O. Silent corticotroph adenomas. Pituitary. 2018;21(2):183–193. [PubMed: 29344907]
- 50.
- Sjostedt E, Bollerslev J, Mulder J, Lindskog C, Ponten F, Casar-Borota O. A specific antibody to detect transcription factor T-Pit: a reliable marker of corticotroph cell differentiation and a tool to improve the classification of pituitary neuroendocrine tumours. Acta Neuropathol. 2017;134(4):675–677. [PubMed: 28823042]
- 51.
- McDonald WC, McDonald KN, Helmer JA, Ho B, Wang A, Banerji N. The Role of T-box Transcription Factor in a Pituitary Adenoma Diagnostic Algorithm. Arch Pathol Lab Med. 2021;145(5):592–598. [PubMed: 32991684]
- 52.
- Cazabat L, Dupuy M, Boulin A, Bernier M, Baussart B, Foubert L, Raffin-Sanson ML, Caron P, Bertherat J, Gaillard S. Silent, but not unseen: multimicrocystic aspect on T2-weighted MRI in silent corticotroph adenomas. Clin Endocrinol (Oxf). 2014;81(4):566–572. [PubMed: 24601912]
- 53.
- Kasuki L, Antunes X, Coelho MCA, Lamback EB, Galvao S, Silva Camacho AH, Chimelli L, Ventura N, Gadelha MR. Accuracy of microcystic aspect on T2-weighted MRI for the diagnosis of silent corticotroph adenomas. Clin Endocrinol (Oxf). 2020;92(2):145–149. [PubMed: 31773787]
- 54.
- Di Ieva A, Davidson JM, Syro LV, Rotondo F, Montoya JF, Horvath E, Cusimano MD, Kovacs K. Crooke's cell tumors of the pituitary. Neurosurgery. 2015;76(5):616–622. [PubMed: 25635886]
- 55.
- Tateno T, Izumiyama H, Doi M, Yoshimoto T, Shichiri M, Inoshita N, Oyama K, Yamada S, Hirata Y. Differential gene expression in ACTH -secreting and non-functioning pituitary tumors. Eur J Endocrinol. 2007;157(6):717–724. [PubMed: 18057378]
- 56.
- Zheng G, Lu L, Zhu H, You H, Feng M, Liu X, Dai C, Yao Y, Wang R, Zhang H, Sun X, Lu Z. Clinical, Laboratory, and Treatment Profiles of Silent Corticotroph Adenomas That Have Transformed to the Functional Type: A Case Series With a Literature Review. Front Endocrinol (Lausanne). 2020;11:558593. [PMC free article: PMC7538591] [PubMed: 33071973]
- 57.
- Araki T, Tone Y, Yamamoto M, Kameda H, Ben-Shlomo A, Yamada S, Takeshita A, Yamamoto M, Kawakami Y, Tone M, Melmed S. Two Distinctive POMC Promoters Modify Gene Expression in Cushing Disease. J Clin Endocrinol Metab. 2021;106(9):e3346–e3363. [PMC free article: PMC8372657] [PubMed: 34061962]
- 58.
- Chinezu L, Vasiljevic A, Trouillas J, Lapoirie M, Jouanneau E, Raverot G. Silent somatotroph tumour revisited from a study of 80 patients with and without acromegaly and a review of the literature. Eur J Endocrinol. 2017;176(2):195–201. [PubMed: 27913611]
- 59.
- Sidhaye A, Burger P, Rigamonti D, Salvatori R. Giant somatotrophinoma without acromegalic features: more "quiet" than "silent": case report. Neurosurgery. 2005;56(5):E1154–discussion E1154. [PubMed: 15854264]
- 60.
- Langlois F, Woltjer R, Cetas JS, Fleseriu M. Silent somatotroph pituitary adenomas: an update. Pituitary. 2018;21(2):194–202. [PubMed: 29305680]
- 61.
- Langlois F, Lim DST, Yedinak CG, Cetas I, McCartney S, Cetas J, Dogan A, Fleseriu M. Predictors of silent corticotroph adenoma recurrence; a large retrospective single center study and systematic literature review. Pituitary. 2018;21(1):32–40. [PubMed: 29032459]
- 62.
- Naritaka H, Kameya T, Sato Y, Furuhata S, Otani M, Kawase T. Morphological characterization and subtyping of silent somatotroph adenomas. Pituitary. 1999;1(3-4):233–241. [PubMed: 11081203]
- 63.
- Kirkman MA, Jaunmuktane Z, Brandner S, Khan AA, Powell M, Baldeweg SE. Active and silent thyroid-stimulating hormone-expressing pituitary adenomas: presenting symptoms, treatment, outcomes, and recurrence. World Neurosurg. 2014;82(6):1224–1231. [PubMed: 24657816]
- 64.
- Cyprich J, Donoho DA, Brunswick A, Hurth K, Carmichael JD, Weiss MH, Zada G. Surgical management of clinically silent thyrotropin pituitary adenomas: A single center series of 20 patients. J Clin Neurosci. 2020;71:70–75. [PubMed: 31668712]
- 65.
- Aydin S, Comunoglu N, Ahmedov ML, Korkmaz OP, Oz B, Kadioglu P, Gazioglu N, Tanriover N. Clinicopathologic Characteristics and Surgical Treatment of Plurihormonal Pituitary Adenomas. World Neurosurg. 2019;130:e765–e774. [PubMed: 31295602]
- 66.
- Erickson D, Scheithauer B, Atkinson J, Horvath E, Kovacs K, Lloyd RV, Young WF Jr. Silent subtype 3 pituitary adenoma: a clinicopathologic analysis of the Mayo Clinic experience. Clin Endocrinol (Oxf). 2009;71(1):92–99. [PubMed: 19170710]
- 67.
- Mete O, Gomez-Hernandez K, Kucharczyk W, Ridout R, Zadeh G, Gentili F, Ezzat S, Asa SL. Silent subtype 3 pituitary adenomas are not always silent and represent poorly differentiated monomorphous plurihormonal Pit-1 lineage adenomas. Mod Pathol. 2016;29(2):131–142. [PubMed: 26743473]
- 68.
- Micko A, Rotzer T, Hoftberger R, Vila G, Oberndorfer J, Frischer JM, Knosp E, Wolfsberger S. Expression of additional transcription factors is of prognostic value for aggressive behavior of pituitary adenomas. J Neurosurg. 2020;134(3):1139–1146. [PubMed: 32302984]
- 69.
- Freda PU, Beckers AM, Katznelson L, Molitch ME, Montori VM, Post KD, Vance ML, Endocrine S. Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2011;96(4):894–904. [PMC free article: PMC5393422] [PubMed: 21474686]
- 70.
- do Carmo Dias Gontijo M, de Souza Vasconcellos L, Ribeiro-Oliveira A Jr. Hook effect and linear range in prolactin assays: distinct confounding entities. Pituitary. 2016;19(4):458–459. [PubMed: 25577219]
- 71.
- Vilar L, Abucham J, Albuquerque JL, Araujo LA, Azevedo MF, Boguszewski CL, Casulari LA, Cunha Neto MBC, Czepielewski MA, Duarte FHG, Faria MDS, Gadelha MR, Garmes HM, Glezer A, Gurgel MH, Jallad RS, Martins M, Miranda PAC, Montenegro RM, Musolino NRC, Naves LA, Ribeiro-Oliveira A Junior, Silva CMS, Viecceli C, Bronstein MD. Controversial issues in the management of hyperprolactinemia and prolactinomas - An overview by the Neuroendocrinology Department of the Brazilian Society of Endocrinology and Metabolism. Archives of endocrinology and metabolism. 2018;62(2):236–263. [PMC free article: PMC10118988] [PubMed: 29768629]
- 72.
- Glezer A, Bronstein MD. Hyperprolactinemia. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2000.
- 73.
- Carosi G, Malchiodi E, Ferrante E, Sala E, Verrua E, Profka E, Giavoli C, Filopanti M, Beck-Peccoz P, Spada A, Mantovani G. Hypothalamic-Pituitary Axis in Non-Functioning Pituitary Adenomas: Focus on the Prevalence of Isolated Central Hypoadrenalism. Neuroendocrinology. 2015;102(4):267–273. [PubMed: 25924873]
- 74.
- Boguszewski CL, de Castro Musolino NR, Kasuki L. Management of pituitary incidentaloma. Best Pract Res Clin Endocrinol Metab. 2019;33(2):101268. [PubMed: 31027975]
- 75.
- Fleseriu M, Hashim IA, Karavitaki N, Melmed S, Murad MH, Salvatori R, Samuels MH. Hormonal Replacement in Hypopituitarism in Adults: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101(11):3888–3921. [PubMed: 27736313]
- 76.
- El-Farhan N, Pickett A, Ducroq D, Bailey C, Mitchem K, Morgan N, Armston A, Jones L, Evans C, Rees DA. Method-specific serum cortisol responses to the adrenocorticotrophin test: comparison of gas chromatography-mass spectrometry and five automated immunoassays. Clin Endocrinol (Oxf). 2013;78(5):673–680. [PubMed: 22994849]
- 77.
- Bonneville F. Silent Corticotroph Adenoma. In: Bonneville J, F B, Cattin F, S N, eds. MRI of the pituitary glan: Springer; 2016.
- 78.
- Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, Trouillas J, Dekkers OM. European Society of E. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. Eur J Endocrinol. 2018;178(1):G1–G24. [PubMed: 29046323]
- 79.
- Boertien TM, Booij J, Majoie C, Drent ML, Pereira AM, Biermasz NR, Simsek S, Veldman RG, Stokkel MPM, Bisschop PH, Fliers E. (68)Ga-DOTATATE PET imaging in clinically non-functioning pituitary macroadenomas. Eur J Hybrid Imaging. 2020;4(1):4. [PMC free article: PMC8218160] [PubMed: 34191241]
- 80.
- Kaltsas GA, Evanson J, Chrisoulidou A, Grossman AB. The diagnosis and management of parasellar tumours of the pituitary. Endocr Relat Cancer. 2008;15(4):885–903. [PubMed: 18809592]
- 81.
- Samejima N, Yamada S, Takada K, Sano T, Ozawa Y, Shimizu T, Usui M, Shishiba Y. Serum alpha-subunit levels in patients with pituitary adenomas. Clin Endocrinol (Oxf). 2001;54(4):479–484. [PubMed: 11318783]
- 82.
- Vieira Neto L, Wildemberg LE, Moraes AB, Colli LM, Kasuki L, Marques NV, Gasparetto EL, de Castro M, Takiya CM, Gadelha MR. Dopamine receptor subtype 2 expression profile in nonfunctioning pituitary adenomas and in vivo response to cabergoline therapy. Clin Endocrinol (Oxf). 2015;82(5):739–746. [PubMed: 25418156]
- 83.
- Greenman Y, Cooper O, Yaish I, Robenshtok E, Sagiv N, Jonas-Kimchi T, Yuan X, Gertych A, Shimon I, Ram Z, Melmed S, Stern N. Treatment of clinically nonfunctioning pituitary adenomas with dopamine agonists. Eur J Endocrinol. 2016;175(1):63–72. [PubMed: 27150495]
- 84.
- Tampourlou M, Ntali G, Ahmed S, Arlt W, Ayuk J, Byrne JV, Chavda S, Cudlip S, Gittoes N, Grossman A, Mitchell R, O'Reilly MW, Paluzzi A, Toogood A, Wass JAH, Karavitaki N. Outcome of Nonfunctioning Pituitary Adenomas That Regrow After Primary Treatment: A Study From Two Large UK Centers. J Clin Endocrinol Metab. 2017;102(6):1889–1897. [PubMed: 28323946]
- 85.
- Lucas JW, Bodach ME, Tumialan LM, Oyesiku NM, Patil CG, Litvack Z, Aghi MK, Zada G. Neurosurgery. 2016;79(4):E533–535. Congress of Neurological Surgeons Systematic Review and Evidence-Based Guideline on Primary Management of Patients With Nonfunctioning Pituitary Adenomas. [PubMed: 27635961]
- 86.
- Huang W, Molitch ME. Management of nonfunctioning pituitary adenomas (NFAs): observation. Pituitary. 2018;21(2):162–167. [PubMed: 29280025]
- 87.
- Murad MH, Fernandez-Balsells MM, Barwise A, Gallegos-Orozco JF, Paul A, Lane MA, Lampropulos JF, Natividad I, Perestelo-Perez L, Ponce de Leon-Lovaton PG, Albuquerque FN, Carey J, Erwin PJ, Montori VM. Outcomes of surgical treatment for nonfunctioning pituitary adenomas: a systematic review and meta-analysis. Clin Endocrinol (Oxf). 2010;73(6):777–791. [PubMed: 20846296]
- 88.
- Reddy R, Cudlip S, Byrne JV, Karavitaki N, Wass JA. Can we ever stop imaging in surgically treated and radiotherapy-naive patients with non-functioning pituitary adenoma? Eur J Endocrinol. 2011;165(5):739–744. [PubMed: 21900406]
- 89.
- Oystese KA, Evang JA, Bollerslev J. Non-functioning pituitary adenomas: growth and aggressiveness. Endocrine. 2016;53(1):28–34. [PubMed: 27066792]
- 90.
- Micko AS, Wohrer A, Wolfsberger S, Knosp E. Invasion of the cavernous sinus space in pituitary adenomas: endoscopic verification and its correlation with an MRI-based classification. J Neurosurg. 2015;122(4):803–811. [PubMed: 25658782]
- 91.
- Buchy M, Lapras V, Rabilloud M, Vasiljevic A, Borson-Chazot F, Jouanneau E, Raverot G. Predicting early post-operative remission in pituitary adenomas: evaluation of the modified knosp classification. Pituitary. 2019;22(5):467–475. [PubMed: 31286328]
- 92.
- Berkmann S, Lattmann J, Schuetz P, Diepers M, Remonda L, Fandino J, Buchfelder M, Mueller B. The Shape grading system: a classification for growth patterns of pituitary adenomas. Acta Neurochir (Wien). 2021;163(11):3181–3189. [PubMed: 34223996]
- 93.
- Losa M, Mortini P, Barzaghi R, Ribotto P, Terreni MR, Marzoli SB, Pieralli S, Giovanelli M. Early results of surgery in patients with nonfunctioning pituitary adenoma and analysis of the risk of tumor recurrence. J Neurosurg. 2008;108(3):525–532. [PubMed: 18312100]
- 94.
- Oh H, Cheun H, Kim YJ, Yoon HK, Kang H, Lee HC, Park HP, Kim JH, Kim YH. Cephalocaudal tumor diameter is a predictor of diabetes insipidus after endoscopic transsphenoidal surgery for non-functioning pituitary adenoma. Pituitary. 2021;24(3):303–311. [PubMed: 33191457]
- 95.
- Kinoshita Y, Taguchi A, Tominaga A, Sakoguchi T, Arita K, Yamasaki F. Predictive factors of postoperative diabetes insipidus in 333 patients undergoing transsphenoidal surgery for non-functioning pituitary adenoma. Pituitary. 2021 [PubMed: 34283369]
- 96.
- Gasco V, Caputo M, Lanfranco F, Ghigo E, Grottoli S. Management of GH treatment in adult GH deficiency. Best Pract Res Clin Endocrinol Metab. 2017;31(1):13–24. [PubMed: 28477728]
- 97.
- Chanson P, Raverot G, Castinetti F, Cortet-Rudelli C, Galland F, Salenave S. French Endocrinology Society non-functioning pituitary adenoma w-g. Management of clinically non-functioning pituitary adenoma. Ann Endocrinol (Paris). 2015;76(3):239–247. [PubMed: 26072284]
- 98.
- Arnold JR, Arnold DF, Marland A, Karavitaki N, Wass JA. GH replacement in patients with non-functioning pituitary adenoma (NFA) treated solely by surgery is not associated with increased risk of tumour recurrence. Clin Endocrinol (Oxf). 2009;70(3):435–438. [PubMed: 19236640]
- 99.
- Pal A, Capatina C, Tenreiro AP, Guardiola PD, Byrne JV, Cudlip S, Karavitaki N, Wass JA. Pituitary apoplexy in non-functioning pituitary adenomas: long term follow up is important because of significant numbers of tumour recurrences. Clin Endocrinol (Oxf). 2011;75(4):501–504. [PubMed: 21521336]
- 100.
- Colao A, Cerbone G, Cappabianca P, Ferone D, Alfieri A, Di Salle F, Faggiano A, Merola B, de Divitiis E, Lombardi G. Effect of surgery and radiotherapy on visual and endocrine function in nonfunctioning pituitary adenomas. J Endocrinol Invest. 1998;21(5):284–290. [PubMed: 9648049]
- 101.
- Brochier S, Galland F, Kujas M, Parker F, Gaillard S, Raftopoulos C, Young J, Alexopoulou O, Maiter D, Chanson P. Factors predicting relapse of nonfunctioning pituitary macroadenomas after neurosurgery: a study of 142 patients. Eur J Endocrinol. 2010;163(2):193–200. [PubMed: 20460423]
- 102.
- Minniti G, Clarke E, Scaringi C, Enrici RM. Stereotactic radiotherapy and radiosurgery for non-functioning and secreting pituitary adenomas. Reports of practical oncology and radiotherapy : journal of Greatpoland Cancer Center in Poznan and Polish Society of Radiation Oncology. 2016;21(4):370–378. [PMC free article: PMC4899479] [PubMed: 27330422]
- 103.
- Ramirez C, Cheng S, Vargas G, Asa SL, Ezzat S, Gonzalez B, Cabrera L, Guinto G, Mercado M. Expression of Ki-67, PTTG1, FGFR4, and SSTR 2, 3, and 5 in nonfunctioning pituitary adenomas: a high throughput TMA, immunohistochemical study. J Clin Endocrinol Metab. 2012;97(5):1745–1751. [PubMed: 22419713]
- 104.
- Cohen-Inbar O, Xu Z, Lee CC, Wu CC, Chytka T, Silva D, Sharma M, Radwan H, Grills IS, Nguyen B, Siddiqui Z, Mathieu D, Iorio-Morin C, Wolf A, Cifarelli CP, Cifarelli DT, Lunsford LD, Kondziolka D, Sheehan JP. Prognostic significance of corticotroph staining in radiosurgery for non-functioning pituitary adenomas: a multicenter study. J Neurooncol. 2017;135(1):67–74. [PMC free article: PMC5817983] [PubMed: 28913674]
- 105.
- Strickland BA, Shahrestani S, Briggs RG, Jackanich A, Tavakol S, Hurth K, Shiroishi MS, Liu CJ, Carmichael JD, Weiss M, Zada G. Silent corticotroph pituitary adenomas: clinical characteristics, long-term outcomes, and management of disease recurrence. J Neurosurg. 2021:1–8. [PubMed: 33962375]
- 106.
- Fountas A, Lavrentaki A, Subramanian A, Toulis KA, Nirantharakumar K, Karavitaki N. Recurrence in silent corticotroph adenomas after primary treatment: A systematic review and meta-analysis. J Clin Endocrinol Metab. 2018 [PubMed: 30590584]
- 107.
- Dekkers OM, Hammer S, de Keizer RJ, Roelfsema F, Schutte PJ, Smit JW, Romijn JA, Pereira AM. The natural course of non-functioning pituitary macroadenomas. Eur J Endocrinol. 2007;156(2):217–224. [PubMed: 17287411]
- 108.
- Galland F, Vantyghem MC, Cazabat L, Boulin A, Cotton F, Bonneville JF, Jouanneau E, Vidal-Trecan G, Chanson P. Management of nonfunctioning pituitary incidentaloma. Ann Endocrinol (Paris). 2015;76(3):191–200. [PubMed: 26054868]
- 109.
- Guo BJ, Yang ZL, Zhang LJ. Gadolinium Deposition in Brain: Current Scientific Evidence and Future Perspectives. Front Mol Neurosci. 2018;11:335. [PMC free article: PMC6158336] [PubMed: 30294259]
- 110.
- Garcia EC, Naves LA, Silva AO, de Castro LF, Casulari LA, Azevedo MF. Short-term treatment with cabergoline can lead to tumor shrinkage in patients with nonfunctioning pituitary adenomas. Pituitary. 2013;16(2):189–194. [PubMed: 22740242]
- 111.
- Nobels FR, de Herder WW, van den Brink WM, Kwekkeboom DJ, Hofland LJ, Zuyderwijk J, de Jong FH, Lamberts SW. Long-term treatment with the dopamine agonist quinagolide of patients with clinically non-functioning pituitary adenoma. Eur J Endocrinol. 2000;143(5):615–621. [PubMed: 11078985]
- 112.
- Pivonello R, Matrone C, Filippella M, Cavallo LM, Di Somma C, Cappabianca P, Colao A, Annunziato L, Lombardi G. Dopamine receptor expression and function in clinically nonfunctioning pituitary tumors: comparison with the effectiveness of cabergoline treatment. J Clin Endocrinol Metab. 2004;89(4):1674–1683. [PubMed: 15070930]
- 113.
- Batista RL, Musolino NRC, Cescato VAS, da Silva GO, Medeiros RSS, Herkenhoff CGB, Trarbach EB, Cunha-Neto MB. Cabergoline in the Management of Residual Nonfunctioning Pituitary Adenoma: A Single-Center, Open-Label, 2-Year Randomized Clinical Trial. Am J Clin Oncol. 2019;42(2):221–227. [PubMed: 30540568]
- 114.
- Greenman Y, Bronstein MD. Cabergoline should be attempted in progressing non-functioning pituitary macroadenoma. Eur J Endocrinol. 2021;185(4):D11–D20. [PubMed: 34288884]
- 115.
- Yavropoulou MP, Tsoli M, Barkas K, Kaltsas G, Grossman A. The natural history and treatment of non-functioning pituitary adenomas (non-functioning PitNETs). Endocr Relat Cancer. 2020;27(10):R375–R390. [PubMed: 32674070]
- 116.
- Even-Zohar N, Greenman Y. Management of NFAs: medical treatment. Pituitary. 2018;21(2):168–175. [PubMed: 29344905]
- 117.
- Taboada GF, Luque RM, Bastos W, Guimaraes RF, Marcondes JB, Chimelli LM, Fontes R, Mata PJ, Filho PN, Carvalho DP, Kineman RD, Gadelha MR. Quantitative analysis of somatostatin receptor subtype (SSTR1-5) gene expression levels in somatotropinomas and non-functioning pituitary adenomas. Eur J Endocrinol. 2007;156(1):65–74. [PubMed: 17218727]
- 118.
- Lee M, Lupp A, Mendoza N, Martin N, Beschorner R, Honegger J, Schlegel J, Shively T, Pulz E, Schulz S, Roncaroli F, Pellegata NS. SSTR3 is a putative target for the medical treatment of gonadotroph adenomas of the pituitary. Endocr Relat Cancer. 2015;22(1):111–119. [PubMed: 25515731]
- 119.
- Gasperi M, Petrini L, Pilosu R, Nardi M, Marcello A, Mastio F, Bartalena L, Martino E. Octreotide treatment does not affect the size of most non-functioning pituitary adenomas. J Endocrinol Invest. 1993;16(7):541–543. [PubMed: 8227984]
- 120.
- Fusco A, Giampietro A, Bianchi A, Cimino V, Lugli F, Piacentini S, Lorusso M, Tofani A, Perotti G, Lauriola L, Anile C, Maira G, Pontecorvi A, De Marinis L. Treatment with octreotide LAR in clinically non-functioning pituitary adenoma: results from a case-control study. Pituitary. 2012;15(4):571–578. [PubMed: 22207350]
- 121.
- Vieria Neto L, Wildemberg LE, Colli LM, Kasuki L, Marques NV, Moraes AB, Gasparetto EL, Takiya CM, Castro M, Gadelha MR. ZAC1 and SSTR2 are downregulated in non-functioning pituitary adenomas but not in somatotropinomas. PLoS One. 2013;8(10):e77406. [PMC free article: PMC3788723] [PubMed: 24098585]
- 122.
- Oystese KA, Casar-Borota O, Normann KR, Zucknick M, Berg JP, Bollerslev J. Estrogen Receptor alpha, a Sex-Dependent Predictor of Aggressiveness in Nonfunctioning Pituitary Adenomas: SSTR and Sex Hormone Receptor Distribution in NFPA. J Clin Endocrinol Metab. 2017;102(9):3581–3590. [PubMed: 28911153]
- 123.
- Zatelli MC, Piccin D, Vignali C, Tagliati F, Ambrosio MR, Bondanelli M, Cimino V, Bianchi A, Schmid HA, Scanarini M, Pontecorvi A, De Marinis L, Maira G. degli Uberti EC. Pasireotide, a multiple somatostatin receptor subtypes ligand, reduces cell viability in non-functioning pituitary adenomas by inhibiting vascular endothelial growth factor secretion. Endocr Relat Cancer. 2007;14(1):91–102. [PubMed: 17395978]
- 124.
- Castellnou S, Vasiljevic A, Lapras V, Raverot V, Alix E, Borson-Chazot F, Jouanneau E, Raverot G, Lasolle H. SST5 expression and USP8 mutation in functioning and silent corticotroph pituitary tumors. Endocrine connections. 2020 [PMC free article: PMC7077525] [PubMed: 32101529]
- 125.
- Bujko M, Kober P, Boresowicz J, Rusetska N, Paziewska A, Dabrowska M, Piascik A, Pekul M, Zielinski G, Kunicki J, Bonicki W, Ostrowski J, Siedlecki JA, Maksymowicz M. USP8 mutations in corticotroph adenomas determine a distinct gene expression profile irrespective of functional tumour status. Eur J Endocrinol. 2019;181(6):615–627. [PubMed: 31581124]
- 126.
- Perez-Rivas LG, Theodoropoulou M, Ferrau F, Nusser C, Kawaguchi K, Stratakis CA, Faucz FR, Wildemberg LE, Assie G, Beschorner R, Dimopoulou C, Buchfelder M, Popovic V, Berr CM, Toth M, Ardisasmita AI, Honegger J, Bertherat J, Gadelha MR, Beuschlein F, Stalla G, Komada M, Korbonits M, Reincke M. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing's Disease. J Clin Endocrinol Metab. 2015;100(7):E997–1004. [PMC free article: PMC4490309] [PubMed: 25942478]
- 127.
- Boertien TM, Drent ML, Booij J, Majoie C, Stokkel MPM, Hoogmoed J, Pereira A, Biermasz NR, Simsek S, Groote Veldman R, Tanck MWT, Fliers E, Bisschop PH. The GALANT trial: study protocol of a randomised placebo-controlled trial in patients with a (68) Ga -DOTATATE PET-positive, clinically non-functioning pituitary macroadenoma on the effect of lanreotide on tumour size. BMJ Open. 2020;10(8):e038250. [PMC free article: PMC7430490] [PubMed: 32792446]
- 128.
- Gulde S, Wiedemann T, Schillmaier M, Valenca I, Lupp A, Steiger K, Yen HY, Bauerle S, Notni J, Luque R, Schmid H, Schulz S, Ankerst DP, Schilling F, Pellegata NS. Gender-Specific Efficacy Revealed by Head-to-Head Comparison of Pasireotide and Octreotide in a Representative In Vivo Model of Nonfunctioning Pituitary Tumors. Cancers (Basel). 2021;13(12) [PMC free article: PMC8235746] [PubMed: 34205778]
- 129.
- Florio T, Barbieri F, Spaziante R, Zona G, Hofland LJ, van Koetsveld PM, Feelders RA, Stalla GK, Theodoropoulou M, Culler MD, Dong J, Taylor JE, Moreau JP, Saveanu A, Gunz G, Dufour H, Jaquet P. Efficacy of a dopamine-somatostatin chimeric molecule, BIM-23A760, in the control of cell growth from primary cultures of human non-functioning pituitary adenomas: a multi-center study. Endocr Relat Cancer. 2008;15(2):583–596. [PubMed: 18509006]
- 130.
- Halem HA, Hochgeschwender U, Rih JK, Nelson R, Johnson GA, Thiagalingam A, Culler MD. TBR-760, a Dopamine-Somatostatin Compound, Arrests Growth of Aggressive Nonfunctioning Pituitary Adenomas in Mice. Endocrinology. 2020;161(8) [PMC free article: PMC7375803] [PubMed: 32591776]
- 131.
- McCormack AI, Wass JA, Grossman AB. Aggressive pituitary tumours: the role of temozolomide and the assessment of MGMT status. Eur J Clin Invest. 2011;41(10):1133–1148. [PubMed: 21496012]
- 132.
- McCormack A, Dekkers OM, Petersenn S, Popovic V, Trouillas J, Raverot G, Burman P. collaborators ESEs. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur J Endocrinol. 2018;178(3):265–276. [PubMed: 29330228]
- 133.
- Widhalm G, Wolfsberger S, Preusser M, Woehrer A, Kotter MR, Czech T, Marosi C, Knosp E. O. (6)-methylguanine DNA methyltransferase immunoexpression in nonfunctioning pituitary adenomas: are progressive tumors potential candidates for temozolomide treatment? Cancer. 2009;115(5):1070–1080. [PubMed: 19156926]
- 134.
- Ilie MD, Raverot G. Treatment Options for Gonadotroph Tumors: Current State and Perspectives. J Clin Endocrinol Metab. 2020;105(10) [PubMed: 32735647]
- 135.
- Dixit S, Baker L, Walmsley V, Hingorani M. Temozolomide-related idiosyncratic and other uncommon toxicities: a systematic review. Anticancer Drugs. 2012;23(10):1099–1106. [PubMed: 22850321]
- 136.
- Ilie MD, Lasolle H, Raverot G. Emerging and Novel Treatments for Pituitary Tumors. J Clin Med. 2019;8(8) [PMC free article: PMC6723109] [PubMed: 31349718]
- 137.
- Nakano-Tateno T, Lau KJ, Wang J, McMahon C, Kawakami Y, Tateno T, Araki T. Multimodal Non-Surgical Treatments of Aggressive Pituitary Tumors. Front Endocrinol (Lausanne). 2021;12:624686. [PMC free article: PMC8033019] [PubMed: 33841328]
- 138.
- Maclean J, Aldridge M, Bomanji J, Short S, Fersht N. Peptide receptor radionuclide therapy for aggressive atypical pituitary adenoma/carcinoma: variable clinical response in preliminary evaluation. Pituitary. 2014;17(6):530–538. [PubMed: 24323313]
- 139.
- Komor J, Reubi JC, Christ ER. Peptide receptor radionuclide therapy in a patient with disabling non-functioning pituitary adenoma. Pituitary. 2014;17(3):227–231. [PubMed: 23740146]
- 140.
- Novruzov F, Aliyev JA, Jaunmuktane Z, Bomanji JB, Kayani I. The use of (68)Ga DOTATATE PET/CT for diagnostic assessment and monitoring of (177)Lu DOTATATE therapy in pituitary carcinoma. Clin Nucl Med. 2015;40(1):47–49. [PubMed: 25275413]
- 141.
- Giuffrida G, Ferrau F, Laudicella R, Cotta OR, Messina E, Granata F, Angileri FF, Vento A, Alibrandi A, Baldari S, Cannavo S. Peptide receptor radionuclide therapy for aggressive pituitary tumors: a monocentric experience. Endocrine connections. 2019;8(5):528–535. [PMC free article: PMC6499924] [PubMed: 30939449]
- 142.
- Bengtsson D, Schroder HD, Andersen M, Maiter D, Berinder K, Feldt Rasmussen U, Rasmussen AK, Johannsson G, Hoybye C, van der Lely AJ, Petersson M, Ragnarsson O, Burman P. Long-term outcome and MGMT as a predictive marker in 24 patients with atypical pituitary adenomas and pituitary carcinomas given treatment with temozolomide. J Clin Endocrinol Metab. 2015;100(4):1689–1698. [PubMed: 25646794]
- 143.
- Tampourlou M, Fountas A, Ntali G, Karavitaki N. Mortality in patients with non-functioning pituitary adenoma. Pituitary. 2018;21(2):203–207. [PMC free article: PMC5849651] [PubMed: 29344906]
- 144.
- Ntali G, Capatina C, Fazal-Sanderson V, Byrne JV, Cudlip S, Grossman AB, Wass JA, Karavitaki N. Mortality in patients with non-functioning pituitary adenoma is increased: systematic analysis of 546 cases with long follow-up. Eur J Endocrinol. 2016;174(2):137–145. [PubMed: 26546611]
- 145.
- Chang EF, Zada G, Kim S, Lamborn KR, Quinones-Hinojosa A, Tyrrell JB, Wilson CB, Kunwar S. Long-term recurrence and mortality after surgery and adjuvant radiotherapy for nonfunctional pituitary adenomas. J Neurosurg. 2008;108(4):736–745. [PubMed: 18377253]
- 146.
- Hammarstrand C, Ragnarsson O, Hallen T, Andersson E, Skoglund T, Nilsson AG, Johannsson G, Olsson DS. Higher glucocorticoid replacement doses are associated with increased mortality in patients with pituitary adenoma. Eur J Endocrinol. 2017;177(3):251–256. [PubMed: 28596421]
- 147.
- Andela CD, Scharloo M, Pereira AM, Kaptein AA, Biermasz NR. Quality of life (QoL) impairments in patients with a pituitary adenoma: a systematic review of QoL studies. Pituitary. 2015;18(5):752–776. [PubMed: 25605584]
- 148.
- Dekkers OM, van der Klaauw AA, Pereira AM, Biermasz NR, Honkoop PJ, Roelfsema F, Smit JW, Romijn JA. Quality of life is decreased after treatment for nonfunctioning pituitary macroadenoma. J Clin Endocrinol Metab. 2006;91(9):3364–3369. [PubMed: 16787991]
- 149.
- Biermasz NR, Joustra SD, Donga E, Pereira AM, van Duinen N, van Dijk M, van der Klaauw AA, Corssmit EP, Lammers GJ, van Kralingen KW, van Dijk JG, Romijn JA. Patients previously treated for nonfunctioning pituitary macroadenomas have disturbed sleep characteristics, circadian movement rhythm, and subjective sleep quality. J Clin Endocrinol Metab. 2011;96(5):1524–1532. [PubMed: 21367934]
- 150.
- Capatina C, Christodoulides C, Fernandez A, Cudlip S, Grossman AB, Wass JA, Karavitaki N. Current treatment protocols can offer a normal or near-normal quality of life in the majority of patients with non-functioning pituitary adenomas. Clin Endocrinol (Oxf). 2013;78(1):86–93. [PubMed: 22640418]
- 151.
- Joustra SD, Kruijssen E, Verstegen MJ, Pereira AM, Biermasz NR. Determinants of altered sleep-wake rhythmicity in patients treated for nonfunctioning pituitary macroadenomas. J Clin Endocrinol Metab. 2014;99(12):4497–4505. [PubMed: 25210880]
- 152.
- Ratnasingam J, Lenders N, Ong B, Boros S, Russell AW, Inder WJ, Ho KKY. Predictors for secondary therapy after surgical resection of nonfunctioning pituitary adenomas. Clin Endocrinol (Oxf). 2017;87(6):717–724. [PubMed: 28626928]
- 153.
- Noh TW, Jeong HJ, Lee MK, Kim TS, Kim SH, Lee EJ. Predicting recurrence of nonfunctioning pituitary adenomas. J Clin Endocrinol Metab. 2009;94(11):4406–4413. [PubMed: 19820025]
- 154.
- Hasanov R, Aydogan BI, Kiremitci S, Erden E, Gullu S. The Prognostic Roles of the Ki-67 Proliferation Index, P53 Expression, Mitotic Index, and Radiological Tumor Invasion in Pituitary Adenomas. Endocr Pathol. 2019;30(1):49–55. [PubMed: 30610566]
- 155.
- Trouillas J, Roy P, Sturm N, Dantony E, Cortet-Rudelli C, Viennet G, Bonneville JF, Assaker R, Auger C, Brue T, Cornelius A, Dufour H, Jouanneau E, Francois P, Galland F, Mougel F, Chapuis F, Villeneuve L, Maurage CA, Figarella-Branger D, Raverot G. members of H, Barlier A, Bernier M, Bonnet F, Borson-Chazot F, Brassier G, Caulet-Maugendre S, Chabre O, Chanson P, Cottier JF, Delemer B, Delgrange E, Di Tommaso L, Eimer S, Gaillard S, Jan M, Girard JJ, Lapras V, Loiseau H, Passagia JG, Patey M, Penfornis A, Poirier JY, Perrin G, Tabarin A. A new prognostic clinicopathological classification of pituitary adenomas: a multicentric case-control study of 410 patients with 8 years post-operative follow-up. Acta Neuropathol. 2013;126(1):123–135. [PubMed: 23400299]
- 156.
- Lelotte J, Mourin A, Fomekong E, Michotte A, Raftopoulos C, Maiter D. Both invasiveness and proliferation criteria predict recurrence of non-functioning pituitary macroadenomas after surgery: a retrospective analysis of a monocentric cohort of 120 patients. Eur J Endocrinol. 2018;178(3):237–246. [PubMed: 29259039]
- 157.
- Hallen T, Olsson DS, Hammarstrand C, Orndal C, Engvall A, Ragnarsson O, Skoglund T, Johannsson G. MCM7 as a marker of postsurgical progression in non-functioning pituitary adenomas. Eur J Endocrinol. 2021;184(4):521–531. [PubMed: 33524001]
- 158.
- Lyu W, Fei X, Chen C, Tang Y. Nomogram predictive model of post-operative recurrence in non-functioning pituitary adenoma. Gland Surg. 2021;10(2):807–815. [PMC free article: PMC7944052] [PubMed: 33708562]
- 159.
- Srirangam Nadhamuni V, Korbonits M. Novel Insights into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms. Endocr Rev. 2020;41(6) [PMC free article: PMC7441741] [PubMed: 32201880]
- 160.
- Neou M, Villa C, Armignacco R, Jouinot A, Raffin-Sanson ML, Septier A, Letourneur F, Diry S, Diedisheim M, Izac B, Gaspar C, Perlemoine K, Verjus V, Bernier M, Boulin A, Emile JF, Bertagna X, Jaffrezic F, Laloe D, Baussart B, Bertherat J, Gaillard S, Assie G. Pangenomic Classification of Pituitary Neuroendocrine Tumors. Cancer Cell. 2020;37(1):123–134 e125. [PubMed: 31883967]
- 161.
- Cooper O, Ben-Shlomo A, Bonert V, Bannykh S, Mirocha J, Melmed S. Silent corticogonadotroph adenomas: clinical and cellular characteristics and long-term outcomes. Horm Cancer. 2010;1(2):80–92. [PMC free article: PMC2921667] [PubMed: 20717480]
- 162.
- Lu JQ, Adam B, Jack AS, Lam A, Broad RW, Chik CL. Immune Cell Infiltrates in Pituitary Adenomas: More Macrophages in Larger Adenomas and More T Cells in Growth Hormone Adenomas. Endocr Pathol. 2015;26(3):263–272. [PubMed: 26187094]
- 163.
- Wang Z, Guo X, Gao L, Deng K, Lian W, Bao X, Feng M, Duan L, Zhu H, Xing B. The Immune Profile of Pituitary Adenomas and a Novel Immune Classification for Predicting Immunotherapy Responsiveness. J Clin Endocrinol Metab. 2020;105(9) [PMC free article: PMC7413599] [PubMed: 32652004]
- Review Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas.[Pituitary. 2018]Review Epidemiology, clinical presentation and diagnosis of non-functioning pituitary adenomas.Ntali G, Wass JA. Pituitary. 2018 Apr; 21(2):111-118.
- Review Management of non-functioning pituitary adenomas: surgery.[Pituitary. 2018]Review Management of non-functioning pituitary adenomas: surgery.Penn DL, Burke WT, Laws ER. Pituitary. 2018 Apr; 21(2):145-153.
- Review Non-functioning pituitary adenomas.[J Endocrinol Invest. 2005]Review Non-functioning pituitary adenomas.Chanson P, Brochier S. J Endocrinol Invest. 2005; 28(11 Suppl International):93-9.
- Review Non-functioning pituitary adenomas: indications for pituitary surgery and post-surgical management.[Pituitary. 2019]Review Non-functioning pituitary adenomas: indications for pituitary surgery and post-surgical management.Esposito D, Olsson DS, Ragnarsson O, Buchfelder M, Skoglund T, Johannsson G. Pituitary. 2019 Aug; 22(4):422-434.
- Review Diagnosis and treatment of pituitary adenomas.[Minerva Endocrinol. 2004]Review Diagnosis and treatment of pituitary adenomas.Chanson P, Salenave S. Minerva Endocrinol. 2004 Dec; 29(4):241-75.
- Non-Functioning Pituitary Adenomas - EndotextNon-Functioning Pituitary Adenomas - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...