

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
The process of growth is complex and is influenced by various factors that act centrally and peripherally. The genetic control of human growth is becoming increasingly clear. Many genes have been identified that contribute to the development and function of the pituitary gland including the somatotrope and the GH/IGF1 axis. Genes encoding “downstream” factors, including the insulin and the insulin receptor, the Short Stature Homeobox and SHP2 affect growth unrelated to growth hormone status, while Aggrecan has been described in cases of short stature with an advanced bone age, as well as in multiple forms of spondyloepiphyseal dysplasia. Defects in these genes have been shown to be responsible for abnormal growth in humans. In this chapter, we describe conditions associated with multiple pituitary hormone deficiency, isolated growth hormone deficiency, and abnormal growth without growth hormone deficiency, discuss the genes that are associated with these conditions, and prepare guidelines for the clinicians to evaluate and treat a child with poor growth. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Human growth starts at conception and proceeds through various identifiable developmental stages. The process of growth depends on both genetic and environmental factors that combine to determine an individual’s eventual height. The genetic control of statural growth is becoming increasingly clear. Many genes have been identified that are required for normal development and function of the pituitary in general, and that control the growth hormone/insulin-like growth factor axis in particular and many more that are involved in numerous cascades of intracellular processes “downstream” of GH/IGF1 action. Mutations of these genes have been shown to be responsible for abnormal growth in humans and animals.
Growth hormone (GH) has been used to treat short children since the 1950’s. Initially only those children with the most pronounced growth failure due to severe growth hormone deficiency (GHD) were considered appropriate candidates, but with time children with growth failure from a range of conditions have been shown to benefit from GH treatment. GH has also been used to treat several catabolic processes, including cystic fibrosis, inflammatory bowel disease, and AIDS wasting. Here we review the physiology of growth, the diagnosis of GH deficiency, treatment options and genetic growth hormone disorders.
GROWTH DISORDERS
Growth failure may be due to genetic mutations, acquired disease and/or environmental deficiencies. Growth failure may result from a failure of hypothalamic growth hormone-releasing hormone (GHRH) production or release, from (genetic or sporadic) mal-development of the pituitary somatotropes, secondary to ongoing chronic illness, malnutrition, intrinsic abnormalities of cartilage and/or bone such as osteochondrodysplasias, and from genetic disorders affecting growth hormone production and responsiveness. Children without any identifiable cause of their growth failure are commonly labeled as having idiopathic short stature (ISS).
Genetic factors affecting growth include pituitary transcription factors (PROP1, POU1F1, HESX1, LHX3, and LHX4), GHRH, the GH secretagogue (GHS), GH, insulin like growth factor-1 (IGF1), insulin like growth factor-2 (IGF2), insulin (INS) and their receptors (GHRHR, GHSR, GHR, IGF1R, IGF2R and INSR) as well as transcription factors controlling GH signaling, including STAT1, STAT3, STAT5a, and STAT5b. Growth is also influenced by other factors such as the Short Stature Homeobox, sex steroids (estrogens and androgens), glucocorticoids and thyroid hormone.
Since the replacement of human pituitary-derived GH with recombinant human GH, much experience has been gained with the use of GH therapy. The Food and Drug Administration (FDA) had expanded GH use for the following conditions for children (1):
- 1.
GH deficiency/insufficiency
- 2.
Chronic renal insufficiency (pretransplantation)
- 3.
Turner syndrome
- 4.
SHOX haploinsufficiency
- 5.
Short stature from Prader-Willi Syndrome (PWS)
- 6.
Children with a history of fetal growth restriction (SGA, IUGR) who have not caught up to a normal height range by age 2 years
- 7.
Children with idiopathic short stature (ISS): height > 2.25 SD below the mean in height and unlikely to catch up in height.
- 8.
Noonan Syndrome
- 9.
Short Bowel Syndrome
FDA approved conditions for GH treatment for adults:
- 1.
Adults with GH deficiency
- 2.
Adults with AIDS wasting
The efficacy of GH treatment has been investigated in children whose height has been compromised due to chronic illnesses such as Crohn’s disease, cystic fibrosis, glucocorticoid-induced suppression of growth in other disorders (asthma and juvenile idiopathic arthritis (JIA), also known as juvenile rheumatoid arthritis (JRA)), and adrenal steroid disorders such as congenital adrenal hyperplasia (CAH). Studies have shown both anabolic effects and improvement of growth velocity after GH treatment in children with glucocorticoid dependent Crohn’s disease (2-4). Improvement in linear growth has also been observed after GH treatment in children with cystic fibrosis and JIA (5-7). The same studies have shown significant improvement in weight gain and body composition, changes that have been variably correlated with improvement in life expectancy and quality of life.
The growth-suppressing effects of glucocorticoids, is also seen in children affected with CAH where high androgens both increase short-term growth velocity and limit the height potential. Most patients with CAH complete their growth prematurely and are ultimately short adults. Lin-Su et al, showed that GH in combination with LHRHa significantly improved their final adult height in children with CAH (8). Larger, long-term prospective studies are needed to determine the safety and efficacy of GH treatment in these populations of children.
The key mediator of GH action in the periphery for both prenatal and postnatal mammalian growth is the IGF system. GH exerts its direct effects at the growth plate and indirect effects via IGF1. Better understanding the role of IGF1 on growth had led to the concept of IGF1 deficiency in addition to GH deficiency. With the introduction of recombinant human (rh) IGF1, today, it is possible to treat conditions due to genetic GH resistance or insensitivity caused by GH receptor defects, and the presence of neutralizing GH antibodies(9).
MULTIPLE PITUITARY HORMONE DEFICIENCY (MPHD)
GH deficiency may occur in combination with other pituitary hormone deficiencies and is often referred to as hypopituitarism, panhypopituitarism or multiple pituitary hormone deficiency (MPHD).
The anterior portion of pituitary gland forms from Rathke's pouch around the third week of gestation (10). It is influenced by the expression of numerous transcription factors and signaling molecules; some of them required for continued normal function of pituitary gland. Mutations have been identified in the genes for several of these pituitary transcription factors and signaling molecules, including GLI2, LHX3, LHX4, HESX1, PROP1, POU1F1, SOX2, PITX2, OTX2 and SOX3 (Table 1). The most frequently mutated gene is PROP1, 6.7% in sporadic and 48.5% in familial cases (11).
The majority of cases of hypopituitarism are idiopathic in origin; however, familial inheritance, which may be either dominant or recessive, accounts for between 5 and 30% of all cases (12). It may present early in the neonatal period or later in childhood. It can be associated with single or multiple pituitary hormone deficiencies, and the endocrinopathy. It may be associated with a number of extrapituitary defects such as optic nerve hypoplasia, anophtalmia, microphtalmia, agenesis of the corpus callosum, and absence of the septum pellucidum.
Table 1.
Transcription Factors Required for Normal Pituitary Development
| Transcription Factors | Function |
|---|---|
| GLI2 | Essential for the forebrain and early stages of the anterior pituitary development |
| LHX3 | Essential for the early development of the anterior pituitary, including the somatotrope, thyrotrope, lactotrope and the gonadotrope (but not the corticotrope) |
| LHX4 | Essential for the proliferation of the anterior pituitary cell types, including the somatotrope, thyrotrope and the corticotrope |
| HESX1 | Essential for the development of the anterior pituitary, including the somatotrope, thyrotrope, lactotrope and the gonadotrope |
| PROP1 | Essential for the development of most cell types of the anterior pituitary, including the somatotrope, the thyrotrope, the lactotrope and the gonadotrope (but not the corticotrope). Also essential for the expression of the PIT1 protein and the extinction of HESX1 in the anterior pituitary |
| POU1F1 (PIT1) | Necessary for somatotrope, lactotrope and thyrotrope development and also for their continued function |
| SOX2 | Essential for the expression of POU1F1 and the development of gonadotrope |
| PITX2 | Necessary for the development of gonadotrope, somatotrope, lactotrope and thyrotrope |
| OTX2 | Transactivates HESX1 and POU1F1 |
| SOX3 | Essential for the early formation of hypothalamic-pituitary axis |
GLI2
GLI2 is a transcription factor molecule, mediating Sonic Hedgehog (SHH) signaling and is necessary for forebrain development as well as for the early stages of pituitary development (13). The clinical phenotype of persons with mutations in GLI2 may vary from asymptomatic individuals to isolated GH deficiency to hypopituitarism in combination with a small anterior pituitary, ectopic posterior pituitary, midfacial hypoplasia, holoprosencephaly, and polydactyly (14-16).

Figure 1.
GLI2
LHX3
LHX3 is a member of the LIM family of HomeoboX transcription factors. The gene LHX3, is located on 9q34.3, comprises 7 exons (including two alternative exon 1's, 1a and 1b) and encodes a protein of 402 amino acids (Figure 2). LHX3 is expressed in the developing Rathke's pouch and is required for the development of most anterior pituitary cell types, including the somatotrope, the thyrotrope, the lactotrope, and the gonadotrope (but notably not the corticotrope). LHX3 binds as a dimer, synergizing with POU1F1 (PIT1). Two unrelated families with MPHD were identified in 2000(17) as harboring mutations in LHX3. The affected members of the family manifested severe growth retardation in association with restricted rotation of the cervical spine. Inheritance is consistent with an autosomal recessive pattern of inheritance and of note one individual was found to have an enlarged pituitary. Recently, a new mutation in LHX3 was described in a child with hypointense pituitary lesion, focal amyotrophy and mental retardation in addition to neck rigidity and growth retardation (18). These clinical findings expand the phenotype associated with mutations in LHX3.

Figure 2.
LHX3
LHX4
LHX4 also has a critical role in the development of anterior pituitary cells, and is co-expressed with LHX3 in Rathke's pouch in an overlapping but not wholly redundant pattern. Raetzman et al showed overlapping functions with PROP1 in early pituitary development, but also observed that their mechanisms of action were not identical. LHX4 is necessary for cell survival and LHX3 expression, with the pituitary hypoplasia seen in LHX4 mutants actually results from increased cell death and reduced differentiation, directly attributable to loss of LHX3; while PROP1 mutants exhibit normal cell proliferation and cell survival but show evidence of defective dorsal-ventral patterning (19). In the absence of both of these genes, no specification of corticotropes, gonadotropes or thyrotropes occurs in the anterior lobe. Although both LHX3 and LHX4 are crucial for the development of pituitary gland, LHX3 is expressed at all stages studied, whereas LHX4 expression is transient at 6 weeks of development (20). LHX4 is located on 1q25 and comprises 6 exons spread over a 45 kb genomic region (Figure 3). An intronic splice site mutation has been described in one family, manifesting GH, TSH and ACTH deficiency, along with cerebellar and skull defects. The mutation is transmitted as an autosomal dominant condition, with complete penetrance. Interestingly, a heterozygous mutant mouse model had no discernable phenotype, while homozygous loss of function in the mouse was fatal (21).

Figure 3.
LHX4
HESX1
HESX1 (HomeoboX gene expressed in Embryonic Stem cells), also referred to as RPX1 (Rathke's Pouch HomeoboX), is necessary for the development of the anterior pituitary. RPX1 comprises 4 exons and encodes a protein of 185 amino acids that features both a homeodomain as well as a repressor domain and is located on chromosome 3p21.2 (Figure 4). The extinguishing of HESX1 requires the appearance of another pituitary transcription factor, PROP1. A mutation has been described in two children of a consanguineous union who had optic nerve hypoplasia, agenesis of the corpus callosum and panhypopituitarism, with an apparent autosomal recessive mode of inheritance (22). This Arg → Cys mutation lies between the repressor and homeodomains but the mutant protein was shown in vitro to be unable to bind to its cognate sequences. A novel homozygous missense mutation (126T) of the critical engrailed homology repressor domain (eh1) of HESX1 was described in a girl born to consanguineous parents (23). Neuroimaging revealed a thin pituitary stalk with anterior pituitary hypoplasia and an ectopic posterior pituitary. Unlike previous cases, she did not have midline or optic nerve abnormalities. Although 126T mutation did not affect the DNA-binding ability of HESX1, it impaired ability of HESX1 to recruit Groucho-related corepressor, thereby leading to partial loss of repression. It appears that HESX1 mutations exhibit variety of clinical phenotypes with no clear genotype-phenotype correlation. Tantalizingly, additional nucleotide variants have been described in individuals with isolated GH deficiency, although it is not convincingly clear that these polymorphisms are actually pathogenic (24,25).

Figure 4.
HESX1
PROP1
PROP1 (the Prophet of PIT-1) encodes a transcription factor required for the development of most pituitary cell lines, including the somatotrope (GH secretion), lactotrope (prolactin (PRL) secretion), thyrotrope (TSH secretion), and the gonadotrope (FSH and LH secretion). Mutation of PROP1, therefore, results in the deficiency of GH, TSH, PRL, FSH and LH although some individuals with PROP1 mutations have been described with ACTH deficiency (26). Since PROP1 does not appear to be required for the development of the corticotrope cell line, the etiology of ACTH deficiency is unclear. It appears that the ACTH deficiency here is a consequence of the compensatory pituitary hyperplasia that develops over time. Significantly, the degree of TSH deficiency appears to be quite variable, even within mutation-identical individuals, suggesting that the general phenotype associated with PROP1 mutations is also quite variable. PROP1 is encoded by three exons and is located on 5q. Many mutations have been described in PROP1-all inherited in an autosomal recessive manner. Although several studies suggest that mutation of PROP1 is the most common cause of familial MPHD, but is less common in sporadic cases of MPHD (11,27). Two recurrent mutations have been described, both involving exonic runs of GA tandem repeats (Figure 5). In both cases, the loss of a tandem unit at either locus results in a frameshift and premature termination, and a protein incapable of transactivation.

Figure 5.
PROP1
POU1F1
POU1F1 encodes the POU1F1 transcription factor, also known as PIT1, which is required for the development and function of three major cell lines of anterior pituitary: somatotropes, lactotropes and thyrotropes. Various mutations in the gene encoding POU1F1 have been described, resulting in a syndrome of multiple pituitary hormone deficiency involving GH, PRL and TSH hormones. POU1F1 is located on 3p11 and consists of six exons encoding 291 amino acids (Figure 6). Many mutations of POU1F1 have been described; some are inherited as autosomal recessive and some as autosomal dominant. There is a wide variety of clinical presentation in patients with POUF1 mutations. Generally, GH and prolactin deficiencies are seen early in life. However, TSH deficiency can be highly variable with presentation later in childhood or normal T4 secretion can be preserved into the 3rd decade (28,29). To date, POU1F1 mutations have been described in a total of 46 patients from 34 families originating in 17 different countries (30). Recessive mutations are generally associated with decreased activation, while dominant mutations have been shown to bind but not transactivate - i.e. act as dominant-negative mutations, rather than through haploinsufficiency. One such mutation is the recurrent Arg271Trp (R271W), located in exon 6, which results from a C T transition at a CpG dinucleotide, i.e. a region predisposed to spontaneous mutagenesis. Another interesting mutation is the Lys216Glu mutation of exon 5. This mutation is unique in that the mutant transcription factor activates both the GH and PRL promoters at levels greater than wild-type (i.e. acts as a superagonist), but down-regulates its own (i.e. the POU1F1) promoter-leading to decreased expression of PIT1. R271W is the most frequent mutation of POU1F1. A recent report describing a novel mutational hot spot (E230K) in Maltese patients suggests a founder effect (29). The same group reported two additional novel mutations within POU1F1 gene; an insertion of a single base pair (ins778A) and a missense mutation (R172Q)(27).

Figure 6.
POU1F1
SOX3
SOX3 encodes a single-exon gene SOX3, an HMG box protein, located on the X chromosome (Xq26.3) in all mammals (31). It is believed to be the gene from which SRY, testis–determining gene evolved (32). Based on sequence homology, SOX, however, is more closely related to SOX1 and SOX2, together comprising the SOXB1 subfamily and are expressed throughout the developing CNS (33,34). In humans, mutations in the SOX3 gene have been implicated in X-linked hypopituitarism and mental retardation. In a single family, a SOX3 gene mutation was shown in affected males who had mental retardation and short stature due to GH deficiency (35). The mutation was an in-frame duplication of 33 bp encoding for an additional 11-alanine, causing an expansion of a polyalanine tract within SOX3. Recently, other mutations including a submicroscopic duplication of Xq27.1 containing SOX3, a novel 7-alanine expansion within the polyalanine tract, and a novel polymorphism (A43T) in the SOX3 gene were described in males with hypopituitarism. Phenotypes of these patients include severe short stature, anterior pituitary hypoplasia, and ectopic posterior pituitary, colossal abnormalities, and infundibular hypoplasia. Although duplications of SOX3 have been implicated in the etiology of X-linked hypopituitarism with mental retardation, in at least one study, none of the affected individuals had mental retardation or learning difficulties (36). Taken together, the data suggests that SOX3 has a critical role in the development of the hypothalamic-pituitary axis in humans, and mutations in SOX3 gene are associated with X-linked hypopituitarism but not necessarily mental retardation(36).
ISOLATED GH DEFICIENCY (IGHD)
Abnormalities either in the synthesis or the activity of GH can cause a wide variation in the clinical phenotype of the patient. Most frequently, it occurs as a sporadic condition of unknown etiology but severe forms of IGHD may result from mutations or deletions in GH1 or GHRHR gene. General clinical features of IGHD deficiency include proportionate growth retardation accompanied by a decreased growth velocity, puppet-like facies, mid-facial hypoplasia, frontal bossing, thin hair, a high-pitched voice, microphallus, moderate trunk obesity, acromicria, delayed bone maturation and dentition. Patients with IGHD appear younger than their chronological age. Puberty may be delayed until late teens, but usually fertility is preserved.
To date, four Mendelian patterns of inheritance for IGHD have been identified on the basis of the type of defect, mode of inheritance, and degree of deficiency.
- 1.
Type 1 GH deficiency is an autosomal recessively inherited condition, which exists as either complete, or partial loss of GH expression.
- a.
Type 1a deficiency is characterized by the complete absence of measurable GH. Infants born with a type 1a defect are generally of normal length and weight, suggesting that, in utero, GH is not an essential growth factor (37,38). Growth immediately after birth and during infancy may also be less dependent on circulating GH levels than during other phases of life. Patients with Type 1a deficiency initially respond to rhGH treatment well. However, about 1/3 of patients develop antibodies to GH which leads to markedly decreased final height as adults (30). The exact prevalence of Type 1a deficiency is not known, and most reported families are consanguineous (30). Mutations in Type 1a have been described in GH1 and GHRHR-including nonsense mutations, microdeletions/frame-shifts, and missense mutations.
- b.
Type 1b deficiency represents a state of partial - rather than an absolute - deficiency of GH, with measurable (but insufficient) serum GH. Therefore, Type 1b is milder than Type 1a deficiency. Patients with Type 1b deficiency do not typically present with mid-facial hypoplasia or microphallus. They also have a good response to GH treatment without developing GH antibodies. Most cases of Type 1b GH deficiency are caused by missense and/or splice site mutations in the GH1 and GHRHR genes (39).
- 2.
Type 2 GH deficiency is an autosomal dominantly inherited disorder with reduced secretion of GH. Patients with Type 2 GHD usually do not have any pituitary abnormality (40). However, recently, it has been shown that their pituitary may become small over time (41). They have a good response to GH treatment. This type of GH deficiency is intuitively less clear, since autosomal dominant conditions generally occur as a result of either haploinsufficiency or secondary to dominant-negative activity. Haploinsufficiency, however, has not been demonstrated in the obligate heterozygote carriers of individuals harboring GH1 deletions, and is therefore an unlikely explanation. Dominant-negative activity is usually associated with multimeric proteins, also making this explanation less intuitive. Type 2 GHD appears to be the most common form of IGHD and many mutations have been identified in GH1 including splicing and missense mutations(42-49). Recent studies suggest that GH1 may not be the only gene involved in Type 2 GHD. Screening 30 families with autosomal dominant IGHD did not show any GH1 mutations, raising the possibility of other gene(s) may be involved (50).
- 3.
Type 3 growth hormone deficiency is inherited in an X-linked recessive manner. There are no candidate genes and no compelling explanations for this condition. There are no reported mutations of the GH-1 gene in Type 3 GHD. In addition to short stature, patients may also have agammaglobulinemia (30).
Table 2 summarizes phenotype of mutations involved in human pituitary transcription factors causing IGHD and MPHD and their mode of inheritance.
Table 2.
Genotype and Phenotype Correlations in Human Pituitary Transcription Factors
| Gene | Phenotype | Mode of Inheritance |
|---|---|---|
| IGHD | ||
| GH-1 | IGHD type 1a/1b IGHD type 2 | AR AD |
| GHRHR | IGHD type 1b | AR |
| MPHD | ||
| LHX3 | Deficiencies of GH, TSH, LH, FSH, PRL, rigid neck, small/normal/or enlarged anterior pituitary | AR |
| LHX4 | Deficiencies of GH, TSH and ACTH, small anterior pituitary, cerebellar and skull defects | AD |
| HESX1 | Hypopituitarism, optic nerve hypoplasia, agenesis of the corpus callosum, ectopic posterior pituitary | AR/AD |
| PROP1 | Hypopituitarism except ACTH deficiency, small/normal/or enlarged anterior pituitary | AR |
| POU1F1 (PIT1) | Deficiencies of GH, TSH, PRL, small or normal anterior pituitary | AR/AD |
| SOX3 | Hypopituitarism, mental retardation, learning difficulties, small anterior pituitary, ectopic posterior pituitary | X-linked recessive |
| OTX2 | Hypopituitarism, microphtalmia | AD |
| GLI2 | Hypopituitarism, small anterior pituitary, ectopic posterior pituitary, holoprosencephaly, polydactily | AD |
AR: Autosomal Recessive; AD: Autosomal Dominant.
HYPOTHALAMIC GH DEFICIENCY
Synthesis and Secretion of GH
GH is synthesized within the somatotropes of the anterior pituitary gland and is secreted into circulation in a pulsatile fashion under tripartite control, stimulated by growth hormone releasing hormone (GHRH), the Growth Hormone Secretagogue (GHS), and Ghrelin and inhibited by somatostatin (SST) (Figure 7). GHRH, GHS and SST secretion are themselves regulated by numerous central nervous system neurotransmitters (Table 3). GH, via a complex signal transduction, exerts direct metabolic effects on target tissues and exerts many of its growth effects through releasing of IGF1 which is mainly produced by the liver and the target tissues (e.g. growth plates). Additional regulation of GH secretion is achieved through feedback control by IGF1 and GH at the pituitary and at the hypothalamus.
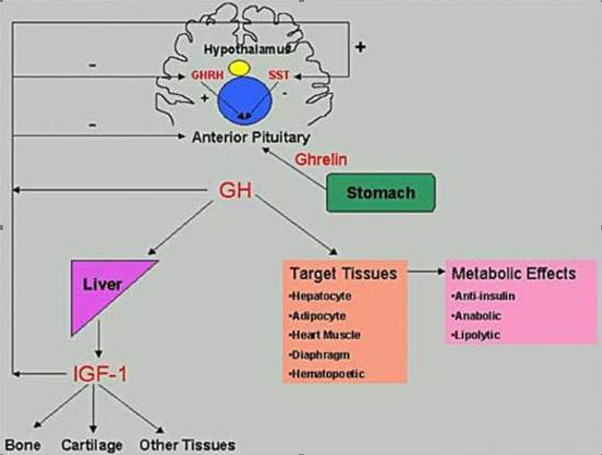
Figure 7.
Hypothalamic-pituitary-peripheral regulation of GH Secretion. SST, somatostatin; GHRH, growth hormone releasing hormone; IGF1, insulin-like growth factor type 1
Table 3.
Neurotransmitters and Neuropeptides Regulating GHRH Secretion from Hypothalamus.
| Dopamine | Gastrin |
| GABA | Neurotensin |
| Substance-P | Calcitonin |
| TRH | Neuropeptide-Y |
| Acetylcholine | Vasopressin |
| VIP | CRHs |
Timing
In addition to the absolute GH levels reached, the timing of the GH pulse is also physiologically important. GH is secreted in episodic pulses throughout the day, and the basal levels of GH are often immeasurably low between these peaks (Figure 8). Figure 8 illustrates normal spontaneous daily GH secretion, while figure 9 represents that of a child with GH deficiency.

Figure 8.
The characteristic pulsatile pattern of GH secretion in normal children. Note the maximal GH secretion during the night.

Figure 9.
GH secretion in a child with GH deficiency. Note the loss (both qualitative and quantitative) of episodic pulses seen in normal children
Approximately 67% or more of the daily production of GH in children and young adults occurs overnight, and most of that during the early nighttime hours that follow the onset of deep sleep. During puberty, there is an increase in GH pulse amplitude and duration, most likely due to estrogens (51). GH secretion is sexually dimorphic, with females having higher secretory burst mass per peak but no difference in the frequency of peaks, or basal GH release (52). In addition, GH secretion is stimulated by multiple physiologic factors (Table 4). Overweight children, independent of pubertal status, have reduced GH levels mainly due to reduced GH burst mass with no change in frequency (53).
Table 4.
Physiologic Factors That Affect GH Secretion
| Factors that stimulate GH secretion | Factors that suppress GH secretion |
|---|---|
| Exercise | Hypothyroidism |
| Stress | Obesity |
| Hypoglycemia | Hyperglycemia |
| Fasting | High carbohydrate meals |
| High protein meals | Excess glucocorticoids |
| Sleep |
Growth Hormone Releasing Hormone
GHRH (also known as Somatocrinin) is the hypothalamic-releasing hormone isolated in 1982 (54) believed to be the chief mediator of GH secretion from the somatotrope. GHRH deficiency is thought to be the most common cause of 'acquired' GHRH deficiency, secondary to (even mild) birth trauma. GHRH includes 5 exons, with transcription of (the non-coding) exon 1 differing on a tissue-specific basis (55). The mature GHRH protein contains 44 amino acids, with an amidated carboxy-terminus (Figure 10). Despite this post-translational modification, much of the GH-secreting ability resides in the (original) amino half, allowing the synthesis of shorter peptides retaining efficacy (e.g. 1-29 GHRH). Despite being cloned in 1985 (56), there are no reports of (spontaneous) mutations in humans or in any animal model. Individuals with mutations in GHRH are predicted to have isolated GH deficiency.

Figure 10.
Growth Hormone Releasing Hormone
Growth Hormone Releasing Hormone Receptor
GHRHR was cloned in 1992(57), described as the cause of isolated GH deficiency (IGHD) in the Little strain of dwarf mouse by 1993 (58,59), mapped in the human by 1994 (60), and demonstrated to be a cause of human GH deficiency in 1996 (39). GHRHR is located on 7p15 (60), comprises 13 exons and encodes a protein of 423 amino acids, belonging to the G-protein coupled, heptahelical transmembrane domain receptors (Figure 11). The initial reports of GHRHR mutations were in geographically isolated (and therefore endogamous) populations in South Asia (39,61,62) and later in Brazil (63). In fact, haplotype analysis of the GHRHR locus in three unrelated families from the Indian subcontinent, carrying the identical E72X nonsense mutation in GHRHR indicated that this represents a common ancient founder mutation (64). An independent analysis of patients with familial isolated GH deficiency from non-consanguineous families revealed that the majority of patients carried the identical E72X mutation, suggesting that E72X mutation can be a reasonable candidate for isolated GH deficiency (65). There are now numerous other reports, making GHRHR one of the most commonly mutated genes in IGHD. Roelfsema et al studied two members of a single family with an inactivating mutation of the GHRHR and noted that the 'normal' pattern of spontaneous GH production was preserved, although the absolute quantity of GH secreted was quite low and the approximate entropy significantly elevated (66); supporting the view that the amplitude of a GH pulse is the result of a GHRH burst, while the timing of GH pulses is the result of a somatostatin trough.

Figure 11.
Growth Hormone Releasing Hormone Receptor
Ghrelin
In 1977 Bowers et al (67) reported on the ability of enkephalins to secrete GH and it was later demonstrated that this secretion was independent of GHRH. This sentinel finding gave rise to a new field of study, that of the growth hormone releasing peptides (GHRP's) or growth hormone secretagogues (GHS's). Twenty-two years later Kojima et al (68) reported the isolation of the endogenous ligand whose actions were mimicked by the enkephalins. They named the hormone Ghrelin, based on the Proto-Indian word for 'grow'. Ghrelin is located on 3p25-26(69) (Figure 12), is processed from a ‘preproGhrelin’ precursor, and is primarily produced by the oxyntic cells of the stomach and to a lesser extent in the arcuate ventro-medial and infundibular nuclei of the hypothalamus (70). Ghrelin also plays a role in regulating food intake. In addition to its GH secreting actions, direct intracerebroventricular injection of Ghrelin in mice has potent orexigenic “appetite stimulating” action, and this action is mediated by NPY, which antagonizes the actions of Leptin.
Several studies have shown that, on a molar basis, Ghrelin is significantly more potent at inducing GH secretion than GHRH (71). Additionally, many of these studies have shown that Ghrelin and GHRH are synergistic, inducing a substantial GH response when given in combination (72-75). Several studies comparing GHRH and Ghrelin demonstrate that 1 ug/kg GHRH results in a GH peak of approximately 25 ng/ml, 1 ug/kg Ghrelin results in a GH peak of approximately 80 ng/ml GH, but when given together, 1ug/kg of GHRH + 1 ug/kg Ghrelin results in a GH peak of approximately 120 ng/ml (75,76). When short normal children were compared to children with neurosecretory GH deficiency, Ghrelin secretion was similar in both groups during daytime but higher Ghrelin levels were detected during the night in short children with neurosecretory GH deficiency. The authors therefore suggest that Ghrelin is not involved in nighttime GH secretion (77), although these findings are also consistent with a relative Ghrelin insensitivity at night. In a group of boys with constitutional delay of puberty, testosterone administration caused the expected increase in GH concentrations but did not affect the 24-hour Ghrelin profile, suggesting that the testosterone-induced GH secretion was not mediated by ghrelin (78). Another study demonstrated a decrease in Ghrelin concentrations following glucagon administration in a group of non-GH-deficient short children, suggesting that Ghrelin does not mediate glucagon-induced GH secretion (79).
A second hormone, Obestatin is also known to be produced from preproGhrelin. Obestatin has anorexigenic effects, opposite those of Ghrelin (80). Several nucleotide changes have been identified in the preproGhrelin locus, and some are associated with body mass index, BMI (81). It is not clear, however, whether these are polymorphisms, or distinct mutations. It is also not clear whether these nucleotide variants exert their effects solely via an altered Ghrelin, a corrupted Obestatin, or a combination of the two. A knockout mouse lacking the preproghrelin locus had no statural or weight phenotype, but this may well be the result of the simultaneous loss of both ghrelin and obestatin. To this point, a transgenic mouse with abnormal ghrelin but normal obestatin did indeed have poor weight gain, explained by either ghrelin deficiency, unopposed obestatin, or both. There are no reports of (spontaneous) mutations in Ghrelin associated with short stature, either in humans or in any animal model, although polymorphisms have been associated with weight/metabolic syndrome. The theoretical phenotype of such an individual would presumably be that of isolated GH deficiency, most likely of post-natal onset and possibly with an abnormally low appetite.

Figure 12.
Ghrelin
Ghrelin Receptor
The receptor for Ghrelin (GHSR) was identified in 1996 by Howard et al (70), prior to the identification of the ligand, and maps to 3q26-27 (Figure 13). Mutations of the GHSR gene have been reported in individuals with isolated GH deficiency (82).
Combining data from numerous investigators, there appear to be differences in the specific roles of these parallel but independent pathways for GH secretion. Given that:
- 1.
Ghrelin induces a larger release of GH than GHRH,
- 2.
Both bolus and continuous GHRH infusion results in a chronic release of GH(83),
- 3.
A bolus of Ghrelin results in GH secretion, but continuous Ghrelin infusion does not; and
- 4.
Ghrelin administration (bolus or continuous) does not cause an increase in GH mRNA;
It is therefore likely that the GHRH/GHRHR arm of the somatotropin pathway serves primarily in the production of de novo GH, and secondarily in the release of (pre-made) GH while Ghrelin/GHSR may serve primarily in the release of stored GH, and only secondarily-if at all-in the production of de novo GH (76,84).

Figure 13.
Ghrelin Receptor
Somatostatin
The somatostatin gene (SST) is located on 3q28, and contains two exons, encoding a 116 amino acid pre-prosomatostatin molecule that is refined down to a 14 amino acid cyclic peptide (as well as a 28 amino acid precursor/isoform)(85) (see figure 14). Pancreatic somatostatin inhibits the release of both insulin and glucagon, while in the CNS somatostatin inhibits the actions of several hypothalamic hormones, including GHRH. For this reason, somatostatin is also known as Growth Hormone Release Inhibiting Hormone. Somatostatin's widespread effects are mediated by five different receptors, all encoded by different genes (rather than through alternative splicing of a single gene). The anti-GHRH actions on the pituitary are primarily mediated by somatostatin receptors (SSTR) 2 and 5, which act by inhibiting cAMP as well as other pathways (86) (see figures 15 and 16). There is a single case report of a nucleotide variant in SSTR5, occurring in a subject with acromegaly. (This individual, however, was also reported as having a mutation in the GSP oncogene; placing the pathological nature of the SSTR5 variant in question). Whereas GHRH induces release of growth hormone stored in secretory vesicles by depolarization of the somatotrope, somatostatin inhibits GH release by hyperpolarizing the somatotrope, rendering it unresponsive to GHRH. There are no reports of mutations in the somatostatin gene, or in SSTR2.
All three of these hypothalamic modifiers of GH secretion act through cell-membrane receptors of the G-protein coupled receptor (GCPR) class. These receptors are characterized by seven membrane-spanning helical domains, an extracellular region that binds (but does not internalize) the ligand hormone, and an intracellular domain that interacts with a G-protein, which contains a catalytic subunit that generates a second messenger (e.g. cyclic AMP or inositol triphosphate).

Figure 14.
Somatostatin

Figure 15.
Somatostatin Receptor 2

Figure 16.
Somatostatin Receptor 5
PITUITARY GH DEFICIENCY
Human Growth Hormone
GH is critical for growth through (most of) childhood as well as for optimal metabolic, neurocognitive, cardiac, musculoskeletal and adipose function throughout life. GH acts through GH receptors on cells of a variety of target tissues. Many, but not all, actions of GH are mediated by insulin-like growth factor 1 (IGF1), also known as Somatomedin-C. IGF1 is released in response to GH and acts as both a hormone and an autocrine/paracrine factor. GH, directly and indirectly through the actions of IGF1, stimulates tissue growth and proliferation, most notably in the epiphyseal growth plates of children, increases lean muscle mass, decreases fat mass, and increases bone mineral density.
Growth hormone is a single-chain polypeptide that contains 191 amino acids with two intramolecular disulfide bonds and the molecular weight of 22,128 Daltons. The GH protein (GHN) is encoded by the GH1 gene located on chromosome 17q22-q24 (Figure 17) in a complex of five genes: two for the growth hormone/growth hormone variant (GH1, GH2), two for chorionic somatomammotropin (CS1, CS2), and one for the somatomammotropin pseudogene (CSL). GH2 encodes the GHV protein that is secreted by the placenta into maternal circulation. GHV has greater lactogenic properties than does GHN and may function to maintain the maternal blood sugar in a desirable range, thus ensuring sufficient nutrition for the fetus.

Figure 17.
Growth Hormone
PERIPHERAL GH RESISTANCE
Growth Hormone Receptor
Growth failure with normal serum GH levels is well known, both at the genetic and the clinical level. Although such cases may be due to defects of GH1 (e.g. bioinactive GH), many such subjects have been shown to have mutations in the GH Receptor (GHR), i.e. Growth Hormone Insensitivity, known as Laron Syndrome. Biochemical hallmarks of this syndrome are increased or normal GH levels with low IGF1 and with absent or decreased response to GH treatment (87).
The growth hormone receptor gene (GHR) is located on 5p13-12 and contains 10 exons which span a physical distance of almost 300 kb of genomic DNA (Figure 18). The GHR consists of a ligand-binding extracellular domain, an 'anchoring' transmembrane domain and an intracellular domain with intrinsic tyrosine kinase activity. A monomeric GHR binds a single GH molecule, which then dimerizes a second GHR, and activates the JAK/STAT and MAPK pathways and is internalized. The internalization leads to extinguishing of the GH signal, and the GHR is recycled for further rounds of activity. Two naturally occurring isoforms of the GHR arise from alternative splicing-one with an alternate exon 3, and the other with an alternate exon 9. The alternative exon 9 isoform yields a protein with only amino acids 1-279, and virtually none of the intracellular domain. This isoform cannot transduce the GH signal and yields higher molar quantities of GHBP (than wild-type GHR), and therefore acts as a GH "sink" (88). The GHR isoform lacking exon 3 has a high prevalence, and may be associated with altered GH signaling, although the direction of the alteration is not clear(89-92).

Figure 18.
Growth Hormone Receptor
Defects in the GH signaling pathway have been demonstrated to be associated with postnatal growth failure. Mutations of Stat5b were reported in patients with severe growth failure. Several mutations of Stat5b gene have been reported. Although patients had a phenotype similar to that of congenital GH deficiency or GHR dysfunction, clinical and biochemical features (including normal serum GHBP concentrations) and immune deficiency(93) distinguish patients with STAT5b defects from patients with GHR defects. It also appears that STAT5b mutations are associated with hyperprolactinemia. It remains unclear whether the hyperprolactinemia is a direct or indirect effect of STAT5b mutations (94).
In humans, the extracellular portion of GHR is enzymatically cleaved and functions as the GH-Binding Protein (GHBP) (95). GHBP presumably serves to maintain GH in an inactive form in the circulation and to prolong the half-life of GH. Serum levels of GHBP are therefore used as a surrogate marker for the presence of GHR, and abnormal levels-both elevated and decreased-may indicate abnormality in the GHR (96,97). Of note is that mutations have also been described in individuals with 'normal' GHBP levels. GHI secondary to GHR mutations are mostly autosomal recessive mutations, but dominant negative mutations have also been described. Individuals with heterozygote mutations in GHR may present with significant short stature (98). Mutations in GHR have also been associated with idiopathic short stature (ISS) (99-101). The original reports of GHR mutation described limited elbow extension and blue sclera, but these findings are not universal.
Many genetic abnormalities have been described in GHR, including nonsense mutations, missense mutations, macrodeletions, microdeletions and splice site changes. Of the latter, one of the most interesting is the "E180E" mutation, wherein an exonic adenosine is converted to a guanine, converting GAA to GAG, which would be predicted to not change the amino acid structure of GHR (both GAA and GAG encode glutamic acid). On this basis, this "silent polymorphism" would be expected to have no phenotype, but in reality, causes GH resistance and extreme short stature by activation of a cryptic splice site. This mutation was noted in Loja and El Oro, Ecuador in two large cohorts. This identical mutation has also been identified in Jews of Moroccan descent, suggesting that this mutation dates back to at least the 1400’s and that the Ecuadorian cohorts, therefore, represent Sephardic Jews who left Spain around the time of the Inquisition at the end of the fifteenth century, CE (102). Another splice site mutation at position 785-3 (C>A in the intron 7) was recently described in a patient and mother with short stature and extremely elevated GHBP (103). The consequence of this novel mutation is a truncated GHR which lacks the transmembrane domain (encoded by exon 8) and the cytoplasmic domain. It was hypothesized by the authors that this GHR variant cannot attach to the cell membrane, and the continual secretion into the circulation results in the elevated levels of serum GHBP detected in the patient and his mother. The presence of the wild-type GHR allele presumably permits some level of normal GH-induced action.
Insulin-Like Growth Factor 1 (IGF1)
Many of growth hormone's physiologic actions are mediated through the insulin-like growth factor, IGF1 (formerly referred to as somatomedin C). Serum IGF1 levels are commonly measured as a surrogate marker of GH status, since IGF1 displays minimal circadian fluctuation in serum concentration. IGF1 plays a critical role in both prenatal and postnatal growth, signaling through the IGF1 as well as the insulin receptor. IGF1 circulates as a ternary complex consisting of IGF1, IGBP3 and ALS. The IGF1 gene is located on 12q22-24.1, consists of six exons and spans over 45 kb of genomic DNA (Figure 19). Alternative splicing produces two distinct IGF1 transcripts, IGF1-A and IGF1-B. Woods et al described a male of a consanguineous union with prenatal (intrauterine) and postnatal growth retardation, sensorineural deafness and mental retardation (104). DNA analysis showed a homozygous partial deletion of the IGF1 gene (104) (131). Subsequently, additional cases have been described (105,106).

Figure 19.
Insulin-Like Growth Factor 1
Mice engineered to completely lack Igf1 (Igf1 knockouts) are born 40% smaller than their normal littermates (107,108). Recent studies of a hepatic-only Igf1 knockout (KO) mouse, however, demonstrate that IGF1 functions primarily in a paracrine or autocrine role, rather than in an endocrine role (109). Liver specific Igf1 knock-out mice, were found to have a 75% reduction in serum Igf1 levels but were able to grow and develop (nearly) normally (109,110) with a mild phenotype developing only late in life (109). A further decrease in serum IGF1 levels of 85% was observed when double gene KO mice were generated lacking both the acid labile subunit (ALS) and hepatic IGF1. Unlike the single hepatic-only IGF1-KO's, these mice showed significant reduction in linear growth as well as 10% decrease in bone mineral density (111). Thus, as illustrated by the combination liver specific IGF1+ALS knock-out mouse model, there likely exists a threshold concentration of circulating IGF1 that is necessary for normal bone growth and suggests that IGF1, IGFBP3, and ALS may play an important role in bone physiology and the pathophysiology of osteoporosis.
In humans, homozygous mutations in ALS result in mild postnatal growth retardation, insulin resistance, pubertal delay, unresponsiveness to GH stimulation tests, elevated basal GH levels, low IGF1 and IGFBP3 levels and undetectable ALS (112-114). Although it is not clear why postnatal growth is mildly affected, it might be due to increased GH secretion due to loss of negative feedback regulation by the low circulating IGF1. Increased GH secretion could then up-regulate the functional GH receptor increasing local IGF1 production, thus protecting linear growth(93) (Figure 20). Over a dozen inactivating mutations of the IGFALS gene have been described in 21 patients with ALS deficiency (115).

Figure 20.
Savage MO Camacho-Hubner C, David A, et al. 2007” Idiopathic short stature: will genetics influence the choice between GH and IGF1 therapy?” Eu J of Endocrine 157:S33 Society of European Journal of Endocrinology (2007). Reproduced by permission. Reprinted with permission(116).
Elevated IGF1 levels has recently been associated with colon, prostate and breast cancer (117-119) and the association was strongest when an elevated IGF1 was combined with a decreased IGFBP3 level. This combination-expected to yield more bioactive IGF1-may merely reflect the tumorigenic process, rather than demonstrate causality. Importantly, GH treatment induces a rise in both IGF1 as well as IGFBP3 (120), and therefore would not be expected to increase cancer risk in normal individuals.
Table 5.
Summary of IGF1 Function in Different Systems and its Effects (121)
| IGF1 Function | IGF1 Deficiency |
| Intrauterine Growth | IUGR |
| Postnatal Growth | Short Stature |
| CNS | Neurodegenerative disease |
| Insulin sensitization/improvement of glucose disposal/beta cell proliferation | Type 1 and Type 2 Diabetes |
| IGF1 Excess | |
| Mitosis/Inhibition of apoptosis | Malignancy |
IGF1 Deficiency
IGF1 deficiency can be classified based on decreased IGF1 synthesis (primary) or decreased IGF1 secondary to decreased or inactive GH (secondary) (122) (see Table 6).
Table 6.
IGF1 Deficiency
| Primary IGF1 Deficiency (normal or elevated GH levels) Defects in IGF1 Production: Mutation in IGF1 gene or bioinactive IGF1 GHR receptor signaling defects (JAK/STAT) Mutations in ALS gene Factors effecting IGF1 production (malnutrition, liver, inflammatory bowel disorders, celiac disease) Defects in IGF1 Action: IGF1 resistance due to receptor or post-receptor defects Factors inhibiting IGF1 binding to IGF1R (increased IGFBPs and presence of IGF1 antibodies) Defects in GH Action: Factors inhibiting (increased GHBPs and presence of GH antibodies) GH receptor defects (decreased GH receptors, GHR antibodies, GHR gene defects) |
| Secondary IGF1 Deficiency (decreased GH levels) Decreased GH production Defects in GH gene Defects in GHRH or GHRH receptor Neocortical/psychological Defects in hypothalamus and pituitary |
Recombinant Human IGF1 (rhIGF1)
rhIGF1 is useful in the treatment of primary IGF1 deficiency resulting from abnormalities of the GH molecule (resulting in a bioinactive GH), the GH receptor (known as Laron syndrome), or GH signaling cascade (123). Studies have shown that rhIGF1 significantly improves height in children unresponsive to rhGH (124,125), and clinical trials clearly demonstrated better response to IGF1 therapy when initiated at an early age (126).
FDA approved conditions for rhIGF1 treatment for children with (127):
- 1.
Severe primary IGF1 deficiency
- 2.
GH gene deletions who have developed neutralizing antibodies to GH
Severe primary IGF1 deficiency is defined by:
- 1.
Height SD score is less than -3SD
- 2.
Basal IGF1 level is below -3SD
- 3.
Normal or elevated GH
The recommended starting dose of rhIGF1 is 40-80 microgram/kg twice daily by subcutaneous injection. If it is tolerated well for at least one week, the dose may be increased by 40 microgram/kg per dose, to the maximum dose of 120 microgram/kg per dose (128).
The most common side effects of IGF1 treatment are pain at injection site and headaches which mostly diminishes after first month of treatment (123). Other less common side effects are lipohypertrophy at the injection site, pseudotumor cerebri, facial nerve palsy and hypoglycemia (126). Another effect of IGF1 treatment is a significant increase in fat mass and BMI(129) —in contradistinction to the lipolytic effect of rhGH treatment. Coarsening of facial feature, increased hair growth, slipped capital femoral epiphysis, scoliosis, hypersensitivity, and allergic reactions including anaphylaxis are other prominent side effects and most commonly are seen during puberty. Growth of lymphoid tissue is a concern which may require tonsillectomy (123).
Insulin-Like Growth Factor 1 Receptor (IGF1R)
The receptor for IGF1 is structurally related to the insulin receptor and similarly has tyrosine kinase activity (Figure 21). IGF1R is located on 15q25-26. The mature (human) IGF1 receptor contains 1337 amino acids and has potent anti-apoptotic activity (130). The IGF1 receptor transduces signals from IGF1, IGF2 and insulin. However, murine data suggest that initially (in the fetus) only the IGF2 signal is operational, while later on in development, both IGF1 and IGF2 (and probably insulin) signal through the IGF1R (131). Hemizygosity for IGF1R has been reported in a single patient (and appears likely in seven others) with IUGR, microcephaly, micrognathia, renal anomalies, lung hypoplasia and delayed growth and development (132). Murine and human studies have shown that mutations in IGF1R result in combined intrauterine and postnatal growth failure (100), confirming the critical role of the IGF system on embryonic, fetal and postnatal growth. A novel heterozygous mutation in the tyrosine kinase domain of the IGF1R gene was recently identified in a family with short stature. The mutation, a heterozygous 19-nucleotide duplication within exon 18 of the IGF1R gene, results in a haploinsuffiency of IGF1R protein due to nonsense mediated mRNA decay (133).

Figure 21.
Insulin-Like Growth Factor 1 Receptor
In summary, IGF1 and IGF1R mutations should be considered if a child presents with the following:
- 1.
Intrauterine and postnatal growth retardation
- 2.
Microcephaly
- 3.
Mental retardation
- 4.
Developmental delay
- 5.
Sensorineural deafness
- 6.
Micrognathia
- 7.
Very low or very high levels of serum IGF1
Insulin-Like Growth Factor 2
IGF2 is thought to be a major prenatal growth hormone and less important in post-natal life.
The human gene, IGF2, is located on 11p15.5 (Figure 22). Chromosome 11p15.5 carries a group of maternally (IGF2) and paternally (H19) imprinted genes that crucial for the fetal growth. Genetic or epigenetic changes in the 11p15.5 region alter the growth (134). IGF2 is maternally imprinted, meaning that the maternal allele is unexpressed. The close proximity of the INS to IGF2-in addition to nearly 50% amino acid identity-suggest that these genes arose through gene duplication events from a common ancestor gene. IGF2 acts via the IGF1 receptor (as well as the insulin receptor). Over-expression of IGF2 results in overgrowth, similar to that seen in Beckwith-Wiedemann Syndrome (which can be due to loss of imprinting, effectively doubling IGF2 expression). A mouse model overexpressing Igf2 demonstrates increased body size, organomegaly, an omphalocele, cardiac, adrenal and skeletal abnormalities, suggestive of Beckwith-Wiedemann and Simpson-Golabi-Behmel syndromes (135). Interestingly, IGF2 expression is normally extinguished by the Wilm's Tumor protein (WT1), providing an explanation for the overgrowth (e.g. hemi-hypertrophy) typically seen in subjects with Wilm's Tumor (136). In contrast, mice without a functional Igf2 (Igf2 knockouts) are born 40% smaller than their normal littermates (identical to Igf1 knockouts).

Figure 22.
Insulin-Like Growth Factor 2
Recent reports on individuals with severe intrauterine growth retardation showed maternal duplication of 11p15 (137). Furthermore, individuals with Silver-Russell-syndrome (SRS, also known as Russell-Silver syndrome) have been found to have an epimutation (demethylation) associated with biallelic expression of H19 and down regulation of IGF2 (138,139). Russell-Silver syndrome is a congenital disorder characterized by intrauterine and postnatal growth retardation, typical facial features (triangular face, micrognathia, frontal bossing, downward slanting of corners of the mouth), asymmetry, and clinodactyly. Other chromosomal abnormalities such as maternal uniparental disomy on chromosome 7 also have been shown in 10% of individuals with SRS (140).
A paternally-derived balanced chromosomal translocation that disrupted the regulatory regions of the predominantly paternally expressed IGF2 gene was described in a woman with short stature, history of severe intrauterine growth retardation (-5.4 SDS), atypical diabetes and lactation failure (141).
Insulin-Like Growth Factor 2 Receptor
A receptor for IGF2, the IGF2R, has been identified, but does not appear to be the mediator of IGF2's growth promoting action. IGF2R is located on 6q26, and encodes a receptor unrelated to the IGF1 or insulin receptor (Figure 23). IGF2R is also the mannose-6-phosphate receptor and serves as a negative modulator of growth (for all IGF's and also insulin). Its main role in vivo is probably as a tumor suppressor gene. While IGF2 is maternally imprinted, mouse Igf2R is paternally imprinted. There is some evidence that (in a temporally-limited fashion) IGF2R is also paternally imprinted in humans. Somatic mutations have been found in hepatocellular carcinoma tissue (heterozygous mutations associated with loss of the other allele), but no germ-line mutations have been identified in individuals with growth abnormalities.

Figure 23.
Insulin-Like Growth Factor 2 Receptor
Insulin
In addition to its glycemic and metabolic roles, insulin functions as a significant growth promoting/anabolic agent. The insulin gene (INS) is located on Chr 11p15.5 and comprises 3 exons (Figure 24). Insulin's role in fetal growth is quite significant, as demonstrated by hyperinsulinemic babies (e.g. infants of diabetic mothers (IDM)). Insulin's growth promoting activity is mediated through a combination of the insulin and the IGF1 receptors. Mutations in the INS gene have been described in subjects with hyperinsulinemia (and/or hyperproinsulinemia) and diabetes mellitus.

Figure 24.
Insulin
Insulin Receptor
The insulin receptor is structurally related to the IGF1 receptor. The gene, INSR, is located on Chr 19p13.2 and contains 22 exons that span over 120 kilobases of genomic DNA (Figure 25). INSR encodes a transmembrane protein with tyrosine kinase activity which is capable of transducing the signals of insulin, IGF1 and IGF2.
Individuals with a mutation in the insulin receptor have been identified and may be the basis for the mythological 'Leprechauns'. They typically have intrauterine growth retardation; small elfin facies with protuberant ears; distended abdomen; relatively large hands, feet, and genitalia; and abnormal skin with hypertrichosis, acanthosis nigricans, and decreased subcutaneous fat. At autopsy, several subjects have been found to have cystic changes in the membranes of gonads and hyperplasia of pancreatic islet cells. Severe mutations generally lead to death within months, but more mild mutations have been found in individuals with insulin resistance, hypoglycemia, acanthosis nigricans, normal subcutaneous tissue and may even be associated with a normal growth pattern! Individuals with even 'mild mutations' have been shown to have a thickened myocardium, enlarged kidneys and ovarian enlargement.

Figure 25.
Insulin Receptor
SHORT STATURE WITH AN ADVANCED BONE AGE
Aggrecan
Aggrecan has also been shown to be involved in human height and the growth process. The aggrecan protein is a major constituent of the extracellular matrix of articular cartilage, where it forms large multimeric aggregates. The gene, ACAN, is located on Chr 15q26.1, comprising 19 exons spread over nearly 72 kilobases of genomic DNA. Exon 1 is approximately 13 kilobases upstream of exons 2-19, which comprise the coding portion of ACAN (142) (Figure 26). ACAN undergoes alternative splicing yielding several isoforms; the predominant isoform being 2132 amino acids long, with three globular domains (G1-3), an ‘interglobular’ (IG) domain, a keratan sulfate (KS) domain and a chondroitin sulfate (CS) domain, largely encoded in a modular fashion.
Domains G1, G2 contain tanden repeat units rich in cysteine, which are necessary for disulfide bridging, the binding of hyaluronic acid and structural integrity, and are separated by the IGD, which provides a level of rigidity. The KS domain contains 11 copies of a six amino acid motif, while the chondroitin sulfate (CS) domain contains over 100 (non-tandem repeated) copies of the dipeptide Serine-Glycine). The G3 domain appears to function in maintaining proper protein folding and subsequent aggrecan secretion. The attachment of hyaluronic acid, keratan and chondroitin sulfate lead to significant water retention, which is largely responsible for the shock-absorbing character of articular cartilage. Aggrecan is also necessary for proper “chondroskeletal morphogenesis” (143), ensuring the proper organization and sequential maturation of the epiphysis.
In 1999, Kawaguchi reported a mutation in ACAN in subjects with lumbar disc herniation(144), then in 2005, both an autosomal dominant form of spondyloephiphyseal dysplasia (SED-Kimberly type) (145) and an autosomal recessive form (SED-Aggrecan type) (146) were shown to arise from mutations in ACAN.
In 2010, cases of autosomal dominant short stature with an advanced bone age were found to have mutations in ACAN, either with or without osteochondritis dissecans and/or (early-onset) osteoarthritis (147-151).
Dateki identified a family of four affected where three members had short stature with an advanced bone age, midface hypoplasia, joint problems and brachydactyly, while the fourth had lumbar disc herniation without other findings(152), attesting to phenotypic heterogeneity, even within a family.

Figure 26.
ACAN
The Short Stature Homeobox-Containing Gene (SHOX) Haploinsufficiency
The Short Stature Homeobox-containing gene (SHOX) was identified in the pseudoautosomal region 1 on the distal end of the X and Y chromosomes at Xp22.3 and Yp11.3 (Figure 27) (153). Mutations in SHOX were observed in 60-100% of Léri-Weill dyschondrosteosis and Langer mesomelic dysplasia (154,155). Turner syndrome is almost always associated with the loss of SHOX gene because of numerical or structural aberration of X chromosome (156). Today it is estimated that SHOX mutations occur with an incidence of roughly 1:1,000 newborns, making mutations of this gene one of the most common genetic defects leading to growth failure in humans.

Genes in pseudoautosomal region 1 do not undergo X inactivation, therefore, healthy individuals express two copies of the SHOX gene, one from each of the sex chromosomes in both 46,XX and 46,XY individuals. The SHOX gene plays an important role in linear growth and is involved in the following:
- 1.
Intrauterine linear skeletal growth
- 2.
Fetal and childhood growth plate in a developmentally specific pattern and responsible for chondrocyte differentiation and proliferation (157).
- 3.
A dose effect: SHOX haploinsufficiency associated with short stature. In contrast, SHOX overdose as seen in sex chromosome polyploidy is associated with tall stature.
A large number of unique mutations (mostly deletions and point mutations) of SHOX have been described (154,156,158). SHOX abnormalities are associated with a broad phenotypic spectrum, ranging from short stature without dysmorphic signs as seen in idiopathic short stature (ISS) to profound Langer’s mesomelic skeletal dysplasia, a form of short stature characterized by disproportionate shortening of the middle segments of the upper arms (ulna) and lower legs (fibula) (159). In contrast to many other growth disorders such as growth hormone deficiency, SHOX deficiency is more common in girls.
Rappold et al developed a scoring system to determine the phenotypic spectrum of SHOX deficiency in children with short stature and identify patients for SHOX molecular testing (158). The authors recommend a careful examination including measurement of body proportions and X-ray of the lower legs and forearm before making the diagnosis of ISS. The scoring system consists of three anthropometric variables (arm span/height ratio, sitting height/height ratio and BMI), and five clinical variables (cubitus valgus, short forearm, bowing of forearm, muscular hypertrophy and dislocation of the ulna at the elbow). Based on the scoring system, authors recommend testing for SHOX deficiency for the individuals with a score greater than four or seven out of a total score of 24 (Table 7).
The recent data show that GH treatment is effective in improving linear growth of patients with SHOX mutations (159).
Table 7.
Scoring system for identifying patients that qualify for short-stature homeobox containing gene (SHOX) testing based on clinical criteria. Reprinted with permission (159)
| Score item | Criterion | Score points |
|---|---|---|
| Arm span/height ratio | <96.5% | 2 |
| Sitting height/height ratio | >55.5% | 2 |
| Body–mass index | >50th percentile | 4 |
| Cubitus valgus | Yes | 2 |
| Short forearm | Yes | 3 |
| Bowing of forearm | Yes | 3 |
| Appearance of muscular hypertrophy | Yes | 3 |
| Dislocation of ulna (at elbow) | Yes | 5 |
| Total | 24 | |
Noonan Syndrome
Noonan syndrome (NS) is a relatively common genetic disorder with the incidence of between 1:1000 and 1:4000 (160). NS is inherited in an autosomal dominant manner, and sporadic cases are not uncommon (50-60%) (161). NS is characterized by short stature, cardiac defects (most commonly pulmonary stenosis and hypertrophic cardiomyopathy), facial dysmorphism (down-slanting, antimongoloid palpebral fissures, ptosis, and low-set posteriorly rotated ears), webbed neck, mild mental retardation, cryptorchidism, feeding difficulties in infancy. The phenotype is variable between affected members of the same family and becomes milder with age (162).
Nearly 50% of patients with NS have gain-of-function mutations in protein tyrosine phosphatase nonreceptor type 11 (PTPN11), the gene encoding the cytoplasmic tyrosine phosphatase SHP-2, which regulates GH signaling by dephosphorylating STAT5b, resulting in down-regulation of GH activity (163). Mutations in four other genes (KRAS, SOS1, NF1 and RAF1) involved in RAS/MAPK signaling systems have been identified in patients with the NS phenotype and related disorders including LEOPORD, Costello, and cardio-facial-cutaneous syndromes (Figure 28) (164).

Although identifying these mutations has contributed to better understanding of the pathogenesis of NS, it appears that the genotype does not completely correlate with the phenotype, e.g. short stature in patients with NS. Several studies have shown that the subjects carrying gain of function mutations of PTPN11 had lower IGF1 levels, poor growth response, and resistance to GH therapy compared to subjects without PTPN11 mutations (165,166). However, data from one large study of individuals with NS did not demonstrate the same correlation between PTPN11 mutations and short stature(160). However, more recent studies showed significant improvement in final adult height in individuals with NS regardless of their mutation type (167,168).
DIAGNOSIS
Diagnosis of GH deficiency during childhood and adolescence is frequently challenging. Children whose height are below the 3rd percentile or -2 SD and have decreased growth velocity require clinical evaluation. Evaluation should begin with a detailed past medical history, family history, diet history, detailed review of prior growth data (including the initial post-natal period) and a thorough physical examination (169). Together, these should help the clinician identify the pattern and cause of growth failure, such as fetal growth restriction (e.g. SGA and IUGR), chronic illness, malnutrition/malabsorption, hypothyroidism, skeletal abnormalities or other identifiable syndromes, such as Turner syndrome. Once growth hormone deficiency is suspected, further testing of the hypothalamic-pituitary axes (including but not limited to the GH-IGF axis) along with radiological evaluation, should be performed (Table 8). It is important to note that the tests cannot be performed simultaneously, or in random order. Certain conditions (e.g. Hypothyroidism and Celiac disease) may mask the presence of others (e.g. GH deficiency), therefore requiring to a step-wise approach with screening tests preceding specific examinations. Since growth failure generally occurs outside of GHD, only those children with signs or symptoms undergo expensive, invasive and non-physiologic GH provocative testing.
Table 8.
Guidelines for Initial Clinical Evaluation of a Child with Growth Failure
| Evaluation | Key elements |
|---|---|
| Birth history | Gestational age, birth weight and length, delivery type, birth trauma, hypoglycemia, prolonged jaundice. |
| Past medical and surgical history | Head trauma, surgery, cranial radiation, CNS infection. |
| Review of systems | Appetite, eating habits, bowel movements. |
| Chronic illness | Anemia, Inflammatory Bowel Disease, cardiovascular disease, renal insufficiency, etc. |
| Family history | Consanguinity, parents and siblings’ heights, family history of short stature, delayed puberty. |
| Physical examination | Body proportions (upper/lower segment ratios, arm span), head circumference, microphallus, dysmorphism, and midline craniofacial abnormalities. |
| Growth pattern | Crossing of percentiles, failure to catch-up. |
| Screening Tests | CBC, BCP, ESR, Celiac screening, TSH and Free T4, UA, IGF1, IGFBP3, Bone age (and a Karyotype for females) |
Growth Charts
The growth pattern is a key element of growth assessment and is best studied by plotting growth data on an appropriate growth chart. US growth charts were developed from cross-sectional data provided by the National Center for Health Statistics and updated in 2000 (170), with body mass index included in this newest set. The supine length should be plotted for children from birth through age 3 years and standing height plotted when the child is old enough to stand, generally after 2 years of age. Ideally, growth data is determined by evaluating subjects at regular (optimally at 3 month) intervals, with the same stadiometer, and with the same individual obtaining the measurements, whenever possible. Three months is the minimal time interval needed between measurements to calculate a reliable growth velocity, and a six to twelve-month interval is optimal. Age and pubertal staging must be considered when evaluating the growth velocity, with the understanding that there is great individual variation in the onset and rate of puberty (171).
Deviations across height percentiles should be noted and evaluated further when confirmed, with the understanding that during the first two years of life, the crossing of length and/or weight percentiles may reflect catch-up or catch-down growth. Crossing percentiles during this period is not always physiological, and must be examined in the context of family, prenatal, birth and medical histories. Additionally, between two and three years of age, statural growth measurement changes from supine to erect, and may also introduce variation. Growth below the normal range (e.g. >-2SD) even without further deviation is consistent with (but not pathognomonic of) GH deficiency. Short stature with a low BMI suggests an abnormality of nutrition/GI tract (e.g. malnutrition, Celiac Disease, etc.), while short stature with an elevated BMI suggests hypothyroidism, Cushing’s syndrome, or a central eating disorder, such as Prader-Willi syndrome, etc.
Figures 29-31 represent growth charts of children studied by the authors who have genetic defects leading to isolated growth hormone deficiency.

Figure 29.
Growth pattern in children with isolated GH deficiency (Type 1A)
Growth failure can manifest as severe growth delay (Figure 29), gradual deceleration (“falling off the curve”) (Figure 30), or alternatively, maintaining a growth pattern parallel to the 3rd percentile, but without catch-up growth (Figure 31).

Figure 30.
Growth pattern in children with isolated GH deficiency (Type 1B)

Figure 31.
Growth pattern in children with isolated GH deficiency (Type 2)
Most children with GH deficiency have normal birth weight and length. However, in most cases, postnatal growth becomes severely compromised. This can be seen even in the first months of life. Although such children may show a normal growth pattern during the first 6 months, growth failure will eventually occur, as GH takes on a more physiologically dominant role and a child’s growth falls below the normal range.
Radiologic Evaluation
The most commonly used system to assess skeletal maturity is to determine the ‘bone age’ of the left hand and wrist, using the method of Greulich and Pyle (172). Children younger than 2 years of age should have their bone age estimated from x-rays of the knee. Tanner and Whitehouse and their colleagues developed a scoring system for each of the hand bones as an alternative method to the method of Greulich and Pyle (173).
Adult height prediction methods estimate adult height by evaluating height at presentation relative to normative values for chronological or bone age. Such methods have been utilized for approximately 60 years (174) and are generally considered accurate in evaluating healthy children with a ‘normal’ growth potential (175,176). Several different methods have been produced and are currently in widespread use, including those of Bayley-Pinneau, the Tanner-Whitehouse-Marshall-Carter and Roche-Wainer-Thissen.
In 1946, Bayley initially described how final height could be estimated from the present height and the bone age, revising the method in 1952 to use the bone age assessment method of Greulich and Pyle (172). They developed what is commonly known as the predicted adult height (PAH) method of Bayley-Pinneau (BP). Tables have been developed for the BP method, listing the proportion of adult height attained at different bone ages, using longitudinal growth data on 192 healthy children in the US. Three tables – average, advanced and retarded – correct for possible differences between CA and BA of more than one year (177). The Bayley-Pinneau PAH method is applicable from age 8 years onwards.
Tanner, Whitehouse, Marshall and Carter developed an adult height prediction model based on current height, the mid-parental height, the age of menarche in girls and the ‘Tanner’ bone age (173). This PAH method (‘TW2’) was developed on the longitudinal data of 211 healthy, British children. TW2 differs from the BP method in that the TW2 lowers the minimal age of prediction to 4 years, and also allows for a quantitative effect of BA, while BP gives a semi-quantitative effect of bone age (i.e. delayed, normal or advanced).
The PAH method of Roche-Wainer-Thissen (RWT) was derived from longitudinal data on approximately 200 “normal” Caucasian American children in southwestern Ohio, at the Fels Research Institute (178). The RWT PAH method assesses the subject’s height, weight, BA and mid-parental height (MPH) and then applies regression techniques to determine the mathematical weighting to be applied to the four variables. The RWT method was designed to allow final height prediction from a single visit, but is only applicable when greater than half of the bones are not fully mature.
Since both the bone age assessments and height prediction methods are created from healthy children (and often children from a single ethnic group and region), their use in ‘other’ populations is potentially inappropriate. In fact, Tanner et al state that their method is applicable to both boys and girls with short stature, but caution that “In clearly pathological children, such as those with endocrinopathies, they do not apply”. Similarly, Roche et al suggest caution when applying the RWT PAH method in ‘non-white and pathological populations’ (178). Zachmann et al reported that the RWT and TW2 methods (which are more BA-reliant) are better when growth potential is normal relative to the BA, however, in conditions with “…abnormal and incorrigible growth patterns…” the BP method was more accurate, stating that with a “non-normal bone maturation to growth potential relationship, the ‘coefficient and regression equations’ (RWT and Tanner) cause an over-prediction of adult height” (179).
As stated above, these methods are based on healthy children and assume that the growth potential is directly proportional to the amount of time left prior to epiphyseal fusion as measured by the bone age. While this is correct for some of the children seen by the pediatric endocrinologist (e.g. healthy children, children with GH deficiency), it is not correct for many others with abnormal growth (e.g. children born SGA, children with idiopathic short stature, Turner syndrome and chronic renal failure). It is likely also inappropriate for children with an abnormal tempo of maturation (e.g. children with Russell-Silver syndrome, precocious puberty and congenital adrenal hyperplasia). In such children, standard growth prediction methods should be used only as ‘general guides’, if at all. Table 9 summarizes these 4 methods.
Table 9.
Summary of Methods Used for PAH
| Methods | Parameters |
|---|---|
| BP | Height, BA, CA |
| TW2 | Height, BA, CA, MPH, the age of menarche in girls |
| RWT | Height, weight, BA, MPH |
| Khamis-Roche | Height, weight, MPH |
Biochemical Evaluation of GH Deficiency
As growth hormone is secreted in a pulsatile manner (usually 6 pulses in 24 hours and mainly during the night) with little serum GH at any given time, several methods have been recommended to assess the adequacy of GH secretion:
- 1.
Stimulation testing: GH provocation utilizing arginine, clonidine, glucagon, L-Dopa, insulin, etc. This practice generally measures pituitary reserve-or GH secretory ability-rather than endogenous secretory status. Trained individuals should perform the GH stimulation test according to a standardized protocol, with special care taken with younger children/infants.
- 2.
GH-dependent biochemical markers: IGF1 and IGFBP3: Values below a cut-off less than -2 SD for IGF1 and/or IGFBP3 strongly suggest an abnormality in the GH axis if other causes of low IGF have been excluded. Age and gender appropriate reference ranges for IGF1 and IGFBP3 are mandatory.
- 3.
24-hour or Overnight GH sampling: Blood sampling at frequent intervals designed to quantify physiologic bursts of GH secretion.
- 4.
IGF generation test: This test is used to assess GH action and for the confirmation of suspected GH insensitivity. GH is given for several days (3-5 days) with serum IGF1 and IGFBP-3 levels measured at the start and end of the test. A sufficient rise in IGF1 and IGFBP-3 levels would exclude severe forms of GH insensitivity (99,171).
Failure to raise the serum GH level to the threshold level in response to provocation suggests the diagnosis of GH deficiency, while a low IGF1 and/or IGFBP3 level is supportive evidence. Although pharmacological GH stimulation tests have known difficulties such as poor reproducibility, arbitrary cut-off limits, varying GH assays, they remain the most easily available and accepted tools to evaluate pituitary GH secretory capacity. GH stimulation test results should be interpreted carefully in conjunction with pubertal status and body weight. Puberty and administration of the sex steroids increase GH response to stimulation tests (180). To prevent false positive results, some centers use sex steroid priming in prepubertal children prior to GH stimulation testing (181). In obese children, the normal regulation of the GH/IGF1 axis is disturbed and GH secretion is decreased. IGF1 levels are very sensitive to the nutritional status, while IGFBP3 are less so. Additionally, the normative range for IGF1 and IGFBP3 values are extremely wide, often with poor discrimination between normal and pathological. Age/pubertal stage and gender-specific threshold values must be utilized for both IGF1 and IGFBP3.
Summary of Diagnosis of GH deficiency
Children with severe GH deficiency can usually be diagnosed easily on clinical grounds, and fail GH stimulation tests. Studies have shown that despite clinical evidence of GH deficiency, some children may pass GH stimulation tests (171). In the case of unexplained short stature, if the child meets most of the following criteria, a trial of GH treatment should be initiated (182):
- 1.
Height >2.25 SD below the mean for age or >2 SD below the mid-parental height percentile,
- 2.
Growth velocity <25th percentile for bone age,
- 3.
Bone age >2 SD below the mean for age,
- 4.
Height prediction is significantly below the mid-parental height,
- 5.
Low serum insulin-like growth factor 1 (IGF1) and/or insulin-like growth factor binding protein 3 (IGFBP3) for bone age and gender
- 6.
Other clinical features suggestive of GH deficiency.
Key elements that may indicate GH deficiency
- 1.
Height more than 2 SD below the mean.
- 2.
Neonatal hypoglycemia, microphallus, prolonged jaundice, or traumatic delivery.
- 3.
Although not required, a peak GH concentration after provocative GH testing of less than 10 ng/ml.
- 4.
Consanguinity and/or a family member with GH deficiency.
- 5.
Midline CNS defects, pituitary hypo- or aplasia, pituitary stalk agenesis, empty sella, ectopic posterior pituitary (‘bright spot’) on MRI.
- 6.
Deficiency of other pituitary hormones: TSH, PRL, LH/FSH and/or ACTH deficiency.
Many practitioners consider GH stimulation tests to be optional in the case of clinical evidence of GH deficiency, in patients with a history of surgery or irradiation of the hypothalamus/pituitary region and growth failure accompanied by additional pituitary hormone deficiencies. Similarly children born SGA, with Turner syndrome, PWS and chronic renal insufficiency do not require GH stimulation testing before initiating GH treatment (182).
TREATMENT
The principal objective of GH treatment in children with GH deficiency is to improve final adult height. Human pituitary-derived GH was first used in children with hypopituitarism over 60 years ago, and abruptly ceased in 1985, after the first cases of Creutzfeld-Jacob disease were recognized. Since 1986, recombinant human GH (rhGH) has been the exclusive form of growth hormone used to treat GH deficiency in the United States and most of the world.
Short stature without overt growth hormone deficiency is very well described, and occurs in Turner Syndrome, renal failure, malnutrition, cardiovascular disease, Prader-Willi syndrome, small for gestational age, inflammatory bowel disease, and osteodystrophies- clearly represents the majority of short/poorly growing children in the world. Although not the focus of this discussion, it is important to realize that - in clinical terms - GH therapy is used to treat growth failure, rather than a biochemical GH deficiency. GH therapy in this setting, in combination with disease-specific treatments, generally improves statural growth and final adult height.
The primary goals of the treatment of a child with GH deficiency are to achieve normal height during childhood and to attain normal adult height. Children should be treated with an adequate dose of rhGH, with the dose tailored to that child’s specific condition. FDA guidelines for GH dose vary according to the indication and are given in Table 10 (182).
Administration of rhGH in the evening is designed to mimic physiologic hGH secretion. Treatment is continued until final height or epiphyseal closure (or both) has been recorded. GH therapy, however, should be continued throughout adulthood in the case of GHD, to optimize the metabolic effects of GH and to achieve normal peak bone mass-albeit at significantly lower “adult” doses. Adult GH replacement should only be started after retesting the individual and again demonstrating a failure to reach the new age-appropriate GH threshold, if appropriate.
Table 10.
GH Dosage
| Indication | Dose (mg/kg/wk) |
|---|---|
| GH Deficiency Children Pre-pubertal Pubertal Adults | 0.16 – 0.35 0.16 – 0.70 0.04 – 0.175 |
| Turner Syndrome | 0.375 |
| Chronic renal insufficiency | 0.35 |
| Prader-Willi Syndrome | 0.24 |
| SGA | 0.48 |
| Idiopathic short stature (ISS) | 0.3 – 0.37 |
| SHOX Deficiency | 0.35 |
| Noonan Syndrome | 0.23 – 0.46 |
The growth response to GH treatment is typically maximal in the first year of treatment and then gradually decreases over the subsequent years of treatment. First year growth response to rhGH is generally 200% of the pre-treatment velocity, and after several years, averages 150% of the baseline. Height improvements of 1 SD are typically achieved in children with GHD after two years of treatment, and between 2 and 2.5 SD after five or seven years.
GH doses are often increased if catch-up growth is inadequate and/or to compensate for the waning effect of rhGH with time. Cohen et al reported a significant improvement in HV when GH dose was adjusted based on IGF1 levels (183). However, GH dose was almost 3 times higher than mean conventional GH dose when IGF1 levels were titrated to the upper limit of normal. The lack of long-term safety data on high doses of GH and high circulating levels of IGF1 levels should be considered. Therefore, weight-based GH dosing is still recommended by many as the standard of care (184).
It is critically important to maximize height with GH therapy before the onset of puberty. Several investigators have advocated modifying puberty or the production of estrogens by the use of GnRH super-analogues (185,186) and aromatase inhibitors (187-190), respectively, in order to expand the therapeutic window for GH treatment, especially in older males.
The response to GH, however, may vary in children(191). Factors may affect the response to GH therapy including
- 1.
The etiology of short stature
- 2.
Age at the start of treatment
- 3.
Height deficit at the start of treatment
- 4.
GH dose and frequency
- 5.
Duration of treatment
- 6.
Genetic factors
Several studies have reported the association between response to GH therapy and a GHR gene polymorphism, the deletion of exon 3 (GHRd3). Although some reports showed better response to GH therapy in GHRd3 carriers with different clinical conditions including GHD, Turner syndrome, SGA, and ISS (89,192-195), many others failed to confirm positive effects of GHRd3 on response to GH treatment (196-198).
Monitoring GH Treatment
Children receiving GH therapy require periodic monitoring. Three-month intervals are commonly chosen to allow for sufficient growth for a meaningful measurement, while minimizing time between dose adjustments/intervention. During follow up visits, height, weight, pubertal status, inspection of injection sites, and a comprehensive clinical exam should be initiated. In clinical practice, there are several parameters to monitor the response to GH treatment; the determination of the growth response (i.e. change in height velocity, ∆HV) being the most important parameter. These points are summarized in Table 11.
Table 11.
Summary of Follow-Up Evaluation
| Parameters | Assessment |
|---|---|
| Bone age | 12-month intervals to assess the predicted height. |
| Thyroid Function Test | 6-month intervals, or immediately, if growth velocity decreases. |
| Serum IGF1 and IGFBP-3 | 12-month intervals. Most useful in maintaining GH dose in ‘safe’ region. They do not necessarily correlate with growth velocity. |
| Metabolic panel, CBC, ESR, HbA1C | 12-month intervals. |
| Dose adjustment | Should be based on weight-change, growth response, pubertal stage, comparison to predicted height at each visit, and IGF-I/IGFBP-3 annually. |
| Adverse Events | Every visit. |
The Safety of GH Treatment
To date, multiple studies have demonstrated the safety of GH therapy (7,169,170,185,199-202). While rhGH treatment is generally considered safe, patients, however, should be monitored closely during treatment. Some of the common side effects seen during GH therapy are scoliosis, slipped capital femoral epiphysis (SCFE), pancreatitis, and pseudotumor cerebri (intracranial hypertension). An analysis of Genentech’s National Cooperative Growth Study (NCGS) identified eleven cases of adrenal insufficiency (AI) resulting in four deaths. All eleven cases of AI occurred in patients with organic GH deficiency (n=8,351), yielding an incidence of 132 per 100,000 in this subgroup, and an overall incidence of AI in NCGS was 20 per 100,000 (203).
Another concern is the use of GH in patients with Prader-Willi syndrome. Early recognition of the syndrome allows earlier intervention to prevent morbidity. Previous studies and data from KIGS showed that earlier initiation of GH treatment in children with PWS significantly improved body composition, muscle tone, growth, and cognition (204).
Fatalities have been reported in patients with Prader-Willi syndrome during or after rhGH therapy (205). Data for children aged 3 years and older showed no statistically significant differences between the GH-treated and untreated groups with respect to cause of death, including respiratory infection or insufficiency (205,206). Although there is no clear evidence that those deaths are related to GH therapy, it was postulated that GH/IGF1 may worsen sleep apnea or hypoventilation via increasing tonsillar/adenoid tissue or worsen pre-existing impaired respiration by increasing volume load (207). However, studies on respiratory function of subjects with Prader-Willi syndrome during rhGH therapy have only demonstrated improved respiratory drive and function (208). In fact, a recent study showed that all subjects tested had abnormal sleep studies/parameters prior to initiating GH treatment, and that GH treatment resulted in an improvement in sleep apnea in the majority of patients with PWS. Importantly, however, a subset had worsening of sleep disturbance shortly after (6 week) starting GH when also developing a respiratory infection (209). Because it is difficult to predict who will worsen with GH treatment, these authors recommend that patients with Prader-Willi syndrome have polysomnography before and 6 weeks after starting rhGH and should be monitored for sleep apnea during upper respiratory tract infections. IGF1 levels should also be monitored.
The data on efficacy and safety of GH treatment in 5220 Turner Syndrome (TS) children during the last 20 years has been reported by NCGS. The incidence of various side effects known to be associated with GH including pseudotumor cerebri, slipped capital femoral epiphysis, and scoliosis was increased in TS patients treated with GH compared with non-TS patients, however, children with TS are known to have a higher incidence of these side effects independent of rhGH treatment (210). Interestingly, type 1 diabetes was increased in GH treated group, most likely unrelated to GH treatment since the predisposition to autoimmune disorders is one of the characteristics of TS. In addition, NCGS data demonstrate a slightly increased incidence of a variety of malignancies in TS, however, this may again be related to the underlying condition, (i.e. not necessarily the rhGH treatment) as girls with TS have been shown to have an increased risk for cancer compared to general population (211). In summary, twenty years of experience in 5220 patients seems reassuring and does not indicate any new rhGH-related safety signals in the TS population (210).
There has been ongoing concern about tumorigenicity of chronically elevated IGF1 levels. It would therefore seem prudent to maintain IGF1 levels in the mid-normal range for age/pubertal stage and gender. Although the long-term consequences of elevated IGF1 levels during childhood are not known, some investigators recommend that dose reductions be considered after the first two years of therapy if IGF1 levels continue to be above the normal range (182). The report from the Safety and Appropriateness of Growth Hormone Treatments in Europe (SAGhE) in 2012, raised many concerns about the long-term safety of rhGH therapy in children. SAGhE is a large database established by eight European countries to evaluate the long-term safety of childhood GH treatment between 1980s and 1990s in 30,000 patients. Preliminary analysis of the patients in France revealed that among patients treated with rhGH, there was a 33% increased relative risk of mortality compared with French general population. They also noted an increased incidence of bone malignancies and cardiovascular disease (212). However, the data from the Belgian, Swedish the Dutch portions of SAGhE did not support or corroborate the findings that were reported from France (213).
Real and Theoretical adverse events of GH therapy are summarized in Table12.
Table 12.
Real and Theoretical Adverse Events of GH Treatment
| Side effects | Comment |
|---|---|
| Slipped capital femoral epiphysis (SCFE) | Unclear whether GH causes SCFE or if it is a result of diathesis and rapid growth induced by the GH. In addition, obesity, trauma, and previous radiation exposure increase the risk for SCFE. At each visit, patients should be evaluated for knee or hip pain/limp. |
| Pseudotumor cerebri | The mechanism is unclear, but it may be a result of GH induced salt and water retention within the CNS. Mostly occurs within the first months of treatment. It is more common in patients with organic GH deficiency, chronic renal insufficiency, and Turner Syndrome (203). Complaints of headache, nausea, dizziness, ataxia, or visual changes should be evaluated immediately. |
| Leukemia | Numerous large studies have not shown any association between rhGH and leukemia in children without predisposing conditions (200,203,214). |
| Recurrence risk of CNS tumors | Extensive studies did not support this possible side effect without risk factors (185,203,215-218) |
| Risk of primary malignancy | Studies have not shown a higher risk of all-site primary malignancy without a history of previous malignancy (219,220) |
| Insulin resisretance | Insulin resistance is associated with GH therapy, though it is generally transient and/or reversible and rarely leads to overt diabetes. Patients with a limited insulin reserve may develop glucose intolerance. HbA1C should be monitored. |
| Pancreatitis | It may occur in patients with Turner syndrome, and associated risk factors (203). |
| Hypothyroidism | Almost 25% of children may develop declines in serum T4 levels, generally reflecting enhanced conversion of T4 to T3, rather than outright hypothyroidism. |
| Transient gynecomastia | These are attributed to anabolic and metabolic effects of GH. |
| Scoliosis | It is more common in Turner syndrome and PWS. Patients should be evaluated for scoliosis at each visit and referred as appropriate |
| Adrenal Insufficiency | GH decreases the conversion of corticosterone to cortisol by a modulating effect on hepatic 11-beta hydroxysteroid dehydrogenase 1. Thus, endogenous cortisol levels can decrease in GHD patients after initiation of GH treatment. Furthermore, GH therapy may unmask previously unsuspected central ACTH deficiency. Whether the patients with hypopituitarism are on GH or not, they have a lifelong risk for adrenal insufficiency. Therefore, they should be monitored closely for adrenal insufficiency and their cortisol dose should be adjusted when GH therapy is started (203). |
| Sleep apnea/sleep disturbance | GH treatment might worsen sleep apnea/sleep disturbance in patients with Prader-Willi Syndrome, especially during a concomitant respiratory infection. |
Transitioning GH Treatment From Childhood to Adulthood
Growing data support the need for continuation of GH treatment in individuals with childhood GH deficiency. GH treatment provides significant benefits in body composition, bone mineralization, lean body mass, lipid metabolism, and quality of life in adults with GH deficiency (221,222). However, identifying appropriate patients for transitioning from childhood to adult GH therapy remains challenging. The majority of children with a diagnosis of GHD and who are treated with GH do not have a permanent GHD and will not require treatment during adulthood. Re-evaluation of GH secretory capacity is recommended after completion of linear growth in adolescents with history of childhood GHD (223). However, such re-evaluation requires cessation of GH treatment for at least one month. Furthermore, there is no established optimal GH stimulation test identified and validated during this transition period. The stimulation test results vary by protocol, and only a few secretagogues (insulin, arginine, and glucagon) are available to confirm GHD. The cut-off values are also more strict; the peak GH level to establish GHD is <6 mcg/L for the insulin tolerance test and ≤ 3 mcg/L for the glucagon test in young adults (224,225). It is in agreement that if a patient has severe GHD secondary to organic defects (hypothalamic-pituitary abnormalities, tumors involving pituitary or hypothalamic area, infiltrative diseases, and cranial irradiation), genetic causes of GHD involving one or more additional pituitary hormone deficiencies and has serum IGF-1 level below the normal range at least one month off therapy, are more likely to have permanent GHD and retesting to confirm GHD is unnecessary (221,225). However, children with idiopathic GHD are less likely to have permanent GHD. In a US study, only one third of patients with idiopathic GHD retested as GHD (226). In that cohort, authors found age <4 at diagnosis and IGFBP-3 below -2.0 SDS were the strongest predictive factors (100% PPV) for permanent GHD. In contrast to previous studies (223), low IGF-1 (< -2.0 SDS) did not have significant power to identify permanent GHD unless IGF-1 level was extremely low (-5.3 SDS) (226).
In summary, current guidelines recommend the measurement of serum IGF-1 levels and a GH stimulation test after cessation of treatment at least one month to determine whether the adolescents with childhood-onset GHD will need ongoing treatment unless they have known organic or genetic defects in the hypothalamic-pituitary region (221,222,225).
CONCLUSION
The genetic control of human growth is becoming increasingly clear. Many genes have been identified that contribute to the development and function of the pituitary gland including the somatotrope and the GH/IGF1 axis. Genes encoding “downstream” factors, including the insulin and the insulin receptor, the Short Stature Homeobox and SHP2 affect growth unrelated to growth hormone status, while Aggrecan has been described in cases of short stature with an advanced bone age, as well as in multiple forms of spondyloepiphyseal dysplasia.
Defects in these genes have been shown to be responsible for abnormal growth in humans. Elucidation of these and new genetic factors will provide us with a better understanding of the physiology of growth, and should lead to the improved diagnosis and treatment of individuals with growth abnormalities.
REFERENCES
- 1.
- Wilson TA, Rose SR, Cohen P, Rogol AD, Backeljauw P, Brown R, Hardin DS, Kemp SF, Lawson M, Radovick S, Rosenthal SM, Silverman L, Speiser P, Drug LWPES, Therapeutics C. Update of guidelines for the use of growth hormone in children: the Lawson Wilkins Pediatric Endocrinology Society Drug and Therapeutics Committee. The Journal of Pediatrics. 2003;143(4):415–421. [PubMed: 14571209]
- 2.
- Savage MO, Simon D, Czernichow PC. Growth hormone treatment in children on chronic glucorticoid therapy. Endocr Dev. 2011;20:194–201. [PubMed: 21164273]
- 3.
- Slonim AE, Bulone L, Damore MB, Goldberg T, Wingertzahn MA, McKinley MJ. A Preliminary Study of Growth Hormone Therapy for Crohn's Disease. New England Journal of Medicine. 2000;342(22):1633–1637. [PubMed: 10833209]
- 4.
- Mauras N, George D, Evans J, Milov D, Abrams S, Rini A, Welch S, Haymond MW. Growth hormone has anabolic effects in glucocorticosteroid-dependent children with inflammatory bowel disease: A pilot study. Metabolism. 2002;51(1):127–135. [PubMed: 11782884]
- 5.
- Touati G, Prieur AM, Ruiz JC, Noel M, Czernichow P. Beneficial effects of one-year growth hormone administration to children with juvenile chronic arthritis on chronic steroid therapy. I. Effects on growth velocity and body composition. The Journal of Clinical Endocrinology and Metabolism. 1998;83(2):403–409. [PubMed: 9467548]
- 6.
- Hardin DS, Stratton R, Kramer JC, de la Rocha SR, Govaerts K, Wilson DP. Growth Hormone Improves Weight Velocity and Height Velocity in Prepubertal Children with Cystic Fibrosis. Horm Metab Res. 1998;30(10):636–641. [PubMed: 9851673]
- 7.
- Hardin DS, Ellis KJ, Dyson M, Rice J, McConnell R, Seilheimer DK. Growth hormone improves clinical status in prepubertal children with cystic fibrosis: results of a randomized controlled trial. J Pediatr. 2001;139(5):636–642. [PubMed: 11713439]
- 8.
- Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, Tansil S, New MI. Treatment with Growth Hormone and LHRH Analogue Improves Final Adult Height in Children with Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab. 2005:jc.2004-2128. [PubMed: 15797962]
- 9.
- Rosenbloom AL. Mecasermin (recombinant human insulin-like growth factor I). Advances in Therapy. 2009;26(1):40. [PubMed: 19198769]
- 10.
- Larsen WJ. Human Embryology. Second ed: Churchhill Livingstone, Inc.
- 11.
- De Rienzo F, Mellone S, Bellone S, Babu D, Fusco I, Prodam F, Petri A, Muniswamy R, De Luca F, Salerno M, Momigliano-Richardi P, Bona G, Giordano M. CPHD ISGoGo. Frequency of genetic defects in combined pituitary hormone deficiency: a systematic review and analysis of a multicentre Italian cohort. Clin Endocrinol (Oxf). 2015;83(6):849–860. [PubMed: 26147833]
- 12.
- Phillips JA, Cogan JD. Genetic basis of endocrine disease. 6. Molecular basis of familial human growth hormone deficiency. J Clin Endocrinol Metab. 1994;78(1):11–16. [PubMed: 8288694]
- 13.
- Roessler E. Du Y-Z, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, Roeder ER, Ming JE, Ruiz i Altaba A, Muenke M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(23):13424–13429. [PMC free article: PMC263830] [PubMed: 14581620]
- 14.
- Roessler E, Ermilov AN, Grange DK, Wang A, Grachtchouk M, Dlugosz AA, Muenke M. A previously unidentified amino-terminal domain regulates transcriptional activity of wild-type and disease-associated human GLI2. Hum Mol Genet. 2005;14(15):2181–2188. [PubMed: 15994174]
- 15.
- França MM, Jorge AAL, Carvalho LRS, Costalonga EF, Vasques GA, Leite CC, Mendonca BB, Arnhold IJP. Novel Heterozygous Nonsense GLI2 Mutations in Patients with Hypopituitarism and Ectopic Posterior Pituitary Lobe without Holoprosencephaly. The Journal of Clinical Endocrinology & Metabolism. 2010;95(11):E384–E391. [PubMed: 20685856]
- 16.
- Demiral M, Demirbilek H, Unal E, Durmaz CD, Ceylaner S, Ozbek MN. Ectopic Posterior Pituitary, Polydactyly, Midfacial Hypoplasia and Multiple Pituitary Hormone Deficiency due to a Novel Heterozygous IVS11-2A>C(C.1957-2A>C) Mutation in GLI2 Gene. J Clin Res Pediatr Endocrinol. 2019 [PMC free article: PMC7499131] [PubMed: 31782289]
- 17.
- Netchine I, Sobrier ML, Krude H, Schnabel D, Maghnie M, Marcos E, Duriez B, Cacheux V, Moers AV, Goossens M, Gruters A, Amselem S. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet. 2000;25(2):182–186. [PubMed: 10835633]
- 18.
- Bhangoo APS, Hunter CS, Savage JJ, Anhalt H, Pavlakis S, Walvoord EC, Ten S, Rhodes SJ. Clinical case seminar: a novel LHX3 mutation presenting as combined pituitary hormonal deficiency. The Journal of Clinical Endocrinology and Metabolism. 2006;91(3):747–753. [PubMed: 16394081]
- 19.
- Raetzman LT, Ward R, Camper SA. Lhx4 and Prop1 are required for cell survival and expansion of the pituitary primordia. Development (Cambridge, England). 2002;129(18):4229–4239. [PubMed: 12183375]
- 20.
- Sobrier ML, Attié-Bitach T, Netchine I, Encha-Razavi F, Vekemans M, Amselem S. Pathophysiology of syndromic combined pituitary hormone deficiency due to a LHX3 defect in light of LHX3 and LHX4 expression during early human development. Gene expression patterns. GEP. 2004;5(2):279–284. [PubMed: 15567726]
- 21.
- Machinis K, Pantel J, Netchine I, Leger J, Camand OJ, Sobrier ML, Dastot-Le Moal F, Duquesnoy P, Abitbol M, Czernichow P, Amselem S. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet. 2001;69(5):961–968. [PMC free article: PMC1274372] [PubMed: 11567216]
- 22.
- Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nature Genetics. 1998;19(2):125–133. [PubMed: 9620767]
- 23.
- Carvalho LR, Woods KS, Mendonca BB, Marcal N, Zamparini AL, Stifani S, Brickman JM, Arnhold IJP, Dattani MT. A homozygous mutation in HESX1 is associated with evolving hypopituitarism due to impaired repressor-corepressor interaction. The Journal of Clinical Investigation. 2003;112(8):1192–1201. [PMC free article: PMC213489] [PubMed: 14561704]
- 24.
- Parks JS, Brown MR, Hurley DL, Phelps CJ, Wajnrajch MP. Heritable disorders of pituitary development. Journal of Clinical Endocrinology and Metabolism. 1999;84(12):4362–4370. [PubMed: 10599689]
- 25.
- Thomas PQ, Dattani MT, Brickman JM, McNay D, Warne G, Zacharin M, Cameron F, Hurst J, Woods K, Dunger D, Stanhope R, Forrest S, Robinson IC, Beddington RS. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Human Molecular Genetics. 2001;10(1):39–45. [PubMed: 11136712]
- 26.
- Al Rosenbloom, Almonte AS, Brown MR, Fisher DA, Baumbach L, Parks JS. Clinical and biochemical phenotype of familial anterior hypopituitarism from mutation of the PROP1 gene. J Clin Endocrinol Metab. 1999;84(1):50–57. [PubMed: 9920061]
- 27.
- Turton JPG, Mehta A, Raza J, Woods KS, Tiulpakov A, Cassar J, Chong K, Thomas PQ, Eunice M, Ammini AC, Bouloux PM, Starzyk J, Hindmarsh PC, Dattani MT. Mutations within the transcription factor PROP1 are rare in a cohort of patients with sporadic combined pituitary hormone deficiency (CPHD). Clinical Endocrinology. 2005;63(1):10–18. [PubMed: 15963055]
- 28.
- Pfäffle RW, DiMattia GE, Parks JS, Brown MR, Wit JM, Jansen M, Van der Nat H, Van den Brande JL, Rosenfeld MG, Ingraham HA. Mutation of the POU-specific domain of Pit-1 and hypopituitarism without pituitary hypoplasia. Science (New York, NY). 1992;257(5073):1118–1121. [PubMed: 1509263]
- 29.
- Turton JPG, Reynaud R, Mehta A, Torpiano J, Saveanu A, Woods KS, Tiulpakov A, Zdravkovic V, Hamilton J, Attard-Montalto S, Parascandalo R, Vella C, Clayton PE, Shalet S, Barton J, Brue T, Dattani MT. Novel Mutations within the POU1F1 Gene Associated with Variable Combined Pituitary Hormone Deficiency. The Journal of Clinical Endocrinology & Metabolism. 2005;90(8):4762–4770. [PubMed: 15928241]
- 30.
- Dattani MT. Growth hormone deficiency and combined pituitary hormone deficiency: does the genotype matter? Clinical Endocrinology. 2005;63(2):121–130. [PubMed: 16060904]
- 31.
- Rizzoti K, Brunelli S, Carmignac D, Thomas PQ, Robinson IC, Lovell-Badge R. SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat Genet. 2004;36(3):247–255. [PubMed: 14981518]
- 32.
- Foster JW, Graves JA. An SRY-related sequence on the marsupial X chromosome: implications for the evolution of the mammalian testis-determining gene. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(5):1927–1931. [PMC free article: PMC43277] [PubMed: 8127908]
- 33.
- Stevanović M, Lovell-Badge R, Collignon J, Goodfellow PN. SOX3 is an X-linked gene related to SRY. Hum Mol Genet. 1993;2(12):2013–2018. [PubMed: 8111369]
- 34.
- Collignon J, Sockanathan S, Hacker A, Cohen-Tannoudji M, Norris D, Rastan S, Stevanovic M, Goodfellow PN, Lovell-Badge R. A comparison of the properties of Sox-3 with Sry and two related genes, Sox-1 and Sox-2. Development. 1996;122(2):509–520. [PubMed: 8625802]
- 35.
- Laumonnier F, Ronce N, Hamel BCJ, Thomas P, Lespinasse J, Raynaud M, Paringaux C, van Bokhoven H, Kalscheuer V, Fryns J-P, Chelly J, Moraine C, Briault S. Transcription Factor SOX3 Is Involved in X-Linked Mental Retardation with Growth Hormone Deficiency. The American Journal of Human Genetics. 2002;71(6):1450–1455. [PMC free article: PMC420004] [PubMed: 12428212]
- 36.
- Woods KS, Cundall M, Turton J, Rizotti K, Mehta A, Palmer R, Wong J, Chong WK, Al-Zyoud M, El-Ali M, Otonkoski T, Martinez-Barbera J-P, Paul Thomas Q, Iain Robinson C, Lovell-Badge R, Karen Woodward J, Mehul Dattani T. Over- and Underdosage of SOX3 Is Associated with Infundibular Hypoplasia and Hypopituitarism. The American Journal of Human Genetics. 2005;76(5):833–849. [PMC free article: PMC1199372] [PubMed: 15800844]
- 37.
- Goosens M, Brauner R, Czernichow P, Duquesnoy P, Rapaport R. Isolated growth hormone (GH) deficiency type 1A associated with a double deletion in the human GH gene cluster. Journal of Clinical Endocrinology and Metabolism. 1986;62(4):712–716. [PubMed: 3005356]
- 38.
- Wagner JK, Eblé A, Hindmarsh PC, Mullis PE. Prevalence of human GH-1 gene alterations in patients with isolated growth hormone deficiency. Pediatric Research. 1998;43(1):105–110. [PubMed: 9432120]
- 39.
- Wajnrajch MP, Gertner JM, Harbison MD, Chua SC, Leibel RL. Nonsense mutation in the human growth hormone releasing hormone receptor causes growth failure analogous to the little (lit) mouse. Nature Genetics. 1996;12:88–90. [PubMed: 8528260]
- 40.
- Binder G, Nagel BH, Ranke MB, Mullis PE. Isolated GH deficiency (IGHD) type II: imaging of the pituitary gland by magnetic resonance reveals characteristic differences in comparison with severe IGHD of unknown origin. Eur J Endocrinol. 2002;147(6):755–760. [PubMed: 12457450]
- 41.
- Mullis PE, Robinson ICAF, Salemi S, Eble A, Besson A, Vuissoz J-M, Deladoey J, Simon D, Czernichow P, Binder G. Isolated Autosomal Dominant Growth Hormone Deficiency: An Evolving Pituitary Deficit? A Multicenter Follow-Up Study. J Clin Endocrinol Metab. 2005;90(4):2089–2096. [PubMed: 15671105]
- 42.
- Cogan JD, Ramel B, Lehto M, Phillips J, Prince M, Blizzard RM, de Ravel TJ, Brammert M, Groop L. A recurring dominant negative mutation causes autosomal dominant growth hormone deficiency--a clinical research center study. The Journal of Clinical Endocrinology and Metabolism. 1995;80(12):3591–3595. [PubMed: 8530604]
- 43.
- Takahashi Y, Shirono H, Arisaka O, Takahashi K, Yagi T, Koga J, Kaji H, Okimura Y, Abe H, Tanaka T, Chihara K. Biologically inactive growth hormone caused by an amino acid substitution. Journal of Clinical Investigation. 1997;100(5):1159–1165. [PMC free article: PMC508291] [PubMed: 9276733]
- 44.
- McCarthy EM, Phillips JA. Characterization of an intron splice enhancer that regulates alternative splicing of human GH pre-mRNA. Human Molecular Genetics. 1998;7(9):1491–1496. [PubMed: 9700205]
- 45.
- Cogan JD, Prince MA, Lekhakula S, Bundey S, Futrakul A, McCarthy EM, Phillips JA. A novel mechanism of aberrant pre-mRNA splicing in humans. Human Molecular Genetics. 1997;6(6):909–912. [PubMed: 9175738]
- 46.
- Hayashi Y, Yamamoto M, Ohmori S, Kamijo T, Ogawa M, Seo H. Inhibition of growth hormone (GH) secretion by a mutant GH-I gene product in neuroendocrine cells containing secretory granules: An implication for isolated GH deficiency inherited in an autosomal dominant manner. Journal of Clinical Endocrinology and Metabolism. 1999;84(6):2134–2139. [PubMed: 10372722]
- 47.
- Kamijo T, Hayashi Y, Seo H, Ogawa M. Hereditary isolated growth hormone deficiency caused by GH1 gene mutations in Japanese patients. Growth Horm IGF Res. 1999;9 Suppl B:31–34. [PubMed: 10549303]
- 48.
- Lee MS, Wajnrajch MP, Kim SS, Plotnick LP, Wang J, Gertner JM, Leibel RL, Dannies PS. Autosomal dominant growth hormone (GH) deficiency type II: the Del32-71-GH deletion mutant suppresses secretion of wild-type GH. Endocrinology. 2000;141(3):883–890. [PubMed: 10698162]
- 49.
- Vivenza D, Guazzarotti L, Godi M, Frasca D, di Natale B, Momigliano-Richiardi P, Bona G, Giordano M. A Novel Deletion in the GH1 Gene Including the IVS3 Branch Site Responsible for Autosomal Dominant Isolated Growth Hormone Deficiency. The Journal of Clinical Endocrinology & Metabolism. 2006;91(3):980–986. [PubMed: 16368751]
- 50.
- Fintini D, Salvatori R, Salemi S, Otten B, Ubertini G, Cambiaso P, Mullis PE. Autosomal-dominant isolated growth hormone deficiency (IGHD type II) with normal GH-1 gene. Hormone Research. 2006;65(2):76–82. [PubMed: 16424673]
- 51.
- Veldhuis JD, Roemmich JN, Rogol AD. Gender and sexual maturation-dependent contrasts in the neuroregulation of growth hormone secretion in prepubertal and late adolescent males and females--a general clinical research center-based study. The Journal of Clinical Endocrinology and Metabolism. 2000;85(7):2385–2394. [PubMed: 10902783]
- 52.
- van den Berg G, Veldhuis JD, Frölich M, Roelfsema F. An amplitude-specific divergence in the pulsatile mode of growth hormone (GH) secretion underlies, the gender difference in mean GH concentrations in men and premenapausal women. Journal of Clinical Endocrinology and Metabolism. 1996;81(7):2460–2467. [PubMed: 8675561]
- 53.
- Roemmich JN, Clark PA, Weltman A, Veldhuis JD, Rogol AD. Pubertal alterations in growth and body composition: IX. Altered spontaneous secretion and metabolic clearance of growth hormone in overweight youth. Metabolism: Clinical and Experimental. 2005;54(10):1374–1383. [PubMed: 16154439]
- 54.
- Rivier J, Spiess J, Thorner M, Vale W. Characterization of a growth hormone-releasing factor from a human pancreatic islet tumour. Nature. 1982;300(5889):276–278. [PubMed: 6292724]
- 55.
- Gonzalez-Crespo S, Boronat A. Expression of the rat growth hormone-releasing hormone gene in placenta is directed by an alternative promoter. Proc Natl Acad Sci U S A. 1991;88(19):8749–8753. [PMC free article: PMC52587] [PubMed: 1924334]
- 56.
- Rogol AD, Blizzard RM, Foley TPJ, Furlanetto R, Selden R, Mayo K, Thorner MO. Growth hormone releasing hormone and growth hormone: genetic studies in familial growth hormone deficiency. Pediatric Research. 1985;19:489–492. [PubMed: 3923425]
- 57.
- Mayo KE. Molecular cloning and expression of a pituitary-specific receptor for growth hormone-releasing hormone. Molecular Endocrinology. 1992;6:1734–1744. [PubMed: 1333056]
- 58.
- Godfrey P, Rahal JO, Beamer WG, Copeland NG, Jenkins NA, Mayo KE. GHRH receptor of little mice contains a missense mutation in the extracellular domain that disrupts receptor function. Nat Genet. 1993;4(3):227–232. [PubMed: 8395283]
- 59.
- Lin SC, Lin CR, Gukovsky I, Lusis AJ, Sawchenko PE, Rosenfeld MG. Molecular basis of the little mouse phenotype and implications for cell type-specific growth. Nature. 1993;364(6434):208–213. [PubMed: 8391647]
- 60.
- Wajnrajch MP, Chua SC, Green ED, Leibel RL. Human growth hormone-releasing hormone receptor (GHRHR) maps to a YAC at Chromosome 7p15. Mammalian Genome. 1994;5(9):595. [PubMed: 8000149]
- 61.
- Baumann G, Maheshwari H. The dwarfs of Sindh: severe growth hormone (GH) deficiency caused by a mutation in the GH-releasing hormone receptor gene. Acta Pediatr Suppl. 1997;423:33–38. [PubMed: 9401536]
- 62.
- Netchine I, Talon P, Dastot F, Vitaux F, Goosens M, Amselem S. Extensive phenotypic analysis of a family with growth hormone (GH) deficiency caused by a mutation in the GH-releasing hormone receptor gene. Journal of Clinical Endocrinology and Metabolism. 1998;83(2):432–436. [PubMed: 9467553]
- 63.
- Salvatori R, Hayashida CY, Aguiar-Oliveira MH, Phillips JA, Souza AH, Gondo RG, Toledo SP, Conceicão MM, Prince M, Maheshwari HG, Baumann G, Levine MA. Familial dwarfism due to a novel mutation of the growth hormone-releasing hormone receptor gene. The Journal of Clinical Endocrinology and Metabolism. 1999;84(3):917–923. [PubMed: 10084571]
- 64.
- Wajnrajch MP, Gertner JM, Harbison MD, Netchine I, Maheshwari HG, Baumann G, Amselem S, Goldstein DB, Leibel RL. Haplotype analysis of the growth hormone releasing hormone receptor locus in three apparently unrelated kindreds from the Indian subcontinent with the identical mutation in the GHRH receptor. American Journal of Medical Genetics. 2003;120A:77–83. [PubMed: 12794696]
- 65.
- Desai MP, Upadhye PS, Kamijo T, Yamamoto M, Ogawa M, Hayashi Y, Seo H, Nair SR. Growth hormone releasing hormone receptor (GHRH-r) gene mutation in Indian children with familial isolated growth hormone deficiency: a study from western India. J Pediatr Endocrinol Metab. 2005;18(10):955–973. [PubMed: 16355809]
- 66.
- Roelfsema F, Biermasz NR, Veldman RG, Veldhuis JD, Frolich M, Stovkis-Brantsma WH, Wit J-M. Growth hormone (GH) secretion in patients with an inactivating defect of the GH-releasing hormone (GHRH) receptor is pulsatile: evidence for a role for non-GHRH inputs into the generation of GH pulses. J Clin Endocrinol Metab. 2001;86(6):2459–2464. [PubMed: 11397840]
- 67.
- MacIntyre I, Szelke M, Royal Postgraduate Medical S, Bowers CY, Chang J, Momany FA, Folkers K. Effects of the enkephalins and enkephalin-analog on release of pituitary hormones in vitro. Molecular endocrinology: proceedings of Endocrinology '77 held at the Royal College of Physicians, London, England on 11-15 July, 1977. New York: Elsevier/North-Holland Biomedical Press %@ 9780444800350%L QP187.A1 M65; 1977.
- 68.
- Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402(6762):656–660. [PubMed: 10604470]
- 69.
- Wajnrajch MP, Gertner JM, Mullis PE, Cogan JD, Dannies PS, Kim S, Saenger P, Moshang T, Phillips JA III, Leibel RL. Arg183His, a New Mutational Hot-Spot in the Growth Hormone Gene Causing Isolated GH Deficiency Type II. Journal of Endocrine Genetics. 2000;1(3):124–134.
- 70.
- Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, Hamelin M, Hreniuk DL, Palyha OC, Anderson J, Paress PS, Diaz C, Chou M, Liu KK, McKee KK, Pong SS, Chaung LY, Elbrecht A, Dashkevicz M, Heavens R, Rigby M, Sirinathsinghji DJS, Dean DC, Melillo D, Patchett AA, Nargund R, Griffin PR, DeMartino JA, Gupta SK, Schaeffer JM, Smith RG, Van der Ploeg LHT. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science. 1996;273:974–977. [PubMed: 8688086]
- 71.
- Takaya K, Ariyasu H, Kanamoto N, Iwakura H, Yoshimoto A, Harada M, Mori K, Komatsu Y, Usui T, Shimatsu A, Ogawa Y, Hosoda K, Akamizu T, Kojima M, Kangawa K, Nakao K. Ghrelin Strongly Stimulates Growth Hormone Release in Humans. J Clin Endocrinol Metab. 2000;85(12):4908–4911. [PubMed: 11134161]
- 72.
- Bercu B, Walker RF. Novel Growth Hormone Secretagogues: Clinical Applications. Endocrinologist. 1997;7(1):51–64.
- 73.
- Giustina A, Bonfanti C, Licini M, Ragni G, Stefana B. Hexarelin, a novel GHRP-6 analog, stimulates growth hormone (GH) release in a GH-secreting rat cell line (GH1) insensitive to GH-releasing hormone. Rugul Pept. 1997;70(1):49–54. [PubMed: 9250581]
- 74.
- Tolis G, Karydis I, Markousis V, Karagiorga M, Mesimeris T, Lenaerts V, Deghenghi R. Growth hormone release by the novel GH releasing peptide Hexarelin in patients with homozygous b-Thalassemia. Journal of Pediatric Endocrinology and Metabolism. 1997;10(1):35–40. [PubMed: 9364340]
- 75.
- Hataya Y, Akamizu T, Takaya K, Kanamoto N, Ariyasu H, Saijo M, Moriyama K, Shimatsu A, Kojima M, Kangawa K, Nakao K. A low dose of ghrelin stimulates growth hormone (gh) release synergistically with gh-releasing hormone in humans. J Clin Endocrinol Metab. 2001;86(9):45524555. [PubMed: 11549707]
- 76.
- Arvat E, Di Vito L, Broglio F, Papotti M, Muccioli G, Dieguez C, Casanueva F, Deghenghi R, Camanni F, Ghigo E. Preliminary evidence that Ghrelin, the natural GH secretagogue (GHS)-receptor ligand, strongly stimulates GH secretion in humans. J Endocrinol Invest. 2000;23(8):493–495. [PubMed: 11021763]
- 77.
- Ghizzoni L, Mastorakos G, Vottero A, Ziveri M, Ilias I, Bernasconi S. Spontaneous growth hormone (GH) secretion is not directly affected by ghrelin in either short normal prepubertal children or children with GH neurosecretory dysfunction. The Journal of Clinical Endocrinology and Metabolism. 2004;89(11):5488–5495. [PubMed: 15531502]
- 78.
- Racine MS, Symons KV, Foster CM, Barkan AL. Augmentation of growth hormone secretion after testosterone treatment in boys with constitutional delay of growth and adolescence: evidence against an increase in hypothalamic secretion of growth hormone-releasing hormone. The Journal of Clinical Endocrinology and Metabolism. 2004;89(7):3326–3331. [PubMed: 15240610]
- 79.
- Hirsh D, Heinrichs C, Leenders B, Wong ACK, Cummings DE, Chanoine J-P. Ghrelin is suppressed by glucagon and does not mediate glucagon-related growth hormone release. Hormone Research. 2005;63(3):111–118. [PubMed: 15775713]
- 80.
- Zhang JV, Ren P-G, Avsian-Kretchmer O, Luo C-W, Rauch R, Klein C, Hsueh AJW. Obestatin, a Peptide Encoded by the Ghrelin Gene, Opposes Ghrelin's Effects on Food Intake. Science. 2005;310(5750):996–999. [PubMed: 16284174]
- 81.
- Ukkola O, Ravussin E, Jacobson P, Snyder EE, Chagnon M, Sjöström L, Bouchard C. Mutations in the Preproghrelin/Ghrelin Gene Associated with Obesity in Humans. J Clin Endocrinol Metab. 2001;86(8):3996–3999. [PubMed: 11502844]
- 82.
- Pantel J, Legendre M, Cabrol S, Hilal L, Hajaji Y, Morisset S, Nivot S, Vie-Luton M-P, Grouselle D, de Kerdanet M, Kadiri A, Epelbaum J, Le Bouc Y, Amselem S. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J Clin Invest. 2006;116:760–768. [PMC free article: PMC1386106] [PubMed: 16511605]
- 83.
- Grossman A, Savage MO, Lytras N, Preece MA, Sueiras-Diaz J, Coy DH, Rees LH, Besser GM. Responses to analogues of growth hormone-releasing hormone in normal subjects, and in growth-hormone deficient children and young adults. Clin Endocrinol (Oxf). 1984;21(3):321–330. [PubMed: 6236914]
- 84.
- Date Y, Murakami N, Kojima M, T. K, Matsukura S, Kangawa K, Nakazato M. Central effects of a novel acylated peptide, ghrelin, on growth hormone release in rats. Biochem Biophys Res Commun. 2000;275(2):477–480. [PubMed: 10964690]
- 85.
- Shen LP, Pictet RL, Rutter WJ. Human somatostatin I: sequence of the cDNA. Proc Natl Acad Sci USA. 1982;79(15):4575–4579. [PMC free article: PMC346717] [PubMed: 6126875]
- 86.
- Rosskopf D, Schurks M, Manthey I, Joisten M, Busch S, Siffert W. Signal transduction of somatostatin in human B lymphoblasts. Am J Physiol Cell Physiol. 2003;284(1):C179–190. [PubMed: 12388115]
- 87.
- Laron Z. Prismatic cases: Laron syndrome (primary growth hormone resistance) from patient to laboratory to patient. The Journal of Clinical Endocrinology and Metabolism. 1995;80(5):1526–1531. [PubMed: 7744997]
- 88.
- Dastot F, Sobrier ML, Duquesnoy P, Duriez B, Goossens M, Amselem S. Alternatively spliced forms in the cytoplasmic domain of the human growth hormone (GH) receptor regulate its ability to generate a soluble GH-binding protein. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(20):10723–10728. [PMC free article: PMC38222] [PubMed: 8855247]
- 89.
- Dos Santos C, Essioux L, Teinturier C, Tauber M, Goffin V, Bougneres P. A common polymorphism of the growth hormone receptor is associated with increased responsiveness to growth hormone. Nat Genet. 2004;36(7):720–724. [PubMed: 15208626]
- 90.
- Binder G, Baur F, Schweizer R, Ranke MB. The d3-Growth Hormone (GH) Receptor Polymorphism Is Associated with Increased Responsiveness to GH in Turner Syndrome and Short Small-for-Gestational-Age Children. J Clin Endocrinol Metab. 2006;91(2):659–664. [PubMed: 16291706]
- 91.
- Jorge AAL, Marchisotti FG, Montenegro LR, Carvalho LR, Mendonca BB, Arnhold IJP. Growth hormone (GH) pharmacogenetics: influence of GH receptor exon 3 retention or deletion on first-year growth response and final height in patients with severe GH deficiency. The Journal of Clinical Endocrinology and Metabolism. 2006;91(3):1076–1080. [PubMed: 16291702]
- 92.
- Pilotta A, Mella P, Filisetti M, Felappi B, Prandi E, Parrinello G, Notarangelo LD, Buzi F. Common polymorphisms of the Growth Hormone (GH) Receptor do not correlate with the growth response to exogenous recombinant human GH in GH deficient children. J Clin Endocrinol Metab. 2006:jc.2005-1308. [PubMed: 16394090]
- 93.
- Rosenfeld RG, Belgorosky A, Camacho-Hubner C, Savage MO, Wit JM, Hwa V. Defects in growth hormone receptor signaling. Trends in endocrinology and metabolism. TEM. 2007;18(4):134–141. [PubMed: 17391978]
- 94.
- Savage MO, Hwa V, David A, Rosenfeld RG, Metherell LA. Genetic Defects in the Growth Hormone-IGF-I Axis Causing Growth Hormone Insensitivity and Impaired Linear Growth. Frontiers in Endocrinology. 2011;2:95. [PMC free article: PMC3356141] [PubMed: 22654835]
- 95.
- Ayling RM, Ross R, Towner P, von Laue S, Finidori J, Moutoussamy S, Buchanan CR, Clayton PE, Norman MR. A dominant-negative mutation of the growth hormone receptor causes familial short stature. Nature Genetics. 1997;16:13–14. [PubMed: 9140387]
- 96.
- Amselem S, Duquesnoy P, Attree O, Novelli G, Bousnina S, Postel-Vinay MC, Goossens M. Laron dwarfism and mutations of the growth hormone-receptor gene. N Engl J Med. 1989;321(15):989–995. [PubMed: 2779634]
- 97.
- Woods KA, Fraser NC, Postel-Vinay MC, Savage MO, Clark AJ. A homozygous splice site mutation affecting the intracellular domain of the growth hormone (GH) receptor resulting in Laron syndrome with elevated GH-binding protein. The Journal of Clinical Endocrinology and Metabolism. 1996;81(5):1686–1690. [PubMed: 8626815]
- 98.
- Woods KA, Dastot F, Preece MA, Clark AJ, Postel-Vinay MC, Chatelain PG, Ranke MB, Rosenfeld RG, Amselem S, Savage MO. Phenotype: genotype relationships in growth hormone insensitivity syndrome. The Journal of Clinical Endocrinology and Metabolism. 1997;82(11):3529–3535. [PubMed: 9360502]
- 99.
- Salerno M, Balestrieri B, Matrecano E, Officioso A, Rosenfeld RG, Di Maio S, Fimiani G, Ursini MV, Pignata C. Abnormal GH receptor signaling in children with idiopathic short stature. J Clin Endocrinol Metab. 2001;86(8):3882–3888. [PubMed: 11502828]
- 100.
- Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, Kiess W. Klammt Jr, Kratzsch Jr, Osgood D, Pfהffle R, Raile K, Seidel B, Smith RJ, Chernausek SD, Group IGRIS. IGF-I Receptor Mutations Resulting in Intrauterine and Postnatal Growth Retardation. New England Journal of Medicine. 2003;349(23):2211–2222. [PubMed: 14657428]
- 101.
- Bonioli E, Tarò M, Rosa CL, Citana A, Bertorelli R, Morcaldi G, Gastaldi R, Coviello DA. Heterozygous mutations of growth hormone receptor gene in children with idiopathic short stature. Growth hormone & IGF research: official journal of the Growth Hormone Research Society and the International IGF Research Society. 2005;15(6):405-410. [PubMed: 16213173]
- 102.
- Rosenbloom AL, Guevara Aguirre J, Rosenfeld RG. The little women of Loja--growth hormone-receptor deficiency in an inbred population of southern Ecuador. N Engl J Med. 1990;323(20):1367–1374. [PubMed: 2233903]
- 103.
- Aalbers AM, Chin D, Pratt KL, Little BM, Frank SJ, Hwa V, Rosenfeld RG. Extreme Elevation of Serum Growth Hormone-Binding Protein Concentrations Resulting from a Novel Heterozygous Splice Site Mutation of the Growth Hormone Receptor Gene. Hormone Research in Paediatrics. 2009;71(5):276–284. [PubMed: 19339792]
- 104.
- Woods KA, Camacho-Hübner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. The New England Journal of Medicine. 1996;335(18):1363–1367. [PubMed: 8857020]
- 105.
- Bonapace G, Concolino D, Formicola S, Strisciuglio P. A novel mutation in a patient with insulin-like growth factor 1 (IGF1) deficiency. Journal of Medical Genetics. 2003;40(12):913–917. [PMC free article: PMC1735341] [PubMed: 14684690]
- 106.
- Walenkamp MJE, Karperien M, Pereira AM, Hilhorst-Hofstee Y, van Doorn J, Chen JW, Mohan S, Denley A, Forbes B, van Duyvenvoorde HA, van Thiel SW, Sluimers CA, Bax JJ. de Laat JaPM, Breuning MB, Romijn JA, Wit JM. Homozygous and heterozygous expression of a novel insulin-like growth factor-I mutation. The Journal of Clinical Endocrinology and Metabolism. 2005;90(5):2855–2864. [PubMed: 15769976]
- 107.
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. 1993;75(1):59–72. [PubMed: 8402901]
- 108.
- Morison IM, Reeve AE. Insulin-like growth factor 2 and overgrowth: molecular biology and clinical implications. Mol Med Today. 1998;4(3):110–115. [PubMed: 9575493]
- 109.
- Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(13):7324–7329. [PMC free article: PMC22084] [PubMed: 10377413]
- 110.
- Sjögren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Törnell J, Isaksson OG, Jansson JO, Ohlsson C. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(12):7088–7092. [PMC free article: PMC22065] [PubMed: 10359843]
- 111.
- Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, LeRoith D. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110(6):771–781. [PMC free article: PMC151128] [PubMed: 12235108]
- 112.
- Domené HM, Bengolea SV, Martínez AS, Ropelato MG, Pennisi P, Scaglia P, Heinrich JJ, Jasper HG. Deficiency of the circulating insulin-like growth factor system associated with inactivation of the acid-labile subunit gene. The New England Journal of Medicine. 2004;350(6):570–577. [PubMed: 14762184]
- 113.
- Hwa V, Haeusler G, Pratt KL, Little BM, Frisch H, Koller D, Rosenfeld RG. Total Absence of Functional Acid Labile Subunit, Resulting in Severe Insulin-Like Growth Factor Deficiency and Moderate Growth Failure. J Clin Endocrinol Metab. 2006;91(5):1826–1831. [PubMed: 16507628]
- 114.
- Domené HM, Scaglia PA, Lteif A, Mahmud FH, Kirmani S, Frystyk J, Bedecarrás P, Gutiérrez M, Jasper HG. Phenotypic effects of null and haploinsufficiency of acid-labile subunit in a family with two novel IGFALS gene mutations. The Journal of Clinical Endocrinology and Metabolism. 2007;92(11):4444–4450. [PubMed: 17726072]
- 115.
- Domené HM, Hwa V, Jasper HG, Rosenfeld RG. Acid-labile subunit (ALS) deficiency. Best Practice & Research Clinical Endocrinology & Metabolism. 2011;25(1):101–113. [PubMed: 21396577]
- 116.
- Savage MO, Camacho-Hübner C, David A, Metherell LA, Hwa V, Rosenfeld RG, Clark AJL. Idiopathic short stature: will genetics influence the choice between GH and IGF-I therapy? European Journal of Endocrinology. 2007;157 Suppl 1:S33–37. [PubMed: 17785695]
- 117.
- Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, Hennekens CH, Pollak M. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science. 1998;279(5350):563–566. [PubMed: 9438850]
- 118.
- Pollak M. Insulin-like growth factor physiology and cancer risk. Eur J Cancer. 2000;36(10):1224–1228. [PubMed: 10882860]
- 119.
- Jernstrom H, Chu W, Vesprini D, Tao Y, Majeed N, Deal C, Pollak M, Narod SA. Genetic factors related to racial variation in plasma levels of insulin-like growth factor-1: implications for premenopausal breast cancer risk. Mol Genet Metab. 2001;72(2):144–154. [PubMed: 11161840]
- 120.
- Cohen P, Clemmons DR, Rosenfeld RG. Does the GH-IGF axis play a role in cancer pathogenesis? Growth Horm IGF Res. 2000;10(6):297–305. [PubMed: 11161960]
- 121.
- Cohen P. Overview of the IGF-I system. Hormone Research. 2006;65 Suppl 1:3–8. [PubMed: 16508327]
- 122.
- Ranke MB. Defining insulin-like growth factor-I deficiency. Hormone Research. 2006;65 Suppl 1:9–14. [PubMed: 16508328]
- 123.
- Savage MO, Camacho-Hübner C, Dunger DB. Therapeutic applications of the insulin-like growth factors. Growth hormone & IGF research: official journal of the Growth Hormone Research Society and the International IGF Research Society. 2004;14(4):301-308. [PubMed: 15231299]
- 124.
- Laron Z, Anin S, Klipper-Aurbach Y, Klinger B. Effects of insulin-like growth factor on linear growth, head circumference, and body fat in patients with Laron-type dwarfism. Lancet (London, England). 1992;339(8804):1258–1261. [PubMed: 1349669]
- 125.
- Ranke MB, Savage MO, Chatelain PG, Preece MA, Rosenfeld RG, Blum WF, Wilton P. Insulin-like growth factor I improves height in growth hormone insensitivity: two years' results. Hormone Research. 1995;44(6):253–264. [PubMed: 8808010]
- 126.
- Ranke MB, Savage MO, Chatelain PG, Preece MA, Rosenfeld RG, Wilton P. Long-term treatment of growth hormone insensitivity syndrome with IGF-I. Results of the European Multicentre Study. The Working Group on Growth Hormone Insensitivity Syndromes. Hormone Research. 1999;51(3):128–134. [PubMed: 10461018]
- 127.
- Collett-Solberg PF, Misra M. Drug, Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine S. The role of recombinant human insulin-like growth factor-I in treating children with short stature. The Journal of Clinical Endocrinology and Metabolism. 2008;93(1):10–18. [PubMed: 18165284]
- 128.
- Laron Z. Insulin-like growth factor-I (lGF-l): safety and efficacy. Pediatric endocrinology reviews: PER. 2004;2 Suppl 1:78–85. [PubMed: 16456486]
- 129.
- Backeljauw PF, Underwood LE. syndrome GCGGhi. Therapy for 6.5-7.5 years with recombinant insulin-like growth factor I in children with growth hormone insensitivity syndrome: a clinical research center study. The Journal of Clinical Endocrinology and Metabolism. 2001;86(4):1504–1510. [PubMed: 11297575]
- 130.
- Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. Embo J. 1986;5(10):2503–2512. [PMC free article: PMC1167146] [PubMed: 2877871]
- 131.
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75(1):73–82. [PubMed: 8402902]
- 132.
- Roback EW, Barakat AJ, Dev VG, Mbikay M, Chretien M, Butler MG. An infant with deletion of the distal long arm of chromosome 15 (q26.1----qter) and loss of insulin-like growth factor 1 receptor gene. Am J Med Genet. 1991;38(1):74–79. [PMC free article: PMC5493390] [PubMed: 1849352]
- 133.
- Fang P, Schwartz ID, Johnson BD, Derr MA, Roberts CT, Hwa V, Rosenfeld RG. Familial short stature caused by haploinsufficiency of the insulin-like growth factor i receptor due to nonsense-mediated messenger ribonucleic acid decay. The Journal of Clinical Endocrinology and Metabolism. 2009;94(5):1740–1747. [PubMed: 19240156]
- 134.
- Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nature Genetics. 2002;32(3):426–431. [PubMed: 12410230]
- 135.
- Eggenschwiler J, Ludwig T, Fisher P, Leighton PA, Tilghman SM, Efstratiadis A. Mouse mutant embryos overexpressing IGF-II exhibit phenotypic features of the Beckwith-Wiedemann and Simpson-Golabi-Behmel syndromes. Genes Dev. 1997;11(23):3128–3142. [PMC free article: PMC316748] [PubMed: 9389646]
- 136.
- McElreavey K, Fellous M. Sex-determining genes. Trends in Endocrinology and Metabolism. 1997;8(9):342–345. [PubMed: 18406823]
- 137.
- Fisher AM, Thomas NS, Cockwell A, Stecko O, Kerr B, Temple IK, Clayton P. Duplications of chromosome 11p15 of maternal origin result in a phenotype that includes growth retardation. Human Genetics. 2002;111(3):290–296. [PubMed: 12215843]
- 138.
- Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, Danton F, Thibaud N, Le Merrer M, Burglen L, Bertrand AM, Netchine I, Le Bouc Y. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet. 2005;37(9):1003–1007. [PubMed: 16086014]
- 139.
- Schönherr N, Meyer E, Eggermann K, Ranke MB, Wollmann HA, Eggermann T. (Epi)mutations in 11p15 significantly contribute to Silver-Russell syndrome: but are they generally involved in growth retardation? European Journal of Medical Genetics. 2006;49(5):414–418. [PubMed: 16603426]
- 140.
- Hannula K, Lipsanen-Nyman M, Kontiokari T, Kere J. A narrow segment of maternal uniparental disomy of chromosome 7q31-qter in Silver-Russell syndrome delimits a candidate gene region. American Journal of Human Genetics. 2001;68(1):247–253. [PMC free article: PMC1234921] [PubMed: 11112662]
- 141.
- Murphy R, Baptista J, Holly J, Umpleby AM, Ellard S, Harries LW, Crolla J, Cundy T, Hattersley AT. Severe intrauterine growth retardation and atypical diabetes associated with a translocation breakpoint disrupting regulation of the insulin-like growth factor 2 gene. The Journal of Clinical Endocrinology and Metabolism. 2008;93(11):4373–4380. [PubMed: 18728168]
- 142.
- Valhmu WB, Palmer GD, Rivers PA, Ebara S, Cheng JF, Fischer S, Ratcliffe A. Structure of the human aggrecan gene: exon-intron organization and association with the protein domains. Biochemical Journal. 1995;309(2):535–542. [PMC free article: PMC1135764] [PubMed: 7626017]
- 143.
- Kiani C, Chen L, Wu YJ, Yee AJ, Yang BB. Structure and function of aggrecan. Cell Research. 2002;12:19. [PubMed: 11942407]
- 144.
- Kawaguchi Y, Osada R, Kanamori M, Ishihara H, Ohmori K, Matsui H, Kimura T. Association between an aggrecan gene polymorphism and lumbar disc degeneration. Spine. 1999;24:2456–2460. [PubMed: 10626307]
- 145.
- Gleghorn L, Ramesar R, Beighton P, Wallis G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am J Hum Genet. 2005;77:484–490. [PMC free article: PMC1226213] [PubMed: 16080123]
- 146.
- Tompson SW, Merriman B, Funari VA, Fresquet M, Lachman RS, Rimoin DL, Nelson SF, Briggs MD, Cohn DH, Krakow D. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am J Hum Genet. 2009;84:72–79. [PMC free article: PMC2668039] [PubMed: 19110214]
- 147.
- Stattin E-L, Wiklund F, Lindblom K, Önnerfjord P, Jonsson B-A, Tegner Y, Sasaki T, Struglics A, Lohmander S, Dahl N, Heinegård D, Aspberg A. A Missense Mutation in the Aggrecan C-type Lectin Domain Disrupts Extracellular Matrix Interactions and Causes Dominant Familial Osteochondritis Dissecans. American Journal of Human Genetics. 2010;86(2):126–137. [PMC free article: PMC2820178] [PubMed: 20137779]
- 148.
- Nilsson O, Guo MH, Dunbar N, Popovic J, Flynn D, Jacobsen C, Lui JC, Hirschhorn JN, Baron J, Dauber A. Short Stature, Accelerated Bone Maturation, and Early Growth Cessation Due to Heterozygous Aggrecan Mutations. The Journal of Clinical Endocrinology and Metabolism. 2014;99(8):E1510–E1518. [PMC free article: PMC4121031] [PubMed: 24762113]
- 149.
- Quintos JB, Guo MH, Dauber A. Idiopathic short stature due to novel heterozygous mutation of the aggrecan gene. J Pediatr Endocrinol Metab. 2015. Preprint. [PMC free article: PMC4501863] [PubMed: 25741789]
- 150.
- Gkourogianni A, Andrew M, Tyzinski L, Crocker M, Douglas J, Dunbar N, Fairchild J, Funari MFA, Heath KE, Jorge AAL, Kurtzman T, LaFranchi S, Lalani S, Lebl J, Lin Y, Los E, Newbern D, Nowak C, Olson M, Popovic J, Průhová Š, Elblova L, Quintos JB, Segerlund E, Sentchordi L, Shinawi M, Stattin E-L, Swartz J. Angel AGd, Cuéllar SD, Hosono H, Sanchez-Lara PA, Hwa V, Baron J, Nilsson O, Dauber A. Clinical Characterization of Patients With Autosomal Dominant Short Stature due to Aggrecan Mutations. The Journal of Clinical Endocrinology & Metabolism. 2017;102(2):460–469. [PMC free article: PMC5413162] [PubMed: 27870580]
- 151.
- van der Steen M, Pfundt R, Maas SJWH, Bakker-van Waarde WM, Odink RJ, Hokken-Koelega ACS. ACAN gene mutations in short children born SGA and response to growth hormone treatment. J Clin Endo Metab. 2017;102:1458–1467. [PubMed: 27710243]
- 152.
- Dateki S, Nakatomi A, Watanabe S, Shimizu H, Inoue Y, Baba H, Yoshiura K, Moriuchi H. Identification of a novel heterozygous mutation of the aggrecan gene in a family with idiopathic short stature and multiple intervertebral disc herniation. J Hum Genet. 2017;62:717–721. [PubMed: 28331218]
- 153.
- Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, Muroya K, Binder G, Kirsch S, Winkelmann M, Nordsiek G, Heinrich U, Breuning MH, Ranke MB, Rosenthal A, Ogata T, Rappold GA. Pseudoautosomal deletions encompassing a novel homeobox gene causes growth failure in idiopathic short stature and Turner syndrome. Nature Genetics. 1997;16:54–62. [PubMed: 9140395]
- 154.
- Huber C, Rosilio M, Munnich A, Cormier-Daire V. High incidence of SHOX anomalies in individuals with short stature. Journal of Medical Genetics. 2006;43(9):735–739. [PMC free article: PMC2564573] [PubMed: 16597678]
- 155.
- Jorge AAL, Nishi MY, Funari MFA, Souza SC, Arnhold IJP, Mendonça BB. Arquivos Brasileiros De Endocrinologia E Metabologia. 2008;52(5):765–773. [Short stature caused by SHOX gene haploinsufficiency: from diagnosis to treatment] [PubMed: 18797583]
- 156.
- Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Hormone Research in Paediatrics. 2011;75(2):81–89. [PubMed: 21325865]
- 157.
- Munns CJF, Haase HR, Crowther LM, Hayes MT, Blaschke R, Rappold G, Glass IA, Batch JA. Expression of SHOX in human fetal and childhood growth plate. The Journal of Clinical Endocrinology and Metabolism. 2004;89(8):4130–4135. [PubMed: 15292358]
- 158.
- Rappold G, Blum WF, Shavrikova EP, Crowe BJ, Roeth R, Quigley CA, Ross JL, Niesler B. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. Journal of Medical Genetics. 2007;44(5):306–313. [PMC free article: PMC2597980] [PubMed: 17182655]
- 159.
- Blum WF, Crowe BJ, Quigley CA, Jung H, Cao D, Ross JL, Braun L, Rappold G. Growth Hormone Is Effective in Treatment of Short Stature Associated with Short Stature Homeobox-Containing Gene Deficiency: Two-Year Results of a Randomized, Controlled, Multicenter Trial. The Journal of Clinical Endocrinology & Metabolism. 2007;92(1):219–228. [PubMed: 17047016]
- 160.
- Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA. The natural history of Noonan syndrome: a long-term follow-up study. Archives of Disease in Childhood. 2007;92(2):128–132. [PMC free article: PMC2083343] [PubMed: 16990350]
- 161.
- Allanson JE. Noonan syndrome. Journal of Medical Genetics. 1987;24(1):9–13. [PMC free article: PMC1049850] [PubMed: 3543368]
- 162.
- Allanson JE, Hall JG, Hughes HE, Preus M, Witt RD. Noonan syndrome: the changing phenotype. American Journal of Medical Genetics. 1985;21(3):507–514. [PubMed: 4025385]
- 163.
- Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nature Genetics. 2001;29(4):465–468. [PubMed: 11704759]
- 164.
- Gelb BD, Tartaglia M. Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Human Molecular Genetics. 2006;15(Spec No 2):R220–226. [PubMed: 16987887]
- 165.
- Ferreira LV, Souza SAL, Arnhold IJP, Mendonca BB, Jorge AAL. PTPN11 (protein tyrosine phosphatase, nonreceptor type 11) mutations and response to growth hormone therapy in children with Noonan syndrome. The Journal of Clinical Endocrinology and Metabolism. 2005;90(9):5156–5160. [PubMed: 15956085]
- 166.
- Limal J-M, Parfait B, Cabrol S, Bonnet D, Leheup B, Lyonnet S, Vidaud M, Le Bouc Y. Noonan syndrome: relationships between genotype, growth, and growth factors. The Journal of Clinical Endocrinology and Metabolism. 2006;91(1):300–306. [PubMed: 16263833]
- 167.
- Raaijmakers R, Noordam C, Karagiannis G, Gregory JW, Hertel NT, Sipilä I, Otten BJ. Response to growth hormone treatment and final height in Noonan syndrome in a large cohort of patients in the KIGS database. Journal of pediatric endocrinology & metabolism. JPEM. 2008;21(3):267–273. [PubMed: 18540254]
- 168.
- Noordam C, Peer PGM, Francois I, De Schepper J, van den Burgt I, Otten BJ. Long-term GH treatment improves adult height in children with Noonan syndrome with and without mutations in protein tyrosine phosphatase, non-receptor-type 11. European Journal of Endocrinology. 2008;159(3):203–208. [PubMed: 18562489]
- 169.
- Growth Hormone Research S. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. The Journal of Clinical Endocrinology and Metabolism. 2000;85(11):3990–3993. [PubMed: 11095419]
- 170.
- Kuczmarski RJ, Ogden C, Grummer-Strawn LM, Guo SS, Wei R, Mei Z, Curtin LR, Roche AF, Johnson CL. CDC Growth Charts: United States. Hyattsville: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics; 2000:1-28.
- 171.
- Sizonenko PC, Clayton PE, Cohen P, Hintz RL, Tanaka T, Laron Z. Diagnosis and management of growth hormone deficiency in childhood and adolescence. Part 1: diagnosis of growth hormone deficiency. Growth hormone & IGF research: official journal of the Growth Hormone Research Society and the International IGF Research Society. 2001;11(3):137-165. [PubMed: 11735230]
- 172.
- Gruelich WW, Pyle SI. Radiographic Atlas of Skeletal Development of the Hand and Wrist. Stanford University Press.
- 173.
- Tanner JM. Assessment of skeletal maturity and prediction of adult height (TW2 method). London ; New York: Academic Press.
- 174.
- Bayley N. Tables for predicting adult height from present height and skeletal age. J Pediat. 1946;28:49. [PubMed: 21012523]
- 175.
- Rosenfield R, Cutler L, Jameson JL, DeGroot L, Burger H. Somatic Growth and Maturation. Endocrinology. Philadelphia: W.B. Saunders Co.; 2001:477-502%@ 0721678408.
- 176.
- Underwood LE, Van Wyk JJ. Normal and aberrant growth. Williams textbook of endocrinology. Philadelphia: W.B. Saunders Co.; 2003:1079-1138.
- 177.
- Bayley N, Pinneau SR. Tables for predicting adult height from skeletal age: revised for use with the Greulich-Pyle hand standards. J Pediatr. 1952;40(4):423–441. [PubMed: 14918032]
- 178.
- Roche AF, Wainer H, Thissen D. The RWT method for the prediction of adult stature. Pediatrics. 1975;56(6):1026–1033. [PubMed: 172855]
- 179.
- Zachmann M, Sobradillo B, Frank M, Frisch H, Prader A. Bayley-Pinneau, Roche-Wainer-Thissen, and Tanner height predictions in normal children and in patients with various pathologic conditions. The Journal of Pediatrics. 1978;93(5):749–755. [PubMed: 712475]
- 180.
- Marin G, Domené HM, Barnes KM, Blackwell BJ, Cassorla FG, Cutler GB. The effects of estrogen priming and puberty on the growth hormone response to standardized treadmill exercise and arginine-insulin in normal girls and boys. The Journal of Clinical Endocrinology and Metabolism. 1994;79(2):537–541. [PubMed: 8045974]
- 181.
- Molina S, Paoli M, Camacho N, Arata-Bellabarba G, Lanes R. Is testosterone and estrogen priming prior to clonidine useful in the evaluation of the growth hormone status of short peripubertal children? Journal of pediatric endocrinology & metabolism. JPEM. 2008;21(3):257–266. [PubMed: 18540253]
- 182.
- Wilson TA, Rose SR, Cohen P, Rogol AD, Backeljauw P, Brown R, Hardin DS, Kemp SF, Lawson M, Radovick S, Rosenthal SM, Silverman L, Speiser P. Update of guidelines for the use of growth hormone in children: The lawson wilkins pediatric endocrinology society drug and therapeutics committee. Journal of Pediatrics. 2003;143(4):415–421. [PubMed: 14571209]
- 183.
- Cohen P, Rogol AD, Howard CP, Bright GM, Kappelgaard A-M, Rosenfeld RG. on behalf of the American Norditropin Study G. Insulin Growth Factor-Based Dosing of Growth Hormone Therapy in Children: A Randomized, Controlled Study. J Clin Endocrinol Metab. 2007;92(7):2480–2486. [PubMed: 17356043]
- 184.
- Baron J. Growth Hormone Therapy in Childhood: Titration Versus Weight-Based Dosing? J Clin Endocrinol Metab. 2007;92(7):2436–2438. [PubMed: 17616638]
- 185.
- Blethen SL, Allen DB, Graves D, August G, Moshang T, Rosenfeld R. Safety of recombinant deoxyribonucleic acid-derived growth hormone: The National Cooperative Growth Study Experience. Journal of Clinical Endocrinology and Metabolism. 1996;81(5):1704–1710. [PubMed: 8626820]
- 186.
- Root AW, Kemp SF, Rundle AC, Dana K, Attie KM. Effect of long-term recombinant growth hormone therapy in children-The National Cooperative Growth Study, USA 1985-1994. Journal of Pediatric Endocrinology and Metabolism. 1998;11(3):403–412. [PubMed: 11517956]
- 187.
- Faglia G, Arosio M, Porretti S. Delayed closure of epiphyseal cartilages induced by the aromatase inhibitor anastrozole. Would it help short children grow up? J Endocrinol Invest. 2000;23(11):721–723. [PubMed: 11194703]
- 188.
- Dunkel L, Wickman S. Novel treatment of delayed male puberty with aromatase inhibitors. Horm Res. 2002;57 Suppl 2:44–52. [PubMed: 12065927]
- 189.
- Zung A, Zadik Z. New approaches in the treatment of short stature. Harefuah. 2002;141(12):1059–1065. [PubMed: 12534205]
- 190.
- Hero M, Norjavaara E, Dunkel L. Inhibition of Estrogen Biosynthesis with a Potent Aromatase Inhibitor Increases Predicted Adult Height in Boys with Idiopathic Short Stature: A Randomized Controlled Trial. J Clin Endocrinol Metab. 2005;90(12):6396–6402. [PubMed: 16189252]
- 191.
- Wassenaar MJ, Dekkers OM, Pereira AM, Wit JM, Smit JW, Biermasz NR, Romijn JA. Impact of the exon 3-deleted growth hormone (GH) receptor polymorphism on baseline height and the growth response to recombinant human GH therapy in GH-deficient (GHD) and non-GHD children with short stature: a systematic review and meta-analysis. J Clin Endocrinol Metab. 2009;94(10):3721–3730. [PubMed: 19584188]
- 192.
- Jorge AA, Marchisotti FG, Montenegro LR, Carvalho LR, Mendonca BB, Arnhold IJ. Growth hormone (GH) pharmacogenetics: influence of GH receptor exon 3 retention or deletion on first-year growth response and final height in patients with severe GH deficiency. J Clin Endocrinol Metab. 2006;91(3):1076–1080. [PubMed: 16291702]
- 193.
- Binder G, Baur F, Schweizer R, Ranke MB. The d3-growth hormone (GH) receptor polymorphism is associated with increased responsiveness to GH in Turner syndrome and short small-for-gestational-age children. J Clin Endocrinol Metab. 2006;91(2):659–664. [PubMed: 16291706]
- 194.
- Tauber M, Ester W, Auriol F, Molinas C, Fauvel J, Caliebe J, Nugent T, Fryklund L, Ranke MB, Savage MO, Clark AJ, Johnston LB, Hokken-Koelega AC. GH responsiveness in a large multinational cohort of SGA children with short stature (NESTEGG) is related to the exon 3 GHR polymorphism. Clin Endocrinol (Oxf). 2007;67(3):457–461. [PMC free article: PMC2040241] [PubMed: 17555507]
- 195.
- Braz AF, Costalonga EF, Montenegro LR, Trarbach EB, Antonini SR, Malaquias AC, Ramos ES, Mendonca BB, Arnhold IJ, Jorge AA. The interactive effect of GHR-exon 3 and -202 A/C IGFBP3 polymorphisms on rhGH responsiveness and treatment outcomes in patients with Turner syndrome. J Clin Endocrinol Metab. 2012;97(4):E671–677. [PubMed: 22278433]
- 196.
- Blum WF, Machinis K, Shavrikova EP, Keller A, Stobbe H, Pfaeffle RW, Amselem S. The growth response to growth hormone (GH) treatment in children with isolated GH deficiency is independent of the presence of the exon 3-minus isoform of the GH receptor. J Clin Endocrinol Metab. 2006;91(10):4171–4174. [PubMed: 16868057]
- 197.
- Pilotta A, Mella P, Filisetti M, Felappi B, Prandi E, Parrinello G, Notarangelo LD, Buzi F. Common polymorphisms of the growth hormone (GH) receptor do not correlate with the growth response to exogenous recombinant human GH in GH-deficient children. J Clin Endocrinol Metab. 2006;91(3):1178–1180. [PubMed: 16394090]
- 198.
- Carrascosa A, Esteban C, Espadero R, Fernández-Cancio M, Andaluz P, Clemente M, Audí L, Wollmann H, Fryklund L, Parodi L. The d3/fl-growth hormone (GH) receptor polymorphism does not influence the effect of GH treatment (66 microg/kg per day) or the spontaneous growth in short non-GH-deficient small-for-gestational-age children: results from a two-year controlled prospective study in 170 Spanish patients. J Clin Endocrinol Metab. 2006;91(9):3281–3286. [PubMed: 16804042]
- 199.
- Lin SC, Lin CR, Gukovsky I, Lusis AJ, Sawchenko PE, Rosenfeld M. Molecular basis of the little mouse phenotype and implications for cell type-specific growth. Nature. 1993;364(6434):208–213. [PubMed: 8391647]
- 200.
- Tuffli GA, Johanson A, Rundle AC, Allen DB. Lack of increased risk for extracranial, nonleukemic neoplasms in recipients of recombinant deoxyribonucleic acid growth hormone. Journal of Clinical Endocrinology & Metabolism. 1995;80(4):1416–1422. [PubMed: 7714117]
- 201.
- Cowell CT, Dietsch S. Adverse events during growth hormone therapy. J Pediatr Endocrinol Metab. 1995;8:243–252. [PubMed: 8821900]
- 202.
- Clayton PE, Cowell CT. Safety issues in children and adolescents during growth hormone therapy-a review. Growth Hormone &IGF Research. 2000;10:306–310. [PubMed: 11161961]
- 203.
- Bell J, Parker KL, Swinford RD, Hoffman AR, Maneatis T, Lippe B. Long-Term Safety of Recombinant Human Growth Hormone in Children. J Clin Endocrinol Metab. 2010;95(1):167–177. [PubMed: 19906787]
- 204.
- Craig ME, Cowell CT, Larsson P, Zipf WB, Reiter EO, Albertsson Wikland K, Ranke MB, Price DA, Board KI. Growth hormone treatment and adverse events in Prader-Willi syndrome: data from KIGS (the Pfizer International Growth Database). Clinical Endocrinology. 2006;65(2):178–185. [PubMed: 16886957]
- 205.
- Tauber M, Diene G, Molinas C, Hébert M. Review of 64 cases of death in children with Prader–Willi syndrome (PWS). American Journal of Medical Genetics Part A. 2008;146A(7):881–887. [PubMed: 18324685]
- 206.
- Eiholzer U. Deaths in children with Prader-Willi syndrome. A contribution to the debate about the safety of growth hormone treatment in children with PWS. Hormone Research. 2005;63(1):33–39. [PubMed: 15604598]
- 207.
- Gerard JM, Garibaldi L, Myers SE, Aceto TJ, Kotagal S, Gibbons VP, Stith J, Weber C. Sleep apnea in patients receiving growth hormone. Clin Pediatr (Phila). 1997;36(6):321–326. [PubMed: 9196230]
- 208.
- Lindgren AC, Hellstrom LG, Ritzen EM, Milerad J. Growth hormone treatment increases CO(2) response, ventilation and central inspiratory drive in children with Prader-Willi syndrome. Eur J Pediatr. 1999;158(11):936–940. [PubMed: 10541953]
- 209.
- Miller J, Silverstein J, Shuster J, Driscoll DJ, Wagner M. Short-term effects of growth hormone on sleep abnormalities in Prader-Willi syndrome. The Journal of Clinical Endocrinology and Metabolism. 2006;91(2):413–417. [PubMed: 16317059]
- 210.
- Bolar K, Hoffman AR, Maneatis T, Lippe B. Long-Term Safety of Recombinant Human Growth Hormone in Turner Syndrome. J Clin Endocrinol Metab. 2008;93(2):344–351. [PubMed: 18000090]
- 211.
- Schoemaker MJ, Swerdlow AJ, Higgins CD, Wright AF, Jacobs PA, Group UKCC. Cancer incidence in women with Turner syndrome in Great Britain: a national cohort study. The Lancet Oncology. 2008;9(3):239–246. [PubMed: 18282803]
- 212.
- Carel J-C, Ecosse E, Landier F, Meguellati-Hakkas D, Kaguelidou F, Rey G, Coste J. Long-term mortality after recombinant growth hormone treatment for isolated growth hormone deficiency or childhood short stature: preliminary report of the French SAGhE study. The Journal of Clinical Endocrinology and Metabolism. 2012;97(2):416–425. [PubMed: 22238382]
- 213.
- Sävendahl L, Maes M, Albertsson-Wikland K, Borgström B, Carel JC, Henrard S, Speybroeck N, Thomas M, Zandwijken G, Hokken-Koelega A. Long-term mortality and causes of death in isolated GHD, ISS, and SGA patients treated with recombinant growth hormone during childhood in Belgium, The Netherlands, and Sweden: preliminary report of 3 countries participating in the EU SAGhE study. J Clin Endo Metab. 2012;97(2):213–217. [PubMed: 22238393]
- 214.
- Maneatis T, Baptista J, Connelly K, Blethen S. Growth hormone safety update from the National Cooperative Growth Study. J Pediatr Endocrinol Metab. 2000;13 Suppl 2:1035–1044. [PubMed: 11086659]
- 215.
- Ergun-Longmire B, Mertens AC, Mitby P, Qin J, Heller G, Shi W, Yasui Y, Robison LL, Sklar CA. Growth Hormone Treatment and Risk of Second Neoplasms in the Childhood Cancer Survivor. J Clin Endocrinol Metab. 2006;91(9):3494–3498. [PubMed: 16822820]
- 216.
- Sklar CA, Mertens AC, Mitby P, Occhiogrosso G, Qin J, Heller G, Yasui Y, Robison LL. Risk of disease recurrence and second neoplasms in survivors of childhood cancer treated with growth hormone: a report from the Childhood Cancer Survivor Study. J Clin Endocrinol Metab. 2002;87(7):3136–3141. [PubMed: 12107213]
- 217.
- Patterson BC, Chen Y, Sklar CA, Neglia J, Yasui Y, Mertens A, Armstrong GT, Meadows A, Stovall M, Robison LL, Meacham LR. Growth Hormone Exposure as a Risk Factor for the Development of Subsequent Neoplasms of the Central Nervous System: A Report From the Childhood Cancer Survivor Study. The Journal of Clinical Endocrinology & Metabolism. 2014;99(6):2030–2037. [PMC free article: PMC4037726] [PubMed: 24606096]
- 218.
- Sklar CA, Antal Z, Chemaitilly W, Cohen LE, Follin C, Meacham LR, Murad MH. Hypothalamic–Pituitary and Growth Disorders in Survivors of Childhood Cancer: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 2018 [PubMed: 29982476]
- 219.
- Child CJ, Zimmermann AG, Jia N, Robison LL, Brämswig JH, Blum WF. Assessment of Primary Cancer Incidence in Growth Hormone-Treated Children: Comparison of a Multinational Prospective Observational Study with Population Databases. Hormone Research in Paediatrics. 2016;85(3):198–206. [PubMed: 26913923]
- 220.
- Raman S, Grimberg A, Waguespack SG, Miller BS, Sklar CA, Meacham LR, Patterson BC. Risk of Neoplasia in Pediatric Patients Receiving Growth Hormone Therapy--A Report From the Pediatric Endocrine Society Drug and Therapeutics Committee. The Journal of Clinical Endocrinology and Metabolism. 2015;100(6):2192–2203. [PMC free article: PMC5393518] [PubMed: 25839904]
- 221.
- Clayton PE, Cuneo RC, Juul A, Monson JP, Shalet SM, Tauber M. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur J Endocrinol. 2005;152(2):165–170. [PubMed: 15745921]
- 222.
- Molitch ME, Clemmons DR, Malozowski S, Merriam GR, Vance ML. Evaluation and Treatment of Adult Growth Hormone Deficiency: An Endocrine Society Clinical Practice Guideline. The Journal of Clinical Endocrinology & Metabolism. 2011;96(6):1587–1609. [PubMed: 21602453]
- 223.
- Tauber M, Moulin P, Pienkowski C, Jouret B, Rochiccioli P. Growth hormone (GH) retesting and auxological data in 131 GH-deficient patients after completion of treatment. The Journal of Clinical Endocrinology and Metabolism. 1997;82(2):352–356. [PubMed: 9024217]
- 224.
- Colao A, Di Somma C, Savastano S, Rota F, Savanelli MC, Aimaretti G, Lombardi G. A reappraisal of diagnosing GH deficiency in adults: role of gender, age, waist circumference, and body mass index. The Journal of Clinical Endocrinology and Metabolism. 2009;94(11):4414–4422. [PubMed: 19773395]
- 225.
- Cook DM, Rose SR. A review of guidelines for use of growth hormone in pediatric and transition patients. Pituitary. 2012;15(3):301–310. [PubMed: 22271255]
- 226.
- Quigley CA, Zagar AJ, Liu CC, Brown DM, Huseman C, Levitsky L, Repaske DR, Tsalikian E, Chipman JJ. United States multicenter study of factors predicting the persistence of GH deficiency during the transition period between childhood and adulthood. International Journal of Pediatric Endocrinology. 2013;2013(1):6. [PMC free article: PMC3605263] [PubMed: 23406437]
- Review Disorders of Growth Hormone in Childhood.[Endotext. 2000]Review Disorders of Growth Hormone in Childhood.Murray PG, Clayton PE. Endotext. 2000
- Review Idiopathic Short Stature and Growth Failure of Unknown Etiology.[Endotext. 2000]Review Idiopathic Short Stature and Growth Failure of Unknown Etiology.Yau M, Lu J, Rapaport R. Endotext. 2000
- Review Pituitary Gigantism.[Endotext. 2000]Review Pituitary Gigantism.George MM, Eugster EA, Chernausek SD. Endotext. 2000
- Review Growth Hormone Stimulation Tests in Assessing Adult Growth Hormone Deficiency.[Endotext. 2000]Review Growth Hormone Stimulation Tests in Assessing Adult Growth Hormone Deficiency.Yuen KCJ. Endotext. 2000
- Review Pediatric Implications of Normal Insulin-GH-IGF Axis Physiology.[Endotext. 2000]Review Pediatric Implications of Normal Insulin-GH-IGF Axis Physiology.Bang P. Endotext. 2000
- Growth and Growth Disorders - EndotextGrowth and Growth Disorders - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...