

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

TAKE-HOME POINTS
- The structure of insulin contains determinants of foldability, trafficking, self-assembly, and receptor binding.
- Insulin is the biosynthetic product of a single-chain precursor, preproinsulin, whose proteolytic processing is coupled to trafficking between cellular compartments.
- The connecting (C) domain of proinsulin is removed by a specialized set of endoproteases and a carboxypeptidase activity, acting mainly within maturing secretory granules.
- Insulin is stored as microcrystalline arrays of zinc insulin hexamers within specialized glucose-regulated secretory vescicles.
- Regulation of insulin secretion is coupled to metabolism and electrophysiologic events involving plasma membrane depolarization and calcium-ion homeostasis.
- The insulin receptor is a transmembrane protein containing an extracellular hormone-binding domain and intracellular tyrosine kinase domain.
- Binding of insulin to the insulin receptor (an (αβ) 2 dimer) is mediated by side chains in both the A- and B chains of insulin.
- The primary hormone-binding site (Site 1) in the extracellular domain of the insulin receptor contains the L1 β -helix of one α -subunit and the C-terminal α CT α -helix of the other α -subunit.
- Dominant mutations in the insulin gene cause monogenic syndromes of diabetes mellitus, prominently including permanent neonatal-onset diabetes, due to toxic misfolding of proinsulin variants.
INTRODUCTION
Insulin plays a central role in the regulation of human metabolism. The hormone is a 51-residue anabolic protein that is secreted by the β-cells in the Islets of Langerhans. Containing two chains (A and B) connected by disulfide bonds, the mature hormone is the post-translational product of a single-chain precursor, designated proinsulin . Extensive studies of the three-dimensional structure of insulin, pioneered by D. C. Hodgkin , have enabled the development of therapeutic analogs for the treatment of the metabolic disorder diabetes mellitus (DM) . The insulin gene is the site of dominant mutations associated with DM . Although such mutations are uncommon, their molecular analysis has provided important insights into the biochemical bases of the hormone’s pathway of biosynthesis and mechanism of receptor binding . The largest class of mutations is associated with the impaired folding of proinsulin, which leads in turn to progressive endoplasmic-reticular (ER) stress, β-cell death and DM, usually with onset in the neonatal period .
Key complementary functions of insulin are (a) stimulation of glucose uptake from the systemic circulation and (b) suppression of hepatic gluconeogenesis, together regulating glucose homeostasis . DM is characterized by decreased glucose tolerance resulting from a relative deficiency of insulin or a lack of sensitivity to the endogenous hormone. Insufficient insulin, or decreased insulin sensitivity, results in hyperglycemia. Long-term exposure of tissues to elevated ambient glucose concentrations is associated with the development of complications, including macro- and microvascular disease. Of particular concern are coronary heart disease, cerebrovascular disease, and the characteristic retinopathy, nephropathy, and neuropathy of this disorder .
The history of the discovery of insulin and its therapeutic utility defined a paradigm for the integration of physiologic and biochemical approaches in experimental medicine . At the end of the 19 th century Von Mering and Minkowski noted that removal of the pancreas led to the development of DM in dogs . In 1916 Schafer first speculated that an antidiabetic hormone, which he named “insuline,” was secreted from pancreatic islets . Barron noted in 1920 that ligation of the pancreatic duct, with destruction of the exocrine pancreas, only resulted in DM if the islets, so named by Langerhans in 1869 , were also destroyed . Subsequently, the work of Banting, Best, Collip and MacCleod in the early 1920′s resulted in the identification of a substance in extracts of pancreas that had the remarkable ability to reduce blood glucose levels in diabetic animals . By 1923 these pancreatic extracts were employed to successfully treat patients with DM. The dramatic clinical utility of insulin encouraged broad public support for medical research .
The modern view of insulin is as a ligand that activates a specific cellular receptor, designated the insulin receptor (IR) . The IR belongs to a superfamily of receptor tyrosine kinase whose activation modulates multiple post-receptor signaling pathways . Insulin thus regulates a host of other cellular processes, such as protein and fat synthesis, RNA and DNA synthesis, as well as cell growth and differentiation . It is, however, the regulation of glucose uptake that is of primary concern in the clinical manifestations of diabetes; therefore, this chapter will begin with a brief description of how plasma glucose homeostasis is achieved.
REGULATION OF PLASMA GLUCOSE BY INSULIN
Specific membrane transporters facilitate the movement of glucose into cells to reduce plasma glucose concentrations in response to insulin stimulation. The transported glucose is subsequently used as metabolic fuel or stored as a complex polymeric structure, designated glycogen. Two major types of glucose transporters are known: Na + -dependent and Na + -independent. Only the Na + -independent transporters possess an insulin-responsive isoform. We describe each in turn.
The Na + -dependent glucose transporter family has been identified in several tissues, particularly in small intestinal epithelium (SGLT1) and the renal proximal tubule (SGLT2) as well as in other kidney tubule cells . These transporters are located on the lumenal side of intestinal and kidney cells and act to absorb glucose against its concentration gradient by coupling the movement of glucose into these cells with the concomitant movement of Na + into the cell. Since Na + is moving down its electrochemical gradient this energy can be used to cotransport glucose into the cells. Thus, this transporter is dependent on the concentrations of extracellular and intracellular sodium ions, which are maintained by a Na + /K + -ATPase ion pump.
The Na + -independent glucose transporter family, consisting of several isoforms, facilitates the movement of glucose down its concentration gradient across a plasma membrane. Although seven isoforms have been identified (designated Glut1-7) , only one will be discussed in detail here, Glut4, because it is the transporter that is in highest concentration in insulin-sensitive tissues, including skeletal muscle, cardiac muscle, and adipose tissue (fat) . Glut4, and to a lesser extent Glut1 enable these cells to increase their uptake of glucose, thereby lowering circulating concentrations. Because the intracellular concentration of glucose is low due to the rapid phosphorylation of glucose to glucose-6-phosphate (not a substrate of the Glut transporters) and its conversion to other metabolic products, the presence of active transporters in the plasma membrane favors the movement of glucose into cells.
Insulin enhances glucose uptake by increasing the number of transporters in the plasma membrane of target cells. This was first demonstrated in adipocytes and subsequently in skeletal and cardiac muscle . Insulin stimulation of such cells mobilizes transporters from intracellular compartments to the plasma membrane to facilitate glucose transport. Translocation of receptors to the plasma membrane has been demonstrated to occur within 30 seconds of insulin stimulation ; as the stimulus dissipates the decrease in the number of plasma membrane receptors declines coincident with a decline in glucose transport . Whereas glucose transport via Glut4 is a passive process (limited only by the chemical potential of the glucose gradient and the V max of the transporters), translocation and reverse reinternalization of receptors are energy-dependent processes . The impaired ability of insulin, on binding and activation of the IR, to signal Glut4 translocation from intracellular stores contributes to postprandial hyperglycemia in Type 2 DM . Animal studies have also demonstrated that insulin resistance is associated with a decreased translocation of glucose transporters to the plasma membrane in muscle cells . In fact, decreased insulin levels in animal models of DM have been shown not only to decrease transporter translocation, but also to attenuate expression of Glut4 in muscle cells. Thus, it appears that insulin provides both a short-term signal to increase glucose-transporter translocation and a long-term signal to maintain a basal level of expression of such transporters in target cells. The combination of acute and basal actions provides a common mechanism in Type 1 DM (characterized by low or vanishing endogenous insulin levels) or Type 2 DM (characterized by insulin resistance) could cause pathologically high plasma glucose levels: loss of regulation and expression of transmembrane glucose transporters. Glut2, expressed on surface of β-cells, contributes to the regulation of insulin secretion . Accordingly, a β-cell specific IR knock-out (KO) model indicated that insulin likely positively regulates its own secretion from the β-cell .
Since its purification and clinical application in 1923 , the central importance of insulin in regulating glucose metabolism and the prevention of DM has stimulated research in attempts to understand the mechanism of action of this peptide hormone. This work has lead to the determination of the three-dimensional structure of insulin , identification of its precursor and the processing and secretion mechanisms that underlie its production . Complementary advances have seen the identification of the IR and of its mechanisms of signal transduction . Recent studies have defined three-dimensional structures of proinsulin and elements of the IR involved in hormone binding . In addition to their fundamental importance, these discoveries have deepened our understanding of the molecular basis of DM and its treatment as discussed below.
INSULIN BIOGENESIS AND MECHANISM OF RELEASE
Insulin was the first peptide hormone discovered. Before Abel crystallized insulin in 1926 and Jensen and Evans in 1935 identified the N-terminal phenylalanine of the B-chain , proving that insulin was indeed a protein, all hormones were believed to be small molecules. With the elucidation of the amino-acid sequence of insulin by Sanger in the mid 1950′s (see Figure 1), it became known that insulin was a two-chain heterodimer consisting of a 21-residue A-chain linked to a 30-residue B chain by two disulfide bonds derived from cysteine residues (A7-B7 and A20-B19). An intrachain disulfide bond also exists within the A-chain (A6-A11).

Figure 1 . Primary structures of porcine insulin and porcine proinsulin. The primary sequence of porcine insulin (a) as determined by Sanger and co-workers ; and proinsulin . The sequence of human insulin is identical to that of porcine insulin except for the change of Ala B30 to Thr B30 in human insulin.
Although this primary structure provided valuable information regarding the amino-acid composition and size (ca. 6,000 Daltons) of the insulin molecule , questions concerning the processes of insulin biosynthesis and secretion were not resolved until the late 1960′s with the discovery of proinsulin . This precursor protein (ca. 9,000 D) contains both the A- and B-chain of insulin in a continuous single chain joined through an intervening segment, designated the C domain . The C domain varies in length among vertebrate species (typically 30-35 residues) and is flanked at each end by dibasic residues (Arg-Arg and Lys-Arg) . Proinsulin is cleaved at those dibasic links by a trypsin-like enzyme to release the mature hormone and a free C-peptide (which lacks the dibasic residues).


Figure 2. (a) Diagrammatic illustration of the processing of insulin. The transcription and translation of the human insulin gene, as well as processing of preproinsulin to insulin is illustrated. (b) Conversion of proinsulin to insulin demonstrating the secondary and tertiary structures of insulin; the flexible C domain in proinsulin and split proinsulins is shown in dotted line. Thick blue arrows indicated predominant path.
Chan et al. subsequently discovered that there was an additional and larger precursor of insulin, preproinsulin. This single-chain polypeptide (ca. 12,000 Daltons) consists of proinsulin extended at the amino-terminus by a 24-residue signal peptide of hydrophobic residues . Such a signal sequence is characteristic of proteins that enter the secretory pathway . The signal peptide of preproinsulin is cleaved coincident with its translocation into the ER , and thus proinsulin itself (together with the oxidative folding machinery of the ER) mediates proper disulfide pairing and three-dimensional protein folding .
The steps involved in the conversion of the information encoded within the 1500 bases of the human insulin gene (as sequenced by Bell and colleagues in 1980 ) into preproinsulin and its subsequent proteolytic conversion to insulin are illustrated in Figure 2A and 2B. The first set of steps occur at the nucleic-acid level: the initial mRNA transcript is modified via excision of the two intervening sequences, the 5′ terminus is capped by 7-methyl guanosine, and the 3’-terminus undergoes polyadenylation to produce a mature mRNA product. This mRNA product encodes preproinsulin, which is translated on the rough endoplasmic reticulum (rER) and subsequently translocated into the RER lumen via a series of interactions of the signal peptide with the signal recognition particle (SRP) and SRP-receptor in the rER membrane . The next set of steps occurs at the protein level. The signal peptide is cleaved in the lumen of the rER by a signal peptidase (located on the lumenal side of the rER membrane). Within the cisternae of the rER, proinsulin undergoes rapid folding and disulfide bond formation to generate the native tertiary structure, the direct precursor of insulin. The signal peptide is rapidly degraded in the rER and is therefore not a normal secretory product of the β-cells .
In the final series of steps proinsulin is transported to the Golgi apparatus where it is packaged into secretory granules and converted to native insulin and C-peptide. The conversion process may begin in the trans Golgi network but continues in the condensing vacuoles (early secretory granules), and the products are stored in mature secretory vesicles, and secreted in equimolar amounts along with small amounts (ca. 2-3%) of proinsulin and intermediate cleavage products . Glucose, in addition to stimulating insulin secretion by β-cells, also activates insulin gene transcription, enhances insulin mRNA stability, and stimulates its translation .
Proinsulin, despite its larger size, shares many of the physical properties of insulin. Proinsulin has been shown, for example, to form dimers and Zn 2+ -coordinated hexamers in a manner similar to insulin , has a comparable isoelectric point and solubility , and cross-reacts with insulin antisera . These findings motivated the hypothesis that the structure of the insulin moiety in proinsulin is similar, if not identical, to that of native insulin . Although the crystal structure of proinsulin has not been determined , presumably due to the flexibility of the C domain, its solution structure has recently been determined by multidimensional NMR methods . The insulin moiety indeed retains the conformation of insulin whereas the C domain is flexible (but not completely disordered). The solution structure rationalizes why proinsulin is a full agonist of insulin and displays 3-5% biological activity ; the binding regions of the insulin moiety are accessible even in the presence of the C domain .
In 1969 it was demonstrated via pulse-chase studies that proteolytic processing of proinsulin occurs in the Golgi apparatus (GA) and/or early secretory vesicles of the β-cells ; and subsequent studies have identified the trans Golgi as the initial compartment wherein proinsulin and its converting enzymes are brought together to form secretory granules . Monoclonal antibodies specific for intact proinsulin also demonstrated that proinsulin is transferred from the rER to the cis- and trans Golgi, where the precursor is concentrated to form prosecretory vesicles . Several studies have demonstrated that shuttling of proinsulin vesicles from the rER to the cis Golgi and through the GA is an energy-requiring process requiring ATP . Subsequent conversion of proinsulin to insulin, initiated in the trans Golgi, accelerates within prosecretory granules as they acidify and mature in the cytosol over a period of 1-3 hours in preparation for secretion. Residual proinsulin and intermediate cleavage products then comprise only 2-3 percent of total stored insulin-related protein. Insulin secretory granules turn over at a much slower rate of many hours or several days normally. In the intracellular pathway taken by proinsulin on budding from the rER, distinct times are required for individual stages of transfer. In rat islets the conversion of proinsulin to insulin begins about 30 min after ribosomal synthesis of preproinsulin and resembles first-order reaction kinetics with half-times of approximately 30-60 minutes .
The conversion of proinsulin to insulin occurs through the joint action of two types of proteases: one with trypsin-like endoprotease activity which cleave after the dibasic residues pairs at each end of the C domain, and another with exopeptidase activity resembling that of carboxypeptidase B to remove the basic residues left after tryptic-like cleavage . Previous studies have also demonstrated that mixtures of pancreatic trypsin and carboxypeptidase B could convert proinsulin to insulin in vitro . Two endoproteases were found within insulinoma secretory granules . Initially called Type I and Type II converting enzymes, each of these acidic endoproteases was found to be dependent on Ca 2+ ions. Type I was active in 1 mM Ca 2+ and cleaved at Arg31-Arg32 in proinsulin (the first two positions of the C domain) whereas Type II required 0.1 mM Ca 2+ and cleaved predominantly at Lys64-Arg65 (the last two positions of the C domain) . Each was also found to have an acidic pH optimum near 6.0.
The discovery in yeast Saccharomyces cerevisciae of an endoprotease, designated Kex2, that cleaves at dibasic residue sites in the yeast α -mating factor and in microbial toxin precursor polypeptides facilitated the search for homologous endoproteases in mammalian β-cells . Kex2 is a homologue of subtilisin, a bacterial serine protease. In yeast this integral membrane protein is localized in the trans Golgi network. Analysis of human insulinoma cDNA via PCR techniques lead to the discovery of PC2, an enzyme having an homologous catalytic domain . PC2 shares 49% amino-acid identity with Kex2, but importantly lacks its transmembrane (TM) segment, indicating that it was likely to be a soluble protease and thus was a candidate for one of the processing endoproteases in β-cell secretory vesicles. Similar screening methods led to the discovery of PC1/3, the second secretory-granule convertase . Subsequent work demonstrated that both PC2 and PC1/3 display optimal activity at pH 5.5, consistent with the internal pH of β-cell secretory granules and are also expressed in the brain and many other neuroendocrine cells. These two proprotein convertases are members of a larger 7-member family of kexin/subtilisin-like endoproteases, which normally function within the secretory pathway in most eukaryotic cells .
Immunocytochemical studies of pancreatic islets β-cells provided evidence for the presence of both PC2 and PC1/3 in β-cells pancreatic islets , and their ability to convert proinsulin to insulin was demonstrated by co-infection with vaccinia viruses expressing PC2 and PC1/3 in Cos7 cells expressing proinsulin . Whereas PC2 mediated cleavage only at the C-peptide/A-chain junction, PC1/3 cleaved at both dibasic sites with a preference for the B-chain/C-peptide site. Subsequent studies also established the identity of PC2 and PC1/3 with the calcium-dependent Type II and Type I insulinoma endoproteases, respectively, as discovered by Davidson and coworkers . The carboxypeptidase B-like exopeptidase, which removes COOH-terminal basic residues after cleavage by PC2 and PC3/PC1, was also found and is known as carboxypeptidase E . Although structurally homologous to pancreatic carboxypeptidases A and B , carboxypeptidase E has several unique features that differentiate it from other carboxypeptidases . In the maturing secretory granule of the B cell, PC2, PC1/3, and carboxypeptidase E work together to convert proinsulin to mature insulin and C-peptide. Studies have demonstrated that PC1/3 first cleaves proinsulin at the B chain-C peptide junction, generating an intermediate product that is preferentially cleaved by PC2 to yield insulin . Thus knockout of PC1/3 results in very high circulating proinsulin levels while PC2 nulls exhibit much lower levels .
The PC family of convertases are all synthesized as inactive precursors with a lengthy N-terminal propeptide that is autocatalytically cleaved at a tetrabasic cleavage site by the proenzyme as it passes from the slightly alkaline ER to the neutral and mildly acidic conditions of the Golgi apparatus and is then removed by a second cleavage within the propeptide which assists in its release and disposal, yielding the active full-length enzyme . In the case of PC1/3 the full length form is active , but undergoes C-terminal autocatalytic truncation to a smaller, more active form within the secretory granules . In the case of PC2 the removal of the propeptide is more complex and requires the presence of neuroendocrine protein 7B2 . Activation of PC2 thus does not occur in the Golgi but rather within the secretory granules where it also functions. The full length enzyme is the active form in the granules within the beta cells and other neuroendocrine tissues. Knockout of 7B2 thus provides a phenocopy of the PC2 null when on the same genetic background, or inbred mouse strain (for review, see Ref ).
Secretory granules in the β-cell undergo maturation in the cytosol. Electron-microscopic (EM) studies have demonstrated that mature granules have a dense crystalline-appearing core with a spacing similar to that of 2-Zn insulin crystals . Newly synthesized insulin is likely to forms crystals with zinc ions that are transported into the maturing secretory granules as demonstrated by a knockout of the zinc transporter (ZnT8) . The crystals reside in the dense core of the β-granules whereas the soluble C-peptide resides in the less dense or clear periphery of the granule . Proinsulin is also known to crystallize with insulin in small amounts, probably as mixed hexamers . Both proinsulin and insulin have the ability to bind zinc and form zinc-coordinated hexamers (two Zn 2+ axial atoms per hexameric unit); the side chain of histidine B10 coordinates axial Zn 2+ ions . The self-assembly and micro-crystallization of zinc-insulin hexamers may be regulated by compartmental pH. The secretory granules possess an intrinsic proton pump, which serves to lower the pH within the granule to pH 5.0-5.5: this is optimal for both prohormone processing and crystallization in vitro . The pH within the rER is less acidic, which promotes thiol-disulfide exchange and hence proinsulin folding with native disulfide pairing.
High ambient glucose concentration in the islets promotes insulin biosynthesis and is the primary regulator of secretion. Elevated glucose concentrations cause an increase in cAMP levels by a mechanism that does not appear to involve activation of adenylate cyclase . cAMP then exerts its effects via a mechanism involving protein kinase A (PKA), leading to the phosphorylation and activation of certain key proteins . Through this complex chain of events, glucose and cAMP (and possibly contributions from the rise in intracellular free calcium and IP3) rapidly increase translation and transcription of insulin mRNA . Insulin mRNA normally turns over slowly, with a half life of approximately 30 hours at normal or below normal levels. However, elevated ambient glucose concentrations increase the half life of insulin mRNA as much as threefold . Calcium dependent exocytosis of secretory granules is the main mechanism of secretion in both glucose-stimulated and basal states . Little or no direct secretion of proinsulin occurs from the rER to the plasma membrane by way of unregulated pathways . Other hormones and chemical substances also play an important role in the regulation of insulin secretion , including glucagon, which is secreted by α -cells in pancreatic islets as discussed in detail in another chapter; glucagon-like peptide (GLP-1: Ref. ); cholecystokinin ; and gastric inhibitory peptide all acting via specific receptors on the β-cell. Inhibitors of insulin secretion include catecholamines (adrenaline and noradrenaline) which interact with adrenergic receptors on the β-cell membrane , and somatostatin which is secreted by -cells of the pancreatic islets . Amylin is also secreted by the β -cell although the regulatory mechanisms for amylin co-secretion are not well understood.
The integrated regulation of insulin secretion in pancreatic β -cells provides an opportunity to develop quantitative data-based computer models relating metabolic sensing to electrophysiological events and to intracellular Ca 2+ dynamics, ultimately leading to exocytosis of the secretory granules . Such analyses provides fundamental insight into the biochemical and biophysical determinants of cytoplasmic transport of organelles .
BIOCHEMISTRY AND ELECTROPHYSIOLOGY OF INSULIN SECRETION
Secretion of insulin from β -cells is not only an important step in the regulation of glucose homeostasis in healthy individuals, but has also been demonstrated to be impaired (for different reasons) in both Type 1 and Type 2 DM . In fact, in the prediabetic state of Type 1 DM as well as in various forms of Type 2 DM, abnormalities in insulin secretion are an integral component of the pathophysiology . In light of its extensive characterization the β-cell also serves as a model of the secretory process for other cell types.
Insulin is stored in large dense core vesicles (LDCV) and released by exocytosis as described above. Such release is a multistep process that consists of the transport of the secretory vesicles to the plasma membrane, then docking, priming, and finally fusion of the vesicle with the plasma membrane. This process is regulated cooperatively by nutrients, other hormones, and neurotransmitters in association with electrical depolarization of the β-cell and release of insulin. Only a small portion of the insulin stored in vesicles in the β-cell is released, however, even under maximum stimulation. This suggests that systemic insulin levels are regulated by secretion rather than by biosynthesis and is not ordinarily limited by the size of storage pools. However the mechanisms that regulate the directed transport of the insulin granules to the plasma membrane are also not well understood.
The best characterized mechanism of coupling glucose metabolism to insulin secretion resides in the electrical excitability of the β-cell. A large number of ion channels, pumps, and transporters contribute to intracellular calcium concentration, as well as other ions, to set the membrane potential (V m ) of the β-cell; this set point is near -70 mV when extracellular glucose is ~3 mM (see Figure 3). In 1968 Dean and Mathews demonstrated that β-cells are electrically excitable and that glucose controlled this excitability . They also showed that the action potentials of β-cells were increased by sulfonylureas. The use of patch-clamp techniques enabled the electrical activity of the β-cell to be studied in detail, and such studies by Ashcroft, Rorsman and others elucidated the key role of ATP-sensitive potassium (KATP) channels in the resting membrane potential of the β-cell as well as the importance of these channels in the mechanism of insulin secretion (for a recent perspective, see ).

Figure 3 . β -cell ion channels. KATP channel conductance predominates in the resting β-cell, maintaining the resting Vm near the electrochemical potential (EK) for K+ (~ -80 mV). These potassium channels belong to the inward rectifier (Kir) subfamily. They obtain their name since these channels conduct K + current into the cell more readily than to the outside of the cell (inward rectification, as a diode). KATP channels are, however, weak inward rectifiers because they pass a significant amount of current in the outward direction. At -70 mV open KATP channels carry a small amount of outward current, which maintains the hyperpolarized resting potential in the β-cells.
A myriad of biochemical and biophysical structure-function studies of recombinant Kir channels has led to a more complete understanding of these channels. The crystal structure of a bacterial Kir analog, the Streptomyces lividens KcSA channel, has been determined ; and the inner pore of a mammalian Kir has likewise been crystallized, and its structure determined . The structures revealed that Kir channels consist of four subunits: each folds into the membrane to define two transmembrane domains (M1 and M2) surrounding a pore loop (P). The four P-loops line the central ion-conducting pore with the M1 and M2 subunits providing outer supports (Figure 4).
Elevation of glucose concentration to >8-10 mM results in depolarization of the β-cell. Glucose is taken up into the β-cell by the GLUT2 transporter and metabolized via glucokinase, glycolysis and in mitochondria to generate ATP. This alters the ATP/ADP ratio, which causes closure of KATP channels and depolarization of the cell via the decreased K + permeability. ATP inhibits KATP channels and ADP opens them . Other nucleotides generated by glucose metabolism (Ap3A: diadenosine triphosphate, and Ap4A: diadenosine tetraphosphate) have been implicated as second messengers mediating the closure of KATP channels, but their significance remains uncertain. Mutations in either the Kir, or SUR1, can result in persistent activation, leading to neonatal hyperinsulinemia and hypoglycemia . It is also known that some Kir channels in β-cells are activated via G-protein coupled receptors as reviewed below.

Figure 4 . Kir channels. KATP channels are unique in the inward rectifier family because they require an auxiliary subunit, the sulfonylurea receptor (SUR1), to function. The SUR1 is a member of the ATP binding cassette (ABC) family of membrane proteins, which includes the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel among others . It was named due to its binding to iodinated glyburide but clearly it is not actually an sufonylurea receptor. KATP channels in the β-cell consists of Kir6.2 subunits surrounded by their accompanying SUR1 subunit (not shown).
Pioneering studies by Katz, Miledi, and Douglas first established intracellular Ca 2+ concentrations as a general coupling factor between membrane depolarization and vesicular exocytosis ; this mechanism operates in β-cells (for review, see ). Voltage dependent Ca 2+ channels (Cav) open on membrane depolarization as caused by Kir channel closure; and it is this Ca 2+ influx that leads in turn to exocytosis of the glucose-regulated secretory granules and insulin secretion.
Cav channels are classified one the basis of a low-voltage threshold (LV: activated at more negative potentials) or high-voltage threshold (HV: activated at relatively depolarized potentials). HV channels can be further divided into subclasses: L, N, P, Q, and R . Insulin secretion is inhibited by dihydropyridine-based calcium-channel blocking agents, which inhibit L-type Cav. Although activators of L-type Cav can stimulate insulin secretion, Cav1.3 KO mice exhibited perturbed islet function resulting in glucose intolerance; these mice were smaller than controls and yet maintained glucose-dependent insulin secretion . This phenotype is likely to be secondary either to upregulation of other Cav1.2 channels or to the existence of other mechanisms of insulin secretion unreliant on voltage gated channels as discussed next. It has been suggested that the neuronal type of Ca channels play a direct role in exocytosis .
Several hormones and neurotransmitters regulate insulin secretion in addition to the voltage-sensitive pathways. Molecules such as epinephrine, galanin, somatostatin, acetylcholine, and glucagon-like peptide (GLP) each contribute to the regulation of insulin secretion by binding to cognate receptors . Cholecystokinin (CCK receptor pathway) and acetylcholine (M3 receptor in β-cells) potentiate insulin secretion via phosphoinositide catabolism with the subsequent mobilization of intracellular calcium ions. These ligands bind to G-protein coupled receptors (GPCRs) that can activate phospholipase-C (PLC) . PLC-catalyzed hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP 2 ) produces inositol 1,4,5-triphosphate (IP 3 ) and diacylglycerol (DAG). Two families of Ca 2+ channels are present on the ER and rER: IP3 receptors and ryanodine receptors (RyR). Each is capable of causing the release of Ca 2+ ions stored in the ER. DAG concomitantly activates protein kinase (PKC). Other potentiators of insulin secretion, such as GLP-1, and glucose-dependent insulinotropic polypeptide (GIP), bind to their respective cognate GPCRs to activate adenyl cyclase and increase intracellular cAMP, which in turn activates protein kinase A (PKA). Stimulation of either PKC or PKA alters second-messenger systems in the β-cell and can chemically modify ion channels to influence insulin secretion. In fact, insulin exocytosis can be induced independently from Ca 2+ fluxes in in vitro studies through intracellular application of GTP or nucleotide analogs GppNHp and GTP S.
Specific proteins are also likely to be involved in the interaction of secretory vesicles with the plasma membrane. Pairing proteins on the vesicle membrane (v-SNARES) have been found to tightly interact with cognate proteins on the target membrane (t-SNARES). Cytosolic cofactors such as N-ethylmaleimide-sensitive factor (NSF) and α /β-SNAP may assist with ATP binding and hydrolysis to cause exocytosis .
In addition to voltage-sensitive pathways and GPCRs, the monomeric G-proteins (such as Rab27 and Rab3 ) may activate second-messenger cascades resulting in exocytosis; and multiple Ca 2+ -binding proteins (such as synaptotagmin ) may further regulate vesicle fusion. It is clear that the process of insulin secretion is highly complex; and research continues to provide new insights into β-cell molecular biology and electrophysiology. Such studies promise to provide a better understanding of insulin action and its deregulation in DM.
INSULIN STRUCTURE
The three-dimensional structure of insulin has been studied in great detail in crystals by X-ray diffraction and in solution by NMR spectroscopy . Such studies have yielded valuable information regarding the folding of proinsulin and function of insulin . As previously mentioned, insulin was first crystallized in rhombohedral form in 1926 ; and almost 10 years later Scott elucidated the importance of zinc ions and other divalent cations in crystallization . In 1969 the structure of hexameric 2-Zn insulin (designated T 6 in modern nomenclature ) was determined by Dorothy C. Hodgkin and coworkers using X-ray methods ; this structure and was later refined to atomic resolution . Currently there are several crystal forms of insulin, defining three structural families of hexamers (T 6 , T 3 R f 3 , and R 6 ) , zinc-free dimers (T 2 ) , and monomeric fragments . These families are shown in schematic form in Figure 5A and as ribbon models in Figure 5B; models of the component T-state protomer and R-state protomer are shown in Figure 6. The solution structure of engineered insulin monomers and dimers resembles the crystallographic T state .

Figure 5 . Structural families of insulin hexamers. A, Schematic representation of the three types of zinc insulin hexamers, designated T 6 , T 3 R f 3 , and R 6 . B, Corresponding ribbon representation of wild-type crystal structures. Axial zinc ions are shown in blue-gray . Coordinates were obtained from Protein Databank entries 4INS , 1TRZ, and 1ZNJ, respectively. Residues B1-B8 exhibit a change in secondary structure as shown in black . T-state protomers are otherwise shown in red , and R-state protomers in blue . For cylinder models of the T- and R-state protomers, see Figure 6 below. This figure is reprinted by Ref with permission of the authors.
The T 6 insulin hexamer (MW ~36000 Da) consists of six molecules of insulin arranged as three dimeric units related by a threefold symmetry axis . The dimers (MW ~12000) possess a pseudo twofold symmetry axis, which is perpendicular to the threefold axis of rotation. Although each protomer within the dimers has similar main-chain structure, they are not identical in the arrangement of certain side chains, breaking the twofold symmetry. The most obvious difference is that the side chain of Phe B25 is folded in towards the hydrophobic core in one protomer but outwards in the other . Two axial Zn 2+ atoms lie on the threefold symmetry axis of the hexamer; each exhibits octahedral coordination by three His B10 residues and three water molecules.
The T 6 insulin structure defines hydrophobic, solvent-exposed, and potential binding surfaces of insulin. This characterization has been supported by NMR-based solution structures of the insulin hexamer , engineered dimer , and engineered monomer . Many additional X-ray structures of insulin , insulin derivatives and insulins of other species, such as the Atlantic hagfish ( Myxine glutinosa ), a variant insulin containing a substitution (His B10 Asn) that prevents zinc binding and hexamer formation . In most of these instances the insulin, or derivative, maintains an overall tertiary structure that corresponds well with the protomers in the T 6 structure. For this reason the T 6 insulin hexamer widely been employed as the prototypic insulin structure. An emerging theme of such studies is that classical crystal structure of the free hormone (T state) depicts spatial relationships pertinent to the folding pathway of proinsulin whereas a key subset of such relationships are altered or broken on receptor binding . The relevance of the crystallographic R state to receptor binding continues to be a source of speculation .

Figure 6 . Classical T and R structures of insulin. Ribbon models of TR dimer based on crystal structures of zinc insulin hexamers. The B chain is shown in black and A chain in green. The position of Gly B8 is shown as a red ball in T-state-specific β -turn (left) or R-state-specific α helix (right). D-amino-acid substitutions at B8 stabilize the T-state but block receptor binding whereas L-amino-acid substitutions destabilize the T-state but can be highly active . The structure of insulin as an engineered monomer in solution resembles the T state. This figure is reprinted from the Supplement to Ref with permission of the author.
Insulin exists primarily as a monomer at low concentrations (~10 -6 M) and forms dimers at higher concentrations at neutral pH . At high concentrations and in the presence of zinc ions, insulin forms hexameric complexes . We shall begin with a discussion of the insulin monomer, which is the circulating state of the molecule in plasma, and then discuss its self-assembly. This section culminates with a description of the IR and evidence for a novel receptor-bound conformation of insulin .
THE INSULIN MONOMER
The A- and B chains of insulin (individually illustrated in Figures 7a and b, respectively) exhibit extensive secondary structure despite their limited lengths (Figure 3). The A chain consists of two α -helical segments (A1-A8 and A12-A19) that are nearly antiparallel. These helices are connected by a non-canonical turn (residues A9-A12), bringing into proximity the N- and C-chain termini. The B chain (Figure 7b) contains central α -helix (residues B9-B19) flanked by disulfide bridges (cystines A7-B7 and A20-B19) and β -turns (B7-B10 and B20-B23); residues B1-B5 are extended in the T state . Each β -turn contains at least one conserved Gly with positive dihedral angle (residues B8, B20, and B23). The B7-B10 β-turn enables the side chain of His B5 to interact with the central region of the A chain I association with cysteine A7-B7. The B20-B23 β -turn orients the C-terminal segment of the B-chain (residues B23-B30) in close proximity and antiparallel to the central B-chain α -helix. Residues B24-B28 have a β-strand structure. The conserved aromatic side chains of Phe B24 and Tyr B26 are in contact with Leu B11 , Val B12 and Leu B15 of the central B-chain α -helix, defining an α -turn- β supersecondary structure.

Figure 7 . The structures of insulin A- and B-chains. The figure illustrates insulin A-chain (a) and B-chain (b) as determined from the three-dimensional X-ray analysis of the T 6 hexamer (2-Zn insulin). Both chains are viewed perpendicular to the threefold symmetry axis of the insulin hexamer (see text).
Joining of the A- and B-chain buries the sulfur atoms of cystines A6-A11 and A20-B19 as well as the nonpolar side chains of Ile A2 , Val A3 , Leu A16 , Tyr A19 , Leu B6 , Leu B11 , and Leu B15 . The edge of the hydrophobic core in part exposes Val A3 , Ile A13 , Val B12 , Val B18 , Phe B24 , and Tyr B26 . Together, these conserved residues contribute to the stability of the native structure. The latter residues also contribute to the surface of the molecule that is partially exposed to solvent in the monomer and involved in dimerization or hexamer assembly discussed below.
The conserved Phe at position B25 is of special interest. Whereas in the T state its main chain amide group hydrogen bonds to the carbonyl oxygen of Tyr A19 , the side chain can adopt different orientations (Figure 8). In molecule I Phe B25 is folded against the hydrophobic core of the same protomer but in molecule II the aromatic ring is displace outward. The actual orientation of Phe B25 in solution is likely an intermediate conformation .Molecules I and II also display notable differences in A-chain structure. Subtle differences between the two molecules of the dimer are also seen in the pattern of hydrogen bonds in the N-terminal A-chain α -helix (A1-A8). These differences are accentuated in the TR transition (below) and may foreshadow the mechanism of induced fit on receptor binding . Such findings illustrate the general principle that insulin (like other globular proteins) exhibits highly organized structure that may nonetheless undergo adjustments on assembly or interactions with ligands or other proteins.

Figure 8 . Structural illustration of the monomer-monomer interface in the insulin dimer. The dimer is viewed along the crystallographic 2-fold axis. The side chains of residues Val B12 , Leu B15 , Phe B24 , Phe B25 , and Tyr B26 which form the core of the insulin dimer are illustrated in the figures (not labeled: Pro B28 ). Four main-chain hydrogen bonds are formed from the main-chain atoms of Phe B24 and Tyr B26 are illustrated as dotted lines. In the panel (b) is a magnified view of the dimer interface in panel (a).
INSULIN SELF-ASSEMBLY
Dimerization. The T 6 insulin hexamer contains three dimers in which molecules I and II form an extensive nonpolar interface (Figure 8a). The C-terminal segments of each B-chain come together to form an antiparallel β-sheet (residues B24-B28 and its dimer-related mates). This sheet, containing four intermolecular main-chain hydrogen bonds, is further stabilized by hydrophobic interactions involving the side chains of Val B12 , Tyr B16 , Phe B24 , Tyr B26 , Pro B28 , and to some extent, Phe B25 (Figure 8b). These residues are shielded from contact with solvent (with the exception of Phe B25 ).Although dimerization is associated with local and non-local damping of conformational fluctuations within the protein (relative to the isolated monomer) , an entropic drive is obtained from desolvation of non-polar surfaces, predicted to liberate bound water molecules into the bulk solution . Dimerization does not require zinc ions and exhibits a dissociation constant K d of approximately 10 -5 M .
Hexamer Formation . In the presence of Zn 2+ , insulin dimers associate to form hexameric units coordinated by two zinc ions within the central axis of the hexamer (Figure 6). These axial Zn 2+ ions are coordinated to the imidazole groups of His B10 (three per zinc ion) and in a particular instance to His B5 . Several hexameric forms of insulin have been crystallized of which the classical T 6 structure (“2-Zn insulin” ) remains the prototype . In the this hexamer three dimers are related by a 3-fold symmetry axis, which is located in the hydrophilic pore at the center of the hexameric unit that connects the two Zn 2+ ions . Each zinc ion is octahedrally coordinated to three His B10 imidazole nitrogens and three water molecules. The three-fold symmetry axis is perpendicular to the approximate two-fold symmetry axis of the dimers. Contacts between dimers in the hexamer are less extensive than contacts between protomers within the dimer . The T 6 hexamer is approximately 50 Å in diameter and 35 Å in height .
Allostery among Hexamers . In crystals and in solution insulin forms three structural families of hexamers (T 6 , T 3 R f 3 , and R 6 ). The equilibrium between these structures is modulated by salt concentration and the binding of phenolic ligands (which favors the R state or frayed R f state). The T 3 R f 3 hexamer (formerly designated 4-Zn insulin with rhombohedral crystal form) and R 6 hexamer are arranged similarly to the classical T 6 hexamer in overall aspects. The local and non-local structural rearrangements among these three families of hexamers are collectively designated the TR transition . Molecular analysis of this transition has provided an influential biophysical model for the propagation of conformational change in protein assemblies . Because elements of the TR transition may also pertain to the mechanism of receptor binding (below), we shall describe salient features of the T 3 R f 3 , and R 6 hexamers in turn.
T 3 R f 3 Hexamers (4-Zn Insulin). The classical “rhombohedral transition” of zinc insulin crystals was induced by high concentrations of sodium chloride . The structural basis of this transition was elucidated by D. C. Hodgkin and coworkers in 1976 . Each dimeric unit consists of one molecule I and one molecule II monomer. Whereas in the hexamer the molecule I trimer (T 3 ) has the same octahedral zinc-ion coordination as in the T 6 hexamer, the molecule II trimer (R f 3 ) exhibits substantial, however, displays structural reorganization. The N-terminal B-chain residues B3-B8 (with “fraying” of flexible terminal residues B1 and B2) forms a continuous extension of the central B9-B19 α -helix. This transition in secondary structure, which entails a movement of more than 25 Å at B1 , is coupled to a change in coordination of the second axial zinc ion from octahedral to tetrahedral; the stoichiometry of zinc ions per hexamer is on average 2.67 . The TR transition also causes a rotation of the A1-A8 α -helix (thus requiring a reorganization of the details of side-chain packing in the hydrophobic core and change in conformation of the A7-B7 disulfide bridge) and small displacement of the B24-B28 β -strand away from the A chain, breaking the main-chain hydrogen bond between Phe B25 and Tyr A19 . Similar T 3 R f 3 hexamers may be induced at lower salt concentrations by phenolic ligands wherein the R f 3 trimer contains three bound phenolic molecules .
R 6 Hexamers. High concentrations of phenolic ligands induce a further conformation change to form the R 6 hexamer . The hexamer contains six (or uncommonly seven) bound phenolic ligands. Crystal forms exist which exhibit rigorous sixfold symmetry or which contain six independent protomers in the asymmetric unit with only quasi-sixfold symmetry. In the R 6 hexamer each protomer contains a continuous B1-B19 α -helix and breakage of the B25-A19 main-chain hydrogen bond association with a small displacement of the B-chain C-terminus segment from the A chain. The specific binding site for the phenolic ligand does not pre-exist in the T 6 structure but is may occur in nascent form as part of an extended conformational equilibrium among the three hexamer types. In this R-state-specific binding pocket two hydrogen bonds engage the phenolic hydroxyl group from the A6 carbonyl oxygen and A11 amide hydrogen. The side chain of His B5 packs against each phenolic molecule. Tetrahedral coordination of the zinc ions resembles that of the salt-induced R f 3 trimer of 4-Zn insulin (above) .
Although the TR transition was originally defined in the crystalline state, spectroscopic studies have verified that an analogous equilibrium exists in solution . This conformational transition of B-chain secondary structure has been studied extensively in solutions of hexameric insulin by monitoring changes in the coordination of Co 2+ (Ni 2+ and Co 2+ also form hexamers) from octahedral to tetrahedral using visible-absorbance spectroscopy , changes in α -helical content detected using circular dichroism , or 2D-NMR . The solution structure of the phenol-stabilized R 6 hexamer resembles the crystal structure .
In addition to the clues provided by the TR transition with respect to the mechanism of receptor binding (next section), the phenol-stabilized R 6 hexamer exhibits augmented thermodynamic and kinetic stability relative to the T6 hexamer. Retarding physical- and chemical degradation of the polypeptide chains, these favorable biophysical properties have been exploited in pharmaceutical formulations to increase the shelf-life of insulin products . Because phenolic ligands were traditionally employed in insulin formulations due to their bacteriostatic properties , their additional role as protein-stabilizing agents and their elegant structural role in the hexamer represents the value of serendipity as a source of therapeutic advance .
INSULIN STRUCTURE-FUNCTION RELATIONSHIPS
It is the insulin monomer that binds to and triggers the insulin receptor. A key unresolved issue is the extent to which the monomer undergoes a change in conformation on binding to the insulin receptor. It is likely that the molecular understanding of how insulin binds were be deepened in the next five years through advances in structural biology of the insulin receptor .
The predominance of structural information as described above pertains to insulin hexamers as described above. Although of biological relevance (insulin undergoes zinc-dependent hexamer self-assembly and micro-crystallization in the secretory granule of the β -cell ), it is unlikely that a substantial amount of hexamer exists in the plasma wherein the concentration of zinc ions is low. Further, although zinc-free dimers are be present in the portal circulation, progressive dilution of the secreted insulin in the systemic circulation would lead to a predominance of monomeric molecules. NMR studies confirm that the conformation of the free monomer in solution resembles that of the T-state crystallographic protomer , but its flexibility raises the possibility that receptor binding is associated with induced fit.
With this caveat in mind, the three-dimensional crystal structure of insulin has nonetheless allowed specific residue positions and side-chain orientations to be related to biological activity. Such analogs have been obtained by synthetic methods , comparison of species variants , and site-directed mutagenesis . Together, such analyses of structure-activity relationships in insulin have yielded an understanding of which residues and positions are necessary for receptor binding . Although such data may be confounded by indirect effects of amino-acid substitutions on the structure of the hormone, overall aspects of the long-sought structure of the hormone-receptor complex have been inferred from photo-cross-linking studies and recently been confirmed in a low-resolution co-crystal structure of insulin bound to a fragment of the receptor ectodomain .
Several assays have been used to determine the binding potency of insulin analogs such as (a) the in vivo mouse convulsion assay, (b) in vitro receptor binding studies of analogs in competition with radio-iodinated insulin, and (c) by the ability of insulin analogs to enhance 14 C-glucose oxidation, or conversion of 3 H-glucose into lipids in adipocytes . Most of these studies show a strong correlation between receptor binding and biological activity , except for high-binding affinity analogs (>120%) which show only 100% activity in vivo probably due to rapid clearance or very low-binding analogs which may accumulate at the cell surface and generate a higher-than-expected activity due to decreased clearance .
Three conserved regions in insulin have been of particular interest in the primary receptor-binding surface of insulin: (i) the N-terminal and C-terminal segments of the A chain (Gly A1 -Ile A2 -Val A3 -Glu A4 and Tyr A19 -Cys A20 -Asn A21 ), (ii) the central α -helix of the B chain (especially Val B12 ) and (iii) and the C-terminal segment of B chain (Phe B24 -Phe B25 -Tyr B26 ). All of these residues are located on or near the surface of insulin and therefore may interact with insulin receptor . This surface is notable for clinical mutations associated with a monogenic syndrome of adult-onset diabetes mellitus. The substitutions are Val A3 Leu (Insulin Wakayama), Phe B24 Ser (Insulin Los Angeles), and Phe B25 Leu (Insulin Chicago) . Whereas the A3 and B25 mutations markedly impair receptor binding, Ser B24 impairs binding by less than tenfold as will be discussed in the final section of this chapter . Evidence for the proximity of these three surfaces to the insulin receptor has been obtained by residue-specific photo-cross-linking studies .
The above surfaces of insulin are classified as its “Site-1” related binding surface by DeMeyts and colleagues in relation to a proposed “Site-2” related surface . Sites 1 and 2 pertain to a proposed architecture and mode of binding of the insulin receptor . The putative Site-2 related surface of insulin, although not rigorously established in the hormone-receptor complex, is proposed to correspond to its hexamer-forming surface, including residues His B10 , Leu B17 , Val B18 , Ser A12 , Leu A13 and Glu A17 . Substitutions in Site 2 affect the kinetic properties of hormone binding disproportionately to effects on affinity. Such kinetic properties (related to the residence time of the hormone-receptor complex) correlate with relative post-receptor signaling pathways; prolonged residence times favor mitogenic signaling relative to metabolic signaling . Although the location of Site 2 in the ectodomain of the insulin receptor is not well defined, such interactions are likely to be of pharmacological interest in relation to the risk of cancer in patients exposed to high doses of insulin .
We discuss in turn structure-activity relationships in the A- and B chains and conclude this section with a brief summary of structural relationships in the ectodomain of the insulin receptor.
A-Chain Analysis. The low-resolution structure of insulin bound to a fragment of the receptor strongly suggests that the A chain retains its native secondary structure (with two α -helices) and tertiary U-shaped structure on receptor binding .
The N-terminal residues of the A chain are conserved among vertebrate insulins and have been extensively investigated for their relevance in ligand receptor interactions. N-acetylation of the A-chain N-terminus results in a reduction of receptor binding to approximately 30%, suggesting the importance of a positively charged free amino group at A1 . Deletion of Gly A1 also results in a reduction in receptor binding to 15% of native hormone. Substitution of Gly A1 by diverse L-amino acids results in analogs having reduced binding of 2-20% whereas D-substitutions are well tolerated . The precise size, shape and hydrophobicity of Ile A2 and Val A3 are stringently required for high-affinity receptor binding . The A2 and A3 side chains adjoin the C-terminal α -helix of the receptor α -subunit (designated α CT; see below) in the low-resolution structure of insulin bound to a receptor fragment . Although the details of side-chain packing were not resolved, a nonpolar interface is implied by the predicted registry of the respective α -helices at this interface. Such contacts are in accord with photo-cross-linking studies .
The invariant C-terminal A-chain residues (Tyr A19 -Cys A20 -Asn A21 ) have also been studied and may have dual roles in structure and function. Whereas the primary role of Tyr A19 is likely to be structural through its long-range packing with the side chain of Ile A2 , the para -OH group of Tyr A19 is exposed to solvent and may contact the receptor. Substitution by Phe or Trp or modification of the ring by mono- or diiodination impairs activity. (The other tyrosine in the A-chain (Tyr A14 ) is not conserved and may be modified with little change in activity .) Removal also impairs activity but substitutions are well tolerated. In the crystal structure the A21 main-chain amide donates a hydrogen bond to the main-chain carbonyl of Gly B23 , and Katsoyannis and colleagues have provided elegant evidence (based on the inductive effect of fluoro-substitutions) that the strength of this hydrogen bond contributes to the efficiency of disulfide pairing in chain combination . The side chain of Asn A21 was not well defined in the low-resolution structure of insulin bound to a receptor fragment .
B-Chain Analysis . The B chain of insulin has been more extensively studied than the A chain, particularly with respect to the TR transition (B1-B8) and receptor-binding determinants in the C-terminal β -strand (B24-B28) . Although neither of these segments was well visualized in the low-resolution structure of insulin bound to a receptor fragment , their absence is likely to reflect technical features of the model system (such as disorder in the co-crystals or absence of the fibronectin-homology receptor domains; see below). Nonetheless, the relative positions of structural elements in the low-resolution co-crystal structure suggests that the B chain (unlike the A chain) undergoes a change in conformation involving its unseen N- and C-terminal segments.
The immediate N-terminal B-chain residues (Phe B1 -Val B2 -Asn B3 -Gln B4 ) can be deleted with only modest reductions in biological activity (60-70% that of insulin in lowering blood glucose in rabbits) . These results suggested that the N-terminal four residues are of limited significance in the hormone-receptor complex. By contrast, successive removal of His B5 results in an analog, des -pentapeptide(B1-B5)insulin, that possesses only 15% activity . Further deletion of Leu B6 results in a compound with < 1% binding affinity; and substitution of Leu B6 by other amino acids (Gly, Ala, and Phe) in full-length insulin results in reduced binding [< 0.1%-10%] in the order presented . Cystine A7-B7 lies on the surface of the free insulin monomer and may in principle contribute to receptor binding.
Evidence for the importance of Gly B8 to the biological activity of insulin has been obtained by non-standard mutagenesis . This position represents the crux of the TR transition. In the T-state Gly B8 lies at the junction between the central α -helix of the B-chain (as part of the B7-B10 β -turn) and its N-terminal segment whereas in the R state Gly B8 (like His B5 ) is part of an extended α -helix. Gly B8 exhibits different dihedral angles in the two states: positive in the T-specific β -turn and negative in the R-specific α -helix. Substitution of Gly B8 by D- (or L-) amino acids leads to stereospecific stabilization (or destabilization) of the T-state. Remarkably, the stabilizing D-substitutions markedly impair receptor binding . This impairment is associated with a shift in the conformational equilibrium among T 6 , T 3 R f 3 , and R 6 hexamers favoring the T-state . The low biological activity of such nonstandard analogs is ascribed to stabilization of a native-like but inactive T-state conformation . In four different low-resolution structures of insulin or insulin analogs bound to a receptor fragment , the inferred dihedral angle of Gly B8 appears to be R-like in three of the structures and T-like in the remaining structure. It is possible that Gly B8 is a site of conformational change in the hormone-receptor complex but not coupled to an R-like α -helical transition of residues B1-B7. An alternative possibility is that a Gly B8 “switch” functions to adjust the conformation of cystine A7-B7 and in turn optimize the spatial relationship between the central B-chain α -helix and N-terminal A-chain α -helix.
Importance of the B-Chain COOH-terminus. The B-chain C-terminal β -strand is the most extensively investigated region of insulin. Deletion analysis has revealed that the C-terminal residues Tyr B26 , Thr B27 , Pro B28 , Lys B29 , and Thr B30 may be removed; the resulting analog des -pentapeptide(B26-B30)-insulin-amide (i.e., lacking a C-terminal carboxylate) is fully active . This and related truncated templates have been widely employed in studies of structure-activity relationships and for synthesis of a pioneering B25-specific photo-cross-linking reagent by Katsoyannis and colleagues . The C-terminal five residues are nonetheless necessary for dimerization and hexamer formation . Crystal and NMR-based structures of such truncated analogs retain native-like structures in the α -helical core of the protein . Despite the dispensability of residues B26-B30 for receptor binding, successive removal of Phe B25 and Phe B25 (with analogous C-terminal amidation) progressively impairs receptor binding (relative affinities 6% and 0.2% binding, respectively) . Photo-cross-linking studies have provided evidence that these side chains contact the insulin receptor and may also provide sites of conformational change .
Phe B24 . The aromatic ring of Phe B24 stabilizes the B20-B23 β -turn and seals one edge of the hydrophobic core adjoining cysteine A20-B19. The functional importance of Phe B24 is indicated by the efficient photo-cross-linking of a para -azido-Phe B24 probe to insulin receptor and by the low activities of diverse analogs, including Tyr B24 and Ala B24 (each impaired by >50-fold) . Surprisingly, however, substitution of Phe B24 by Gly is well tolerated , and D-amino-acid substitutions can even enhance receptor binding . Although a range of NMR and fluorescence studies of such anomalous analogs have been reported with varying results (which may in part reflect protein flexibility rather than stable elements of structure) , NMR studies of a super-active engineered monomer containing D-Ala B24 have suggested that the main-chain at position B24 may be a site of a conformational change on receptor binding , leading to detachment of the B24-B28 β -strand from the α -helical core of the hormone as long envisaged . Such detachment is supported by the low-resolution co-crystal structure of a model hormone-receptor complex . Interestingly, D-amino-acid substitutions at position B24 impair (rather than enhance) the binding of truncated insulin analogs lacking residues B26-B30 . A possible explanation for this finding is that chiral perturbation is not needed to displace the truncated β -strand whereas the D-side chain itself is less favorably oriented in such analogs.
Phe B25 . Photo-reactive insulin analogs containing para -azido-Phe or para -benzoyl-Phe at position B25 efficiently cross-link to the receptor . For this reason and due to the marked inactivity of Phe B25 Leu (Insulin Chicago) , substitutions of Phe B25 has been extensively studied. High activity requires a trigonal (sp2 hybridized) -carbon as in aromatic side chains rather than a tetrahedral (sp3 hybridized) -carbon as in Leu . Tolerance of β -napthyl-alanine nonetheless suggests that there is room at the hormone-receptor interface for a larger aromatic side chain. Interestingly, the activities of certain low-binding analogs, such as [homo-Phe B25 ]-insulin, are in part rescued when B26-B30 is removed. These experiments suggest that PheB25 may make two contributions to receptor binding: the specific docking of its side chain and as a further site (in addition to B24) of main-chain conformational change leading to detachment of the B-chain C-terminal segment.
STRUCTURE OF THE INSULIN RECEPTOR
Nearly forty-five years after the structure of insulin was first solved, it is known only in part how insulin binds to its receptor, progress being slowed by the biochemical and structural complexity of the insulin receptor, a heavily-glycosylated disulfide-linked (αβ) 2 dimer.
In the biosynthesis of the insulin receptor each pro-receptor monomer is proteolytically cleaved into an N-terminal α -chain and C-terminal β -chain linked by a single disulfide bond. The extracellular portion of the (αβ) 2 dimer includes both α -chains as part of each transmembrane β-chain. Each receptor monomer consists of several structural domains: from the N-terminus, a leucine‑rich repeat domain L1 (residues 1-157), a cysteine‑rich region (CR, residues 158-310) a second leucine‑rich repeat domain L2 (residues 311-470), and three fibronectin type-III domains: FnIII‑1 (residues 471-595), FnIII‑2 (residues 596-808) and FnIII‑3 (residues 809-906). FnIII‑2 contains a ~120‑residue insert domain (ID, residues 638-756) which contains the α / β cleavage site. C‑terminal of the FnIII‑3 domain lies a single transmembrane α -helix, followed by a ~40‑residue intracellular juxtamembrane region (JM), a tyrosine kinase (TK) catalytic domain and a ~100‑residue C‑tail.
Crystallographic studies of the free ectodomain by C. Ward and colleagues have shown that the extracellular domain assembles as an inverted “V”; in each protomer the L1-CR-L2 domains forming one leg and the three FnIII domains the other (Figure 9). One protomer is related to the other in the dimer by a two-fold rotation about the axis of the inverted “V”, resulting in the L1-CR-L2 leg of one monomer being packed against the three FnIII domains of the other. As discussed above, current models envisage that hormone binding is mediated by two adjoining structural elements termed Site 1 and Site 2 . Both sites are required for high-affinity hormone binding and negative cooperativity .

Figure 9 . Structure of the free ectodomain of the insulin receptor. The original structure was determined by Ward and colleagues and refined to include the α CT element (magenta within dashed ovals) by Lawrence and colleagues . The disulfide-linked dimer of intact a subunits and truncated β subunits (( αβ ) 2 ) was stabilized by F ab immunoglobulin fragments. The dimeric structure adopts an inverted-V conformation. Abbreviations: α CT, C-terminal α -helical element of the α -subunit; CR, cysteine-rich domain; FnIII-1,2, and 3, fibronectin homology domains 1, 2 and 3; ID α , the portion of the insert domain within the α -subunit; and L1 and L2, large domains 1 and 2. We thank M. C. Lawrence for preparation of the figure (reprinted from the web-based Supplement to Ref with permission).
The primary hormone-binding surface is provided by Ste 1, containing two distinct regions: (i) the central of the three β ‑sheets that make up the L1 domain, and (ii) the last 16 residues of the α -chain (the so-called α CT segment). In the intact ectodomain the L1 and α CT elements belong to different α -subunits in the (αβ) 2 dimer. Co-crystals diffracting to 3.9 Å have been obtained of insulin or truncated insulin analogs bound to an L1-CR fragment of the ectodomain together with a synthetic α CT peptide . Interpretation of the electron-density map was aided by modeling based on the known structure of the L1-CR-L2 fragment and the lack of significant structural changes in the complex. Three tubes of electron density were observed fitting the known lengths and orientations of the three α -helices in insulin (the central B-chain α -helix and two A-chain α -helices). The resulting model (Figure 10) is remarkable for its unexpected features. Whereas extensive interactions had been predicted between insulin and L1, the major interface is between insulin and α CT. The role of L1 is to bind and orient the opposite face of α CT. Further, the position and orientation of α CT on the nonpolar surface of L1 differed from its position and orientation in the free ectodomain . Such repositioning is significant as the bound location of α CT is such as to displace residues B26-B30 from their positions in classical structures of insulin. As a further surprise, with the exception of the B-chain C-terminal segment (not visualized in the reported structure), it is the A chain (and not the B chain) that provides the more extensive receptor-binding surface. The key A-chain recognition element comprises A1-A4 as anticipated by mutagenesis and chemical modification as discussed above.

Figure 10 . Structure of the “micro-receptor” complex between insulin and a fragment of the insulin receptor . This ternary complex contains an L1-CR fragment of the receptor α -subunit, a synthetic α CT peptide, and insulin. Abbreviations: α CT, C-terminal α -helical element of the α -subunit; CR, cysteine-rich domain; InsA and InsB, respective A- and B chains of insulin; and L1, large domain 1. We thank M. C. Lawrence for preparation of the figure (reprinted from Ref with permission).
The current low-resolution structure rationalizes a wealth of prior biochemical data but leaves unanswered many questions. How and why the B chain changes conformation on receptor binding and how such changes may be propagated to effect signal transduction are not clear. Further, because residues B1-B7 were not visualized, the relevance of an R-like transition in secondary structure could not be definitively evaluated. A possible location for Site 2 was suggested when the structure of the insulin receptor ectodomain dimer was determined : loops at the junction of the FnIII‑1 and FnIII‑2 domains from the protomer opposite to that contributing the L1 domain to Site 1. This model is consistent with the "cross-linking" trans binding mechanism of receptor activation . To date no co-crystals have been reported with receptor fragments containing Site 2.
MUTATIONS IN THE INSULIN GENE
The classical insulopathies (Insulins Chicago, Los Angeles and Wakayama) accumulated in the blood stream as mutant insulins. These all have point mutations that lower receptor binding and biological activity. Rare clinical syndromes result from mutations elsewhere affecting key steps in biosynthesis within the β -cell .
A monoallelic genetic mutation resulting in substitution of His B10 by Asp leads to the secretion of a mutant proinsulin (319): Asp B10 -proinsulin (Proinsulin Providence) due to its selectively increased export via constitutive pathways of a significant proportion of the prohormone (approx. 1/3); this fraction thus escapes processing but also undergoes increased endosomal pathway-mediated degradation, resulting in circulating hyperproinsulinemia in a mouse model . No defect was observed in the processing of the fraction of the Asp B10 proinsulin that remained within the regulated secretory pathway. Asp B10 -insulin also exhibits enhanced binding affinity and this property, in its prohormone may be related to its possible mis-sorting, i.e. via insulin receptor mediated intracellular trafficking. Expression in cultured β-cells has confirmed that the mutant proinsulin can be fully processed at normal rates, but a large fraction of unprocessed mutant prohormone is released via an unregulated-constitutive pathway . Proinsulin processing is also impaired when Arg65 of the C-domain dibasic pair (Lys64-Arg65) is mutated to His65 or Leu65 (Proinsulin Boston/Denver/Tokyo and Proinsulin Kyoto, respectively) . In all these cases the defect lies in the inability of processing enzymes (PC3/PC1 and PC2) to cleave at the mutated cleavage site, which leads to the secretion of a partially cleaved intermediate form of proinsulin.
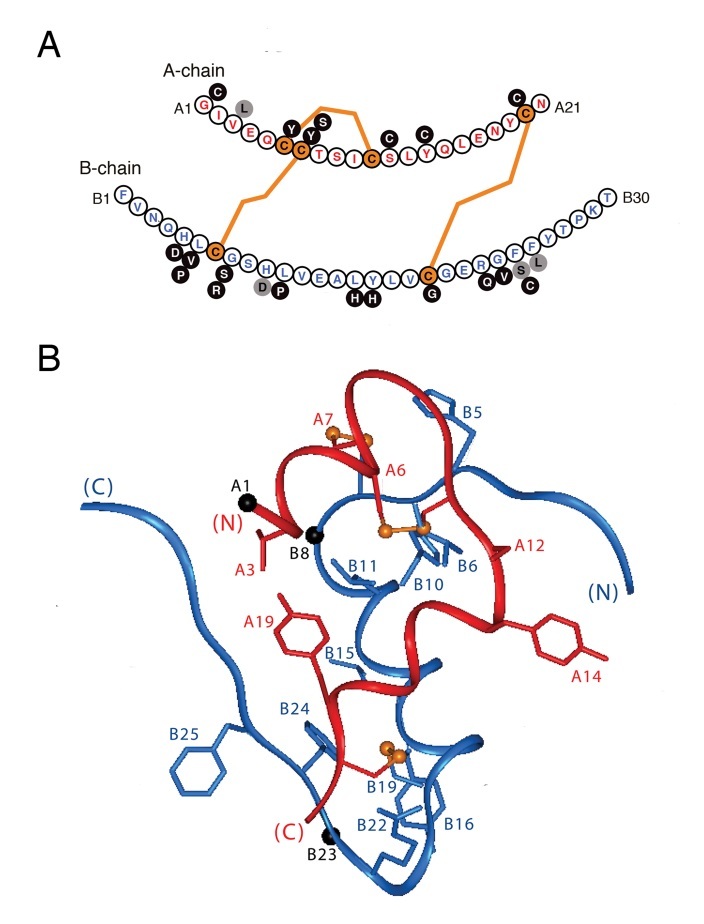
Figure 11 . Mutations in the insulin moiety of proinsulin associated with neonatal diabetes mellitus. (A) Such mutations may be classified as cysteine-related or non-cysteine-related. The latter provide probes for structural determinants of the efficiency of disulfide pairing in the redox-coupled folding of nascent proinsulin (for review, see ).
It has recently been found that the majority of dominant negative mutations in the insulin gene affecting biosynthesis and folding of the hormone lead to permanent neonatal-onset DM with impaired secretion of the variant or wild-type insulin or proinsulin . Impaired β -cell function often develops prior to maturation of the immune system and so presents as an auto-antigen negative form of apparent Type 1 DM. Dominant mutations in the insulin gene define the second most common genetic cause of permanent neonatal DM (relative to heterozygous activating mutation in a subunit of the β -cell voltage-gated potassium channel, either KCNJ11 (encoding the Kr6.2 subunit) or ABCC8 (encoding the Sur1 subunit) ). Rarely the β-cells survive for years or even decades in the cases of apparently less damaging insulin mutations, as mentioned below.
Mutations in the insulin gene causing neonatal diabetes occur in each region of preproinsulin, including its signal peptide, A-, B- and C domains. The majority result in addition or removal of a cysteine, leading in either case to an odd number of potential pairing sites: the resulting imbalance leads in general to misfolding and aggregation . Mutations identified in the insulin moiety of proinsulin are shown in Figure 11A (sequence) and Figure 11B (structure). One human mutation encodes the same “Akita” substitution (Cys A7 Tyr) as in the Ins2 gene of the Mody4 mouse ; this dominant murine substitution thus provides a relevant model of progressive β -cell failure . The variant murine proinsulin in vitro undergoes partial unfolding with increased aggregation . Analogous perturbations were characterized in human insulin- and proinsulin analogs lacking cystine A7-B7 . Heterozygous expression of a variant Ins2 allele encoding Cys A6 Ser (identified in an N-ethyl-N-nitrosourea screen) likewise induces DM . The identification of identical human and murine mutations at position A7 suggests that the pathogenesis of neonatal DM in these patients is likewise similar to that of the Akita mouse . Biochemical studies of clinical variants in β -cells or neurosecretory cell lines have revealed perturbations in disulfide pairing, which range from severe or mild depending on the site of mutation and the properties of the substituted side chain .
Akita β -cells exhibit an early defect in the folding and trafficking of both wild-type and variant proinsulins with elevated markers of ER stress, electron-dense deposits in abnormal ER and GA, mitochondria swelling, and progressive loss of β -cell mass . Extension of such findings to human variants is supported by mouse models of human mutant proinsulin constructs .

Figure 12 . Structural features of insulin contributing to foldability. Stereo stick representations of selected regions of the insulin protomer (2-Zn molecule 1; Protein Databank identifier 4INS). Unless otherwise indicated, A-chain is shown in gray and B-chain in black. Selected A-chain side chains are highlighted in color (below); golden balls indicate sulfur atoms of disulfide bridges. (A) Environment of solvent-exposed cystine A7-B7 showing key role of the phi dihedral angle of Gly B8 (arrow), predicted to influence orientation of thiolate B7 to favor disulfide pairing. The side chains of His B5 and Leu B6 are shown in red. (B) Environment of internal cystine A6-A11 highlighting packing of Leu B6 and Ile B11 (red). (C) Environment of internal cystine A20-B19 highlighting packing of A- and B-chain side chains in hydrophobic core: (magenta) Leu A16 and Tyr A19 ; (red) Val B12 , Leu B15 , Gly B23 (ball at C α position), and Phe B24 . Important structural sites not shown: the inter-chain A3-related crevice (lined by Ile A2 , Tyr B26 and Pro B28 ) and T-state-specific tertiary contacts between the N-terminal arm of the B-chain (residues B1-B5) and the A-chain. This figure is adapted from Ref with permission of the author.
Whereas an odd number of cysteines presumably induces a severe block to folding, non-cysteine-related mutations are also observed in such patients (Figure 11). Such sites of mutation identify residues whose side chain or main-chain conformation contributes to the efficiency of folding. Selected structural relationships in the native state presumed to direct specific disulfide pairing in the oxidative folding pathway of proinsulin are illustrated in Figure 12. The folding process may be visualized as a series of trajectories on free-energy landscapes of progressive steepness with successive disulfide pairing (Figure 13A). This pathway highlights the importance of cysteine A20-B19 as a key initial folding intermediate associated with partial formation of native-like super-secondary structure as shown at left in Figure 13B . Competing subsequent routes to the native state are likely to yield a variety of partial folds with accessible unpaired cysteines . Aberrant disulfide bond formation between such partial folds may account for the interruption of wild-type proinsulin folding in the presence of a non-foldable clinical variant.

Figure 13 . Energy landscape view of proinsulin folding and disulfide pairing. (A) Formation of successive disulfide bridges may be viewed as enabling a sequence of folding trajectories on a succession of steeper funnel-shaped free-energy landscapes. (B) Preferred pathway of disulfide pairing begins with cystine A20-B19 (left), whose pairing is directed by a nascent hydrophobic core formed by the central B-domain α -helix (residues B9-B19), part of the C-terminal B-chain β -strand (B24-B26), and part of the C-terminal A-domain α -helix (A16-A20). Alternative pathways mediate formation of successive disulfide bridges (middle panel) en route to the native state (right). The mechanism of disulfide pairing is perturbed by clinical mutations associated with misfolding of proinsulin. Sites of non-cysteine-related mutations causing neonatal DM (Figure 11 above) highlight native structural features critical to foldability. Figure is reprinted from Ref with permission of the author.
Among these, a few better-tolerated mutations present later in life as auto-antibody-negative presumed Type 1 DM or Type 2 DM. One such mutation, presenting in the second decade as maturity-onset diabetes of the young (MODY), is due to substitution of Arg B22 by Gln. This mutation alters a solvent-exposed site in the B21-B24 β -turn not required for receptor binding . DM presenting in the third decade of life is associated with substitution of Phe B24 by Ser . This mutation causes only a mild impairing of receptor binding but like Gln B22 imposes ER stress at a level intermediate between over-expression of wild-type proinsulin and neonatal variants.
Chronic elevation of more moderate ER stress in β -cells due to subtle mutations in proinsulin (such as substitution of Arg B22 by Gln and the classical SerB24 allele of Insulin Chicago) presumably leads to a slower loss of β -cell mass . Such milder mutations are associated with onset of diabetes later in childhood or early adulthood. Evidence is also accumulating that subtle perturbations of wild-type insulin biosynthesis may contribute to the pathogenesis of non-syndromic Type 2 diabetes as in the Akita mouse . A newly recognized biophysical contribution to such β -cell dysfunction is “molecular crowding” in the ER due to over-expression of nascent proinsulin in the face of peripheral insulin resistance , a concept that may have therapeutic implications .
REFERENCES
1. Steiner, D.F. 1967. Evidence for a precursor in the biosynthesis of insulin. Trans. N. Y. Acad. Sci. 30:60-68.
2. Adams, M.J., Blundell, T.L., Dodson, E.J., Dodson, G.G., Vijayan, M., Baker, E.N., Hardine, M.M., Hodgkin, D.C., Rimer, B., and Sheet, S. 1969. Structure of rhombohedral 2 zinc insulin crystals. Nature 224:491-495.
3. Blundell, T.L., Cutfield, J.F., Cutfield, S.M., Dodson, E.J., Dodson, G.G., Hodgkin, D.C., Mercola, D.A., and Vijayan, M. 1971. Atomic positions in rhombohedral 2-zinc insulin crystals. Nature 231:506-511.
4. Brange, J., Ribel, U., Hansen, J.F., Dodson, G., Hansen, M.T., Havelund, S., Melberg, S.G., Norris, F., Norris, K., and Snel, L. 1988. Monomeric insulins obtained by protein engineering and their medical implications. Nature 333:679-682.
5. Brange, J., and Vølund, A. 1999. Insulin analogs with improved pharmacokinetic profiles. Adv. Drug Deliv. Rev. 35:307-335.
6. Hirsch, I.B. 2005. Insulin analogues. N. Engl. J. Med. 352:174-183.
7. Bell, G.I., Pictet, R.L., Rutter, W.J., Cordell, B., Tischer, E., and Goodman, H.M. 1980. Sequence of the human insulin gene. Nature 284:26-32.
8. Steiner, D.F., Tager, H.S., Chan, S.J., Nanjo, K., Sanke, T., and Rubenstein, A.H. 1990. Lessons learned from molecular biology of insulin-gene mutations. Diabetes Care 13:600-609.
9. Stoy, J., Edghill, E.L., Flanagan, S.E., Ye, H., Paz, V.P., Pluzhnikov, A., Below, J.E., Hayes, M.G., Cox, N.J., Lipkind, G.M., et al. 2007. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. U. S. A. 104:15040-15044.
10. Weiss, M.A. 2013. Diabetes mellitus due to the toxic misfolding of proinsulin variants. FEBS Lett. 587:1942-1950.
11. Greeley, S.A., Tucker, S.E., Naylor, R.N., Bell, G.I., and Philipson, L.H. 2010. Neonatal diabetes mellitus: a model for personalized medicine. Trends Endocrinol. Metab. 21:464-472.
12. Liu, M., Hodish, I., Haataja, L., Lara-Lemus, R., Rajpal, G., Wright, J., and Arvan, P. 2010. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol. Metab. 21:652-659.
13. De Meyts, P., and Whittaker, J. 2002. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat. Rev. Drug Discov. 1:769-783.
14. Greeley, S.A., Naylor, R.N., Philipson, L.H., and Bell, G.I. 2011. Neonatal diabetes: an expanding list of genes allows for improved diagnosis and treatment. Curr. Diab. Rep. 11:519-532.
15. Scott, E.L. 1912. On the influence of intravenous injections of an extract of the pancreas on experimental pancreatic diabetes. Am. J. Physiol. 29:306-310.
16. Duvillie, B., Cordonnier, N., Deltour, L., Dandoy-Dron, F., Itier, J.M., Monthioux, E., Jami, J., Joshi, R.L., and Bucchini, D. 1997. Phenotypic alterations in insulin-deficient mutant mice. Proc. Natl. Acad. Sci. U. S. A. 94:5137-5140.
17. Jonsson, J., Carlsson, L., Edlund, T., and Edlund, H. 1994. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 371:606-609.
18. Schӓfer, E.A. 1916. The Endocrine Organs: An introduction to the study of internal secretion . London: Longmans, Green, and Co. 27 pp.
19. Bliss, M., editor. 1982. The discovery of insulin . Chicago, Illinois: University of Chicago Press. 93-99 pp.
20. Nandi, A., Kitamura, T., Kahn, C.R., and Accili, D. 2004. Mouse models of insulin resistance. Physiol. Rev. 84:623-647.
21. Leslie, R.D.G., and Robbins, D.C. 1995. Diabetes: Clinical Science in Practice : Cambridge University Press. 492 pp.
22. Von Mering, J., and Minkowski, O. 1890. Diabetes mellitus nach pankreas exterpation. Arch. Exp. Path. Pharmacol. 26:371.
23. Barron, M. 1920. The relation of the islets of Langerhans to diabetes with special reference to cases of pancreatic lithiasis. In Surgery, gynecology, and obstetrics . Chicago: The Surgical Publishing Company of Chicago. 437-448.
24. Langerhans, P. 1869. Beitrage zur Mikroskopischen Anatomie der Bauchspeicheldruse. In Institute of Pathology . Berlin: University of Berlin. 32.
25. Banting, F.G., and Best, C.H. 1922. The internal secretion of the pancreas. J. Lab. Clin. Med. 7.
26. Ullrich, K.J. 1979. Sugar, amino acid, and Na + cotransport in the proximal tubule. Annu. Rev. Physiol. 41:181-195.
27. Kahn, C.R., Neville, D.M., and Roth, J. 1973. Insulin receptor interaction in the obese hyperglycemic mouse. A model of insulin resistance. J. Biol. Chem. 248:244-250.
28. Petruzzelli, L., Herrera, R., and Rosen, O.M. 1984. Insulin receptor is an insulin-dependent tyrosine protein kinase: copurification of insulin-binding activity and protein kinase activity to homogeneity from human placenta. Proc. Natl. Acad. Sci. U. S. A. 81:3327-3331.
29. Ullrich, A., Bell, J.R., Chen, E.Y., Herrera, R., Petruzelli, L.M., Dull, T.J., Gray, A., Coussens, L., Liao, Y.C., Tsubokawa, M., et al. 1985. Human insulin receptor and its relationship to the tyrosine kinase family of oncogenes. Nature 313:756-761.
30. Ward, C.W., and Lawrence, M.C. 2011. Landmarks in insulin research. Front. Endocrin. 2:1-11.
31. De Meyts, P. 2008. The insulin receptor: a prototype for dimeric, allosteric membrance receptors? Trends Biochem. Sci. 33:376-384.
32. Hopfer, U. 1987. Membrane transport mechanisms for hexoses and and amino acids in the small intestine. In Physiology of the Gastrointestinal Tract . L.R. Johnson, editor. New York: Raven Press. 1499-1526.
33. Haase, W., and Koepsell, H. 1989. Electron microscopic immunohistochemical localization of components of Na+-cotransporters along the rat nephron. Eur. J. Cell Biol. 48:360-374.
34. Rosholt, M.N., and King, P.A. 1995. Diabetes: Clinical science in practice. In Diabetes: Clinical science in practice . R.D.G. Leslie, and D.C. Robbins, editors. Cambridge, MA: Cambridge University Press. 77-95.
35. Fukumoto, H., Kayano, T., Buse, J.B., Edwards, Y., Pilch, P.F., Bell, G.I., and Seino, S. 1989. Cloning and characterization of the major insulin-responsive glucose transporter expressed in human skeletal muscle and other insulin-responsive tissues. J. Biol. Chem. 264:7776-7779.
36. James, D.E., Strube, M., and Mueckler, M. 1989. Molecular cloning and characterization of an insulin-regulatable glucose transporter Nature (Lond) 338:83-87
37. Charron, M.J., and Kahn, B.B. 1990. Divergent molecular mechanisms for insulin-resistant glucose transport in muscle and adipose cells in vivo . J. Biol. Chem. 265:7994-8000.
38. Cushman, S.W., and Wardzala, L.J. 1980. Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J. Biol. Chem. 255:4758-4762.
39. Kono, T., Suzuki, K., Dansey, L.E., Robinson, F.W., and Blevins, T.L. 1981. Energy-dependent and protein synthesis-independent recycling of the insulin-sensitive glucose transport mechanism in fat cells. J. Biol. Chem. 256:6400-6407.
40. Rosholt, M.N., King, P.A., and Horton, E.S. 1994. High-fat diet reduces glucose transporter responses to both insulin and exercise. Am. J. Physiol. 266:R95-101.
41. King, P.A., Betts, J.J., Horton, E.D., and Horton, E.S. 1993. Exercise, unlike insulin, promotes glucose transporter translocation in obese Zucker rat muscle. Am. J. Physiol. 265:R447-452.
42. Wheeler, T.J. 1988. Translocation of glucose transporters in response to anoxia in heart. J. Biol. Chem. 263:19447-19454.
43. Karnieli, E., Chernow, B., Hissin, P.J., Simpson, I.A., and Foley, J.E. 1986. Insulin stimulates glucose transport in isolated human adipose cells through a translocation of intracellular glucose transporters to the plasma membrane: a preliminary report. Horm. Metab. Res. 18:867-868.
44. Clark, A.E., Holman, G.D., and Kozka, I.J. 1991. Determination of the rates of appearance and loss of glucose transporters at the cell surface of rat adipose cells. Biochem. J. 278 ( Pt 1):235-241.
45. Baron, A.D., Brechtel, G., Wallace, P., and Edelman, S.V. 1988. Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am. J. Physiol. 255:E769-774.
46. Barnard, R.J., Lawani, L.O., Martin, D.A., Youngren, J.F., Singh, R., and Scheck, S.H. 1992. Effects of maturation and aging on the skeletal muscle glucose transport system. Am. J. Physiol. 262:E619-626.
47. Bourey, R.E., Koranyi, L., James, D.E., Mueckler, M., and Permutt, M.A. 1990. Effects of altered glucose homeostasis on glucose transporter expression in skeletal muscle of the rat. J. Clin. Invest. 86:542-547.
48. Klip, A., Ramlal, T., Bilan, P.J., Cartee, G.D., Gulve, E.A., and Holloszy, J.O. 1990. Recruitment of GLUT-4 glucose transporters by insulin in diabetic rat skeletal muscle. Biochem. Biophys. Res. Commun. 172:728-736.
49. Unger, R.H. 1991. Diabetic hyperglycemia: link to impaired glucose transport in pancreatic β cells. Science 251:1200-1205.
50. Thorens, B., Wu, Y.J., Leahy, J.L., and Weir, G.C. 1992. The loss of GLUT2 expression by glucose-unresponsive β cells of db/db mice is reversible and is induced by the diabetic environment. J. Clin. Invest. 90:77-85.
51. Kulkarni, R.N., Bruning, J.C., Winnay, J.N., Postic, C., Magnuson, M.A., and Kahn, C.R. 1999. Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96:329-339.
52. Kitamura, T., Kahn, C.R., and Accili, D. 2003. Insulin receptor knockout mice. Annu. Rev. Physiol. 65:313-332.
53. Braun, M., Ramracheya, R., and Rorsman, P. 2012. Autocrine regulation of insulin secretion. Diabetes Obes. Metab. 14 Suppl 3:143-151.
54. Baker, E.N., Blundell, T.L., Cutfield, J.F., Cutfield, S.M., Dodson, E.J., Dodson, G.G., Hodgkin, D.M., Hubbard, R.E., Isaacs, N.W., and Reynolds, C.D. 1988. The structure of 2Zn pig insulin crystals at 1.5 Å resolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 319:369-456.
55. Steiner, D.F. 1977. The Banting Memorial Lecture 1976. Insulin today. Diabetes 26:322-340.
56. Steiner, D.F. 1978. On the role of the proinsulin C-peptide. Diabetes 27 Suppl 1:145-148.
57. Dodson, G., and Steiner, D. 1998. The role of assembly in insulin's biosynthesis. Curr. Opin. Struct. Biol. 8:189-194.
58. Steiner, D.F. 2000. New aspects of proinsulin physiology and pathophysiology. J. Pediatr. Endocrinol. Metab. 13:229-239.
59. Seidah, N.G., and Prat, A. 2007. The proprotein convertases are potential targets in the treatment of dyslipidemia. J. Mol. Med. 85:685-696.
60. Yang, Y., Hua, Q.X., Liu, J., Shimizu, E.H., Choquette, M.H., Mackin, R.B., and Weiss, M.A. 2010. Solution structure of proinsulin: connecting domain flexibility and prohormone processing. J. Biol. Chem. 285:7847-7851.
61. Smith, B.J., Huang, K., Kong, G., Chan, S.J., Nakagawa, S., Menting, J.G., Hu, S.-Q., Whittaker, J., Steiner, D.F., Katsoyannis, P.G., et al. 2010. Structural resolution of a tandem hormone-binding element in the insulin receptor and its implications for design of peptide agonists. Proc. Natl. Acad. Sci. U. S. A. 107:6771-6776.
62. Menting, J.G., Whittaker, J., Margetts, M.B., Whittaker, L.J., Kong, G.K., Smith, B.J., Watson, C.J., Zakova, L., Kletvikova, E., Jiracek, J., et al. 2013. How insulin engages its primary binding site on the insulin receptor. Nature 493:241-245.
63. Abel, J.J. 1926. Crystalline Insulin. Proc. Natl. Acad. Sci. U. S. A. 12:132-136.
64. Murnaghan, J.H., and Talalay, P. 1967. John Jacob Abel and the crystallization of insulin. Perspect. Biol. Med. 10:334-380.
65. Jensen, H., and Evans, J., E. A. . 1935. Studies on crystalline insulin: xviii. the nature of the free amino groups in insulin and the isolation of phenylalanine and proline from crystalline insulin J. Biol. Chem. 108 1-9.
66. Ryle, A.P., Sanger, F., Smith, L.F., and Kitai, R. 1955. The disulphide bonds of insulin. Biochem. J. 60:541-556.
67. Sanger, F. 1959. Chemistry of insulin; determination of the structure of insulin opens the way to greater understanding of life processes. Science 129:1340-1344.
68. Chance, R.E., Ellis, R.M., and Bromer, W.W. 1968. Porcine proinsulin: characterization and amino acid sequence. Science 161:165-167.
69. Harfenist, E.J., and Craig, L.C. 1952. The molecular weight of insulin. J. Am. Chem. Soc. 74 J. Am. Chem. Soc.,.
70. Steiner, D.F., and Oyer, P.E. 1967. The biosynthesis of insulin and a probable precursor of insulin by a human islet cell adenoma. Proc. Natl. Acad. Sci. U. S. A. 57:473-480.
71. Steiner, D.F., Cunningham, D., Spigelman, L., and Aten, B. 1967. Insulin biosynthesis: evidence for a precursor. Science 157:697-700.
72. Steiner, D.F., Clark, J.L., Nolan, C., Rubenstein, A.H., Margoliash, E., Aten, B., and Oyer, P.E. 1969. Proinsulin and the biosynthesis of insulin. Recent Prog. Horm. Res. 25:207-282.
73. Clark, J.L., Cho, S., Rubenstein, A.H., and Steiner, D.F. 1969. Isolation of a proinsulin connecting peptide fragment (C-peptide) from bovine and human pancreas. Biochem. Biophys. Res. Commun. 35:456-461.
74. Brown, H., Sangar, F., and Kitai, R. 1955. The structure of pig and sheep insulins. Biochem. J. 60:555-565.
75. Chan, S.J., Keim, P., and Steiner, D.F. 1976. Cell-free synthesis of rat preproinsulins: characterization and partial amino acid sequence determination. Proc. Natl. Acad. Sci. U. S. A. 73:1964-1968.
76. Lomedico, P.T., Chan, S.J., Steiner, D.F., and Saunders, G.F. 1977. Immunological and chemical characterization of bovine preproinsulin. J. Biol. Chem. 252:7971-7978.
77. Steiner, D.F., Chan, S.J., Welsh, J.M., Nielsen, D., Michael, J., Tager, H.S., and Rubenstein, A.H. 1986. Models of peptide biosynthesis: the molecular and cellular basis of insulin production. Clin. Invest. Med. 9:328-336.
78. Sanders, S.L., and Schekman, R. 1992. Polypeptide translocation across the endoplasmic reticulum membrane. J. Biol. Chem. 267:13791-13794.
79. Kreil, G. 1981. Transfer of proteins across membranes. Annu. Rev. Biochem. 50:317-348.
80. Steiner, D.F., editor. 2001. The prohormone convertases and precursor processing in protein biosynthesis : Academic Press, New York. 163-198 pp.
81. Liu, M., Lara-Lemus, R., Shan, S.O., Wright, J., Haataja, L., Barbetti, F., Guo, H., Larkin, D., and Arvan, P. 2012. Impaired cleavage of preproinsulin signal peptide linked to autosomal-dominant diabetes. Diabetes 61:828-837.
82. Walter, P., Gilmore, R., and Blobel, G. 1984. Protein translocation across the endoplasmic reticulum. Cell 38:5-8.
83. Patzelt, C., Chan, S.J., Duguid, J., Hortin, G., Keim, P., Heinrikson, R.L., and Steiner, D.F. 1978. In Regulatory proteolytic enzymes and their inhibitors . S. Magnusson, M. Ottensen, B. Foltmann, K. Dano, and H. Neurath, editors. New York: Pergamon. p 69 Pergaman, New Yor.
84. Steiner, D.F., Clark, J.L., Nolan, C., Rubenstein, A.H., Margoliash, E., Melani, F., and Oyer, P.E., editors. 1970. The biosynthesis of insulin and some speculation regarding the pathogenesis of human diabetes . New York: John Wiley & Sons. 57 pp.
85. Steiner, D.F., Kemmler, W., Clark, J.L., Oyer, P.E., and Rubenstein, A.H., editors. 1972. The biosynthesis of insulin . Baltimore: Williams & Wilkins. 175-198 pp.
86. Frank, B.H., and Veros, A.J. 1968. Physical studies on proinsulin-association behavior and conformation in solution. Biochem. Biophys. Res. Commun. 32:155-160.
87. Rubenstein, A.H., Melani, F., Pilkis, S., and Steiner, D.F. 1969. Proinsulin. Secretion, metabolism, immunological and biological properties. Postgrad. Med. J. 45:Suppl:476-481.
88. Fullerton, W.W., Potter, R., and Low, B.W. 1970. Proinsulin: Crystallization and preliminary x-ray diffraction studies. Proc. Natl. Acad. Sci. U. S. A. 66:1213-1219.
89. Freychet, P., Brandenburg, D., and Wollmer, A. 1974. Receptor-binding assay of chemically modified insulins. Comparison with in vitro and in vivo bioassays. Diabetologia 10:1-5.
90. Orci, L., Ravazzola, M., Amherdt, M., Madsen, O., Vassalli, J.D., and Perrelet, A. 1985. Direct identification of prohormone conversion site in insulin-secreting cells. Cell 42:671-681.
91. Orci, L., Glick, B.S., and Rothman, J.E. 1986. A new type of coated vesicular carrier that appears not to contain clathrin: its possible role in protein transport within the Golgi stack. Cell 46:171-184.
92. Wattenberg, B.W., and Rothman, J.E. 1986. Multiple cytosolic components promote intra-Golgi protein transport. Resolution of a protein acting at a late stage, prior to membrane fusion. J. Biol. Chem. 261:2208-2213.
93. Nagamatsu, S., Bolaffi, J.L., and Grodsky, G.M. 1987. Direct effects of glucose on proinsulin synthesis and processing during desensitization. Endocrinology 120:1225-1231.
94. Kemmler, W., Peterson, J.D., and Steiner, D.F. 1971. Studies on the conversion of proinsulin to insulin. I. Conversion in vitro with trypsin and carboxypeptidase B. J. Biol. Chem. 246:6786-6791.
95. Nolan, C., Margoliash, E., Peterson, J.D., and Steiner, D.F. 1971. The structure of bovine proinsulin. J. Biol. Chem. 246:2780-2795.
96. Oyer, P.E., Cho, S., Peterson, J.D., and Steiner, D.F. 1971. Studies on human proinsulin. Isolation and amino acid sequence of the human pancreatic C-peptide. J. Biol. Chem. 246:1375-1386.
97. Busse, W.D., and Gattner, H.G. 1973. Selective cleavage of one disulfide bond in insulin: preparation and properties of insulin A7-B7-di-S-sulfonate. Hoppe Seylers Z Physiol. Chem. 354:147-155.
98. Tager, H.S., Emdin, S.O., Clark, J.L., and Steiner, D.F. 1973. Studies on the conversion of proinsulin to insulin. II. Evidence for a chymotrypsin-like cleavage in the connecting peptide region of insulin precursors in the rat. J. Biol. Chem. 248:3476-3482.
99. Davidson, H.W., Rhodes, C.J., and Hutton, J.C. 1988. Intraorganellar calcium and pH control proinsulin cleavage in the pancreatic β cell via two distinct site-specific endopeptidases. Nature 333:93-96.
100. Rhodes, C.J., Lincoln, B., and Shoelson, S.E. 1992. Preferential cleavage of des-31,32-proinsulin over intact proinsulin by the insulin secretory granule type II endopeptidase. Implication of a favored route for prohormone processing. J. Biol. Chem. 267:22719-22727.
101. Julius, D., Brake, A., Blair, L., Kunisawa, R., and Thorner, J. 1984. Isolation of the putative structural gene for the lysine-arginine-cleaving endopeptidase required for processing of yeast prepro-α-factor. Cell 37:1075-1089.
102. Fuller, R.S., Sterne, R.E., and Thorner, J. 1988. Enzymes required for yeast prohormone processing. Annu. Rev. Physiol. 50:345-362.
103. Mizuno, K., Nakamura, T., Ohshima, T., Tanaka, S., and Matsuo, H. 1988. Yeast KEX2 genes encodes an endopeptidase homologous to subtilisin-like serine proteases. Biochem. Biophys. Res. Commun. 156:246-254.
104. Smeekens, S.P., and Steiner, D.F. 1990. Identification of a human insulinoma cDNA encoding a novel mammalian protein structurally related to the yeast dibasic processing protease Kex2. J. Biol. Chem. 265:2997-3000.
105. Smeekens, S.P., Avruch, A.S., LaMendola, J., Chan, S.J., and Steiner, D.F. 1991. Identification of a cDNA encoding a second putative prohormone convertase related to PC2 in AtT20 cells and islets of Langerhans. Proc. Natl. Acad. Sci. U. S. A. 88:340-344.
106. Seidah, N.G., Marcinkiewicz, M., Benjannet, S., Gaspar, L., Beaubien, G., Mattei, M.G., Lazure, C., Mbikay, M., and Chretien, M. 1991. Cloning and primary sequence of a mouse candidate prohormone convertase PC1 homologous to PC2, Furin, and Kex2: distinct chromosomal localization and messenger RNA distribution in brain and pituitary compared to PC2. Mol. Endocrinol. 5:111-122.
107. Shennan, K.I., Smeekens, S.P., Steiner, D.F., and Docherty, K. 1991. Characterization of PC2, a mammalian Kex2 homologue, following expression of the cDNA in microinjected Xenopus oocytes. FEBS Lett. 284:277-280.
108. Bailyes, E.M., Shennan, K.I., Seal, A.J., Smeekens, S.P., Steiner, D.F., Hutton, J.C., and Docherty, K. 1992. A member of the eukaryotic subtilisin family (PC3) has the enzymic properties of the type 1 proinsulin-converting endopeptidase. Biochem. J. 285 (Pt 2):391-394.
109. Zhou, A., Webb, G., Zhu, X., and Steiner, D.F. 1999. Proteolytic processing in the secretory pathway. J. Biol. Chem. 274:20745-20748.
110. Seidah, N.G., and Prat, A. 2012. The biology and therapeutic targeting of the proprotein convertases. Nat Rev Drug Discov 11:367-383.
111. Smeekens, S.P., Montag, A.G., Thomas, G., Albiges-Rizo, C., Carroll, R., Benig, M., Phillips, L.A., Martin, S., Ohagi, S., Gardner, P., et al. 1992. Proinsulin processing by the subtilisin-related proprotein convertases furin, PC2, and PC3. Proc. Natl. Acad. Sci. U. S. A. 89:8822-8826.
112. Tanaka, S., Kurabuchi, S., Mochida, H., Kato, T., Takahashi, S., Watanabe, T., and Nakayama, K. 1996. Immunocytochemical localization of prohormone convertases PC1/PC3 and PC2 in rat pancreatic islets. Arch. Histol. Cytol. 59:261-271.
113. Skidgel, R.A. 1988. Basic carboxypeptidases: regulators of peptide hormone activity. Trends Pharmacol. Sci. 9:299-304.
114. Fricker, L.D., Evans, C.J., Esch, F.S., and Herbert, E. 1986. Cloning and sequence analysis of cDNA for bovine carboxypeptidase E. Nature 323:461-464.
115. Furuta, M., Carroll, R., Martin, S., Swift, H.H., Ravazzola, M., Orci, L., and Steiner, D.F. 1998. Incomplete processing of proinsulin to insulin accompanied by elevation of Des-31,32 proinsulin intermediates in islets of mice lacking active PC2. J. Biol. Chem. 273:3431-3437.
116. Steiner, D.F. 2011. Adventures with insulin in the islets of Langerhans. J Biol Chem 286:17399-17421.
117. Zhu, X., Zhou, A., Dey, A., Norrbom, C., Carroll, R., Zhang, C., Laurent, V., Lindberg, I., Ugleholdt, R., Holst, J.J., et al. 2002. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc. Natl. Acad. Sci. U. S. A. 99:10293-10298.
118. Furuta, M., Yano, H., Zhou, A., Rouille, Y., Holst, J.J., Carroll, R.J., Ravazzola, M., Orci, L., Furuta, H., and Steiner, D.F. 1997. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc. Natl. Acad. Sci. U. S. A. 94:6646-6651.
119. Muller, L., Zhu, X., and Lindberg, I. 1997. Mechanism of the facilitation of PC2 maturation by 7B2: involvement in ProPC2 transport and activation but not folding. J Cell Biol 139:625-638.
120. Westphal, C.H., Muller, L., Zhou, A., Zhu, X., Bonner-Weir, S., Schambelan, M., Steiner, D.F., Lindberg, I., and Leder, P. 1999. The neuroendocrine protein 7B2 is required for peptide hormone processing in vivo and provides a novel mechanism for pituitary Cushing's disease. Cell 96:689-700.
121. Greider, M.H., Howell, S.L., and Lacy, P.E. 1969. Isolation and properties of secretory granules from rat islets of Langerhans. II. Ultrastructure of the β granule. J. Cell Biol. 41:162-166.
122. Lange, R.H. 1974. Crystalline islet B-granules in the grass snake (Natrix natrix (L.)): tilting experiments in the electron microscope. J. Ultrastruct. Res. 46:301-307.
123. Michael, J., Carroll, R., Swift, H.H., and Steiner, D.F. 1987. Studies on the molecular organization of rat insulin secretory granules. J. Biol. Chem. 262:16531-16535.
124. Lemaire, K., Ravier, M.A., Schraenen, A., Creemers, J.W., Van de Plas, R., Granvik, M., Van Lommel, L., Waelkens, E., Chimienti, F., Rutter, G.A., et al. 2009. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc. Natl. Acad. Sci. U. S. A. 106:14872-14877.
125. Steiner, D.F. 1973. Cocrystallization of proinsulin and insulin. Nature 243:528-530.
126. Frank, B.H., and Veros, A.J. 1970. Interaction of zinc with proinsulin. Biochem. Biophys. Res. Commun. 38:284-289.
127. Grant, P.T., Coombs, T.L., and Frank, B.H. 1972. Differences in the nature of the interaction of insulin and proinsulin with zinc. Biochem. J. 126:433-440.
128. Howell, S.L., Tyhurst, M., Duvefelt, H., Andersson, A., and Hellerstrom, C. 1978. Role of zinc and calcium in the formation and storage of insulin in the pancreatic β-cell. Cell Tissue Res. 188:107-118.
129. Kemmler, W., Steiner, D.F., and Borg, J. 1973. Studies on the conversion of proinsulin to insulin. 3. Studies in vitro with a crude secretion granule fraction isolated from rat islets of Langerhans. J. Biol. Chem. 248:4544-4551.
130. Steiner, D.F., and Rubenstein, A. 1973. In Proceedings of the 8th Midwest Conference on Endocrinology and Metabolism . F.M. Matschinsky, R. Fertel, J. Kotler-Brajtburg, S. Stillings, J. Ellerman, F. Raybaud, J.H. Thurston, R.P. Breitenbach, and X.J. Mussachia, editors: University of Missouri. 43-59.
131. Prentki, M., and Matschinsky, F.M. 1987. Ca 2+ , cAMP, and phospholipid-derived messengers in coupling mechanisms of insulin secretion. Physiol Rev 67:1185-1248.
132. Hughes, S.J., and Ashcroft, S.H.J. 1992. In Nutrient regulation of insulin secretion . P.R. Flatt, editor. Colchester, England: Portland Press. 271-289
133. Nielsen, D.A., Welsh, M., Casadaban, M.J., and Steiner, D.F. 1985. Control of insulin gene expression in pancreatic β-cells and in an insulin-producing cell line, RIN-5F cells. I. Effects of glucose and cyclic AMP on the transcription of insulin mRNA. J. Biol. Chem. 260:13585-13589.
134. Welsh, M., Nielsen, D.A., MacKrell, A.J., and Steiner, D.F. 1985. Control of insulin gene expression in pancreatic β-cells and in an insulin-producing cell line, RIN-5F cells. II. Regulation of insulin mRNA stability. J. Biol. Chem. 260:13590-13594.
135. Rabinovitch, A., Blondel, B., Murray, T., and Mintz, D.H. 1980. Cyclic adenosine-3',5'-monophosphate stimulates islet B cell replication in neonatal rat pancreatic monolayer cultures. J. Clin. Invest. 66:1065-1071.
136. Swenne, I. 1982. Effects of cyclic AMP on DNA replication and protein biosynthesis in fetal rat islets of Langerhans maintained in tissue culture. Biosci. Rep. 2:867-876.
137. Tanese, T., Lazarus, N.R., Devrim, S., and Recant, L. 1970. Synthesis and release of proinsulin and insulin by isolated rat islets of Langerhans. J. Clin. Invest. 49:1394-1404.
138. Rhodes, C.J., and Halban, P.A. 1987. Newly synthesized proinsulin/insulin and stored insulin are released from pancreatic B cells predominantly via a regulated, rather than a constitutive, pathway. J. Cell Biol. 105:145-153.
139. Kelly, R.B. 1985. Pathways of protein secretion in eukaryotes. Science 230:25-32.
140. Valverde, I., Garcia-Morales, P., Ghiglione, M., and Malaisse, W.J. 1983. The stimulus-secretion coupling of glucose-induced insulin release. LIII. Calcium-dependency of the cyclic AMP response to nutrient secretagogues. Horm. Metab. Res. 15:62-68.
141. Holst, J.J., Orskov, C., Nielsen, O.V., and Schwartz, T.W. 1987. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 211:169-174.
142. Rajan, S., Torres, J., Thompson, M.S., and Philipson, L.H. 2012. SUMO downregulates GLP-1-stimulated cAMP generation and insulin secretion. Am. J. Physiol. Endocrinol. Metab. 302:E714-723.
143. Howell, S.L., Jones, P.M., and Persaud, S.J. 1994. Regulation of insulin secretion: the role of second messengers. Diabetologia 37 Suppl 2:S30-35.
144. Berggren, P.O., Rorsman, P., and Efendic, S. 1992. In Nutrient regulation of insulin secretion . P.R. Flatt, editor. Colchester, England: Portland Press. 289-318
145. Persaud, S.J., Jones, P.M., and Howell, S.L. 1989. Effects of Bordetella pertussis toxin on catecholamine inhibition of insulin release from intact and electrically permeabilized rat islets. Biochem. J. 258:669-675.
146. Rorsman, P., and Braun, M. 2013. Regulation of insulin secretion in human pancreatic islets. Annu. Rev. Physiol. 75:155-179.
147. Fridlyand, L.E., and Philipson, L.H. 2011. Coupling of metabolic, second messenger pathways and insulin granule dynamics in pancreatic b -cells: a computational analysis. Prog. Biophys. Mol. Biol. 107:293-303.
148. Fridlyand, L.E., Jacobson, D.A., and Philipson, L.H. 2013. Ion channels and regulation of insulin secretion in human b -cells: a computational systems analysis. Islets 5:1-15.
149. Tabei, S.M., Burov, S., Kim, H.Y., Kuznetsov, A., Huynh, T., Jureller, J., Philipson, L.H., Dinner, A.R., and Scherer, N.F. 2013. Intracellular transport of insulin granules is a subordinated random walk. Proc. Natl. Acad. Sci. U. S. A. 110:4911-4916.
150. Polonsky, K.S. 1995. Lilly Lecture 1994. The β-cell in diabetes: from molecular genetics to clinical research. Diabetes 44:705-717.
151. Taylor, S.I., Accili, D., and Imai, Y. 1994. Insulin resistance or insulin deficiency. Which is the primary cause of NIDDM? Diabetes 43:735-740.
152. Turner, R.C., Hattersley, A.T., Shaw, J.T., and Levy, J.C. 1995. Type II diabetes: clinical aspects of molecular biological studies. Diabetes 44:1-10.
153. Dean, P.M., and Matthews, E.K. 1968. Electrical activity in pancreatic islet cells. Nature 219:389-390.
154. Dean, P.M., and Matthews, E.K. 1970. Glucose-induced electrical activity in pancreatic islet cells. J. Physiol. 210:255-264.
155. Ashcroft, F.M., Harrison, D.E., and Ashcroft, S.J. 1984. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature 312:446-448.
156. Cook, D.L., and Hales, C.N. 1984. Intracellular ATP directly blocks K + channels in pancreatic B-cells. Nature 311:271-273.
157. Cook, D.L., Ikeuchi, M., and Fujimoto, W.Y. 1984. Lowering of pHi inhibits Ca2+-activated K+ channels in pancreatic B-cells. Nature 311:269-271.
158. McTaggart, J.S., Clark, R.H., and Ashcroft, F.M. 2010. The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. J. Physiol. 588:3201-3209.
159. Doyle, D.A., Morais Cabral, J., Pfuetzner, R.A., Kuo, A., Gulbis, J.M., Cohen, S.L., Chait, B.T., and MacKinnon, R. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280:69-77.
160. Nishida, M., and MacKinnon, R. 2002. Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell 111:957-965.
161. Ashcroft, F.M., and Rorsman, P. 1990. ATP-sensitive K + channels: a link between B-cell metabolism and insulin secretion. Biochem. Soc. Trans. 18:109-111.
162. Seino, S., Iwanaga, T., Nagashima, K., and Miki, T. 2000. Diverse roles of K ATP channels learned from K ir 6.2 genetically engineered mice. Diabetes 49:311-318.
163. Huopio, H., Shyng, S.L., Otonkoski, T., and Nichols, C.G. 2002. K ATP channels and insulin secretion disorders. Am. J. Physiol. Endocrinol. Metab. 283:E207-216.
164. Stoffel, M., Espinosa, R., 3rd, Powell, K.L., Philipson, L.H., Le Beau, M.M., and Bell, G.I. 1994. Human G-protein-coupled inwardly rectifying potassium channel (GIRK1) gene (KCNJ3): localization to chromosome 2 and identification of a simple tandem repeat polymorphism. Genomics 21:254-256.
165. Reuveny, E., Slesinger, P.A., Inglese, J., Morales, J.M., Iniguez-Lluhi, J.A., Lefkowitz, R.J., Bourne, H.R., Jan, Y.N., and Jan, L.Y. 1994. Activation of the cloned muscarinic potassium channel by G protein β γ subunits. Nature 370:143-146.
166. Philipson, L.H., Kuznetsov, A., Toth, P.T., Murphy, J.F., Szabo, G., Ma, G.H., and Miller, R.J. 1995. Functional expression of an epitope-tagged G protein-coupled K + channel (GIRK1). J. Biol. Chem. 270:14604-14610.
167. Aguilar-Bryan, L., and Bryan, J. 1999. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20:101-135.
168. Katz, B., and Miledi, R. 1965. The effect of calcium on acetylcholine release from motor nerve terminals. Proc. R. Soc. Lond. B Biol. Sci. 161:496-503.
169. Douglas, W.W. 1968. Stimulus-secretion coupling: the concept and clues from chromaffin and other cells. Br. J. Pharmacol. 34:451-474.
170. Wollheim, C.B., and Sharp, G.W. 1981. Regulation of insulin release by calcium. Physiol. Rev. 61:914-973.
171. Rorsman, P., Braun, M., and Zhang, Q. 2012. Regulation of calcium in pancreatic a - and b -cells in health and disease. Cell Calcium 51:300-308.
172. Tsien, R.W., Ellinor, P.T., and Horne, W.A. 1991. Molecular diversity of voltage-dependent Ca 2+ channels. Trends Pharmacol. Sci. 12:349-354.
173. Namkung, Y., Skrypnyk, N., Jeong, M.J., Lee, T., Lee, M.S., Kim, H.L., Chin, H., Suh, P.G., Kim, S.S., and Shin, H.S. 2001. Requirement for the L-type Ca 2+ channel α 1D subunit in postnatal pancreatic β cell generation. J. Clin. Invest. 108:1015-1022.
174. Braun, M., Ramracheya, R., Bengtsson, M., Zhang, Q., Karanauskaite, J., Partridge, C., Johnson, P.R., and Rorsman, P. 2008. Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes 57:1618-1628.
175. Straub, S.G., and Sharp, G.W. 2012. Evolving insights regarding mechanisms for the inhibition of insulin release by norepinephrine and heterotrimeric G proteins. Am. J. Physiol. Cell Physiol. 302:C1687-1698.
176. Sutton, R.B., Fasshauer, D., Jahn, R., and Brunger, A.T. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature 395:347-353.
177. Regazzi, R., Ravazzola, M., Iezzi, M., Lang, J., Zahraoui, A., Andereggen, E., Morel, P., Takai, Y., and Wollheim, C.B. 1996. Expression, localization and functional role of small GTPases of the Rab3 family in insulin-secreting cells. J. Cell Sci. 109 ( Pt 9):2265-2273.
178. Mizuta, M., Kurose, T., Miki, T., Shoji-Kasai, Y., Takahashi, M., Seino, S., and Matsukura, S. 1997. Localization and functional role of synaptotagmin III in insulin secretory vesicles in pancreatic β-cells. Diabetes 46:2002-2006.
179. Peking, I.R.G. 1971. Insulin's crystal structure at 2.5A resolution. Peking Rev. 40:11-16.
180. Blundell, T.L., Dodson, G.G., Hodgkin, D.C., and Mercola, D.A. 1972. Insulin: the structure in the crystal and its reflection in chemistry and biology. Adv. Protein Chem. 26:279-402.
181. Bentley, G., Dodson, E., Dodson, G., Hodgkin, D., and Mercola, D. 1976. Structure of insulin in 4-zinc insulin. Nature 261:166-168.
182. Derewenda, U., Derewenda, Z., Dodson, E.J., Dodson, G.G., Reynolds, C.D., Smith, G.D., Sparks, C., and Swenson, D. 1989. Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature 338:594-596.
183. Smith, G.D., Pangborn, W.A., and Blessing, R.H. 2003. The structure of T6 human insulin at 1.0 A resolution. Acta Crystallogr. D Biol. Crystallogr. 59:474-482.
184. Hua, Q.X., Shoelson, S.E., Kochoyan, M., and Weiss, M.A. 1991. Receptor binding redefined by a structural switch in a mutant human insulin. Nature 354:238-241.
185. Knegtel, R.M., Boelens, R., Ganadu, M.L., and Kaptein, R. 1991. The solution structure of a monomeric insulin. A two-dimensional 1 H-NMR study of des -(B26-B30)-insulin in combination with distance geometry and restrained molecular dynamics. Eur. J. Biochem. 202:447-458.
186. Jørgensen, A.M., Kristensen, S.M., Led, J.J., and Balschmidt, P. 1992. Three-dimensional solution structure of an insulin dimer. A study of the B9(Asp) mutant of human insulin using nuclear magnetic resonance, distance geometry and restrained molecular dynamics. J. Mol. Biol. 227:1146-1163.
187. Olsen, H.B., Ludvigsen, S., and Kaarsholm, N.C. 1996. Solution structure of an engineered insulin monomer at neutral pH. Biochemistry 35:8836-8845.
188. Hua, Q.X., Hu, S.Q., Frank, B.H., Jia, W., Chu, Y.C., Wang, S.H., Burke, G.T., Katsoyannis, P.G., and Weiss, M.A. 1996. Mapping the functional surface of insulin by design: structure and function of a novel A-chain analogue. J. Mol. Biol. 264:390-403.
189. Sorensen, M.D., Bjorn, S., Norris, K., Olsen, O., Petersen, L., James, T.L., and Led, J.J. 1997. Solution structure and backbone dynamics of the human α3-chain type VI collagen C-terminal Kunitz domain. Biochemistry 36:10439-10450.
190. Ludvigsen, S., Olsen, H.B., and Kaarsholm, N.C. 1998. A structural switch in a mutant insulin exposes key residues for receptor binding. J. Mol. Biol. 279:1-7.
191. Xu, B., Hua, Q.X., Nakagawa, S.H., Jia, W., Chu, Y.C., Katsoyannis, P.G., and Weiss, M.A. 2002. Chiral mutagensis of insulin's hidden receptor-binding surface: structure of an allo -isoleucine A2 analogue. J. Mol. Biol. 316:435-441.
192. Hua, Q.X., Xu, B., Huang, K., Hu, S.Q., Nakagawa, S., Jia, W., Wang, S., Whittaker, J., Katsoyannis, P.G., and Weiss, M.A. 2009. Enhancing the activity of insulin by stereospecific unfolding. Conformational life cycle of insulin and its evolutionary origins. J. Biol. Chem. 284:14586-14596.
193. Žáková, L., Kletvíková, E., Veverka, V., Lepsík, M., Watson, C.J., Turkenburg, J.P., Jirácek, J., and Brzozowski, A.M. 2013. Structural integrity of the B24 site in human insulin is important for hormone functionality. J. Biol. Chem. 288:10230-10240.
194. Liang, D.C., Chang, W.R., and Wan, Z.L. 1994. A proposed interaction model of the insulin molecule with its receptor. Biophys. Chem. 50:63-71.
195. Scott, D.A. 1934. Crystalline insulin. Biochem. J. 28:1592-1602 1591.
196. Scott, D.A., and Fisher, A.M. 1935. Crystalline insulin. Biochem. J. 29:1048-1054.
197. Brader, M.L., and Dunn, M.F. 1991. Insulin hexamers: new conformations and applications. Trends Biochem. Sci. 16:341-345.
198. Badger, J., Harris, M.R., Reynolds, C.D., Evans, A.C., Dodson, E.J., Dodson, G.G., and North, A.C. 1991. Structure of the pig insulin dimer in the cubic crystal. Acta Crystallogr. B Struct. Sci. 47:127-136.
199. Badger, J. 1992. Flexibility in crystalline insulins. Biophys. J. 61:816-819.
200. Bi, R.C., Dauter, Z., Dodson, E., Dodson, G., Giordano, F., and Reynolds, C. 1984. Insulin structure as a modified and monomeric molecule. Biopolymers 23:391-395.
201. Liang, D.C., Stuart, D., B., D.J., Todd, R., You, J.M., and Luo, M.Z. 1985. X-ray studies of des-pentapeptide (B26-B30) insulin at 2.4 Å resolution: The DPI molecule and its structural relationship with insulin. Sci. Sin. 28:472-484.
202. Bao, S.J., Xie, D.L., Zhang, J.P., Chang, W.R., and Liang, D.C. 1997. Crystal structure of des heptapeptide(B24-B30)insulin at 1.6 Å resolution: implications for receptor binding. Proc. Natl. Acad. Sci. U. S. A. 94:2975-2980.
203. Wan, Z., Huang, K., Whittaker, J., and Weiss, M.A. 2008. The structure of a mutant insulin uncouples receptor binding from protein allostery. An electrostatic block to the TR transition. J. Biol. Chem. 283:21198-21210.
204. Derewenda, U., Derewenda, Z.S., Dodson, G.G., and Hubbard, R.E. 1990. Insulin structure. In Insulin—the handbook of experimental pharmacology . P. Cuatrecasas, and S. Jacobs, editors. Berlin: Springer-Verlag. 23-39.
205. Jacoby, E., Hua, Q.X., Stern, A.S., Frank, B.H., and Weiss, M.A. 1996. Structure and dynamics of a protein assembly. 1 H-NMR studies of the 36 kDa R 6 insulin hexamer. J. Mol. Biol. 258:136-157.
206. Chang, X., Jorgensen, A.M., Bardrum, P., and Led, J.J. 1997. Solution structures of the R6 human insulin hexamer. Biochemistry 36:9409-9422.
207. Jørgensen, L.N., Dideriksen, L.H., and Drejer, K. 1992. Carcinogenic effect of the human insulin analog B-10 Asp in female rats. Diabetologia 35 Suppl 1:A3.
208. Hua, Q.X., and Weiss, M.A. 1990. Toward the solution structure of human insulin: sequential 2D 1 H NMR assignment of a des -pentapeptide analogue and comparison with crystal structure. Biochemistry 29:10545-10555.
209. Derewenda, U., Derewenda, Z., Dodson, E.J., Dodson, G.G., Bing, X., and Markussen, J. 1991. X-ray analysis of the single chain B29-A1 peptide-linked insulin molecule. A completely inactive analogue. J. Mol. Biol. 220:425-433.
210. Smith, G.D., and Dodson, G.G. 1992. Structure of a rhombohedral R6 insulin/phenol complex. Proteins 14:401-408.
211. Dodson, E.J., Dodson, G.G., Lewitova, A., and Sabesan, M. 1978. Zinc-free cubic pig insulin: crystallization and structure determination. J. Mol. Biol. 125:387-396.
212. Cutfield, J.F., Cutfield, S.M., Dodson, E.J., Dodson, G.G., Emdin, S.F., and Reynolds, C.D. 1979. Structure and biological activity of hagfish insulin. J. Mol. Biol. 132:85-100.
213. Nakagawa, S.H., Zhao, M., Hua, Q.X., Hu, S.Q., Wan, Z.L., Jia, W., and Weiss, M.A. 2005. Chiral mutagenesis of insulin. Foldability and function are inversely regulated by a stereospecific switch in the B chain. Biochemistry 44:4984-4999.
214. Hua, Q.X., Nakagawa, S.H., Hu, S.Q., Jia, W., Wang, S., and Weiss, M.A. 2006. Toward the active conformation of insulin. Stereospecific modulation of a structural switch in the B chain. J. Biol. Chem. 281:24900-24909.
215. Nakagawa, S.H., Hua, Q.X., Hu, S.Q., Jia, W., Wang, S., Katsoyannis, P.G., and Weiss, M.A. 2006. Chiral mutagenesis of insulin. Contribution of the B20-B23 β-turn to activity and stability. J. Biol. Chem. 281:22386-22396.
216. Weiss, M.A. 2009. Proinsulin and the genetics of diabetes mellitus. J. Biol. Chem. 284:19159-19163.
217. Frank, B.H., Pekar, A.H., and Veros, A.J. 1972. Insulin and proinsulin conformation in solution. Diabetes 21:486-491.
218. Goldman, J., and Carpenter, F.H. 1974. Zinc binding, circular dichroism, and equilibrium sedimentation studies on insulin (bovine) and several of its derivatives. Biochemistry 13:4566-4574.
219. Milthorpe, B.K., Nichol, L.W., and Jeffrey, P.D. 1977. The polymerization pattern of zinc(II)-insulin at pH 7.0. Biochim. Biophys. Acta 495:195-202.
220. McKern, N.M., Lawrence, M.C., Streltsov, V.A., Lou, M.Z., Adams, T.E., Lovrecz, G.O., Elleman, T.C., Richards, K.M., Bentley, J.D., Pilling, P.A., et al. 2006. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature 443:218-221.
221. Kruger, P., Strassburger, W., Wollmer, A., van Gunsteren, W.F., and Dodson, G.G. 1987. The simulated dynamics of the insulin monomer and their relationship to the molecule's structure. Eur. Biophys. J. 14:449-459.
222. Wodak, S.J., Alard, P., Delhaise, P., and Renneboog-Squilbin, C. 1985. Simulation of conformational changes in 2 Zn insulin. J. Mol. Biol. 181:317-322.
223. Weiss, M.A., Hua, Q.X., Lynch, C.S., Frank, B.H., and Shoelson, S.E. 1991. Heteronuclear 2D NMR studies of an engineered insulin monomer: assignment and characterization of the receptor-binding surface by selective 2 H and 13 C labeling with application to protein design. Biochemistry 30:7373-7389.
224. Tidor, B., and Karplus, M. 1994. The contribution of vibrational entropy to molecular association. The dimerization of insulin. J. Mol. Biol. 238:405-414.
225. Rupley, J.A. 1967. The binding and cleavage by lysozyme of N-acetylglucosamine oligosaccharides. Proc. R. Soc. Lond. B Biol. Sci. 167:416-428.
226. Jeffrey, P.D., Milthorpe, B.K., and Nichol, L.W. 1976. Polymerization pattern of insulin at pH 7.0. Biochemistry 15:4660-4665.
227. Pocker, Y., and Biswas, S.B. 1981. Self-association of insulin and the role of hydrophobic bonding: a thermodynamic model of insulin dimerization. Biochemistry 20:4354-4361.
228. Cutfield, J.F., Cutfield, S.M., Dodson, E.J., dodson, G.G., Reynolds, C.D., and Vallely, D. 1981. In Structural studies on molecules of biological interest . G.G. Dodson, J. Glusker, and D. Sayre, editors. Oxford: Oxford University Press. 527-546.
229. Chothia, C., Lesk, A.M., Dodson, G.G., and Hodgkin, D.C. 1983. Transmission of conformational change in insulin. Nature 302:500-505.
230. Ciszak, E., and Smith, G.D. 1994. Crystallographic evidence for dual coordination around zinc in the T 3 R 3 human insulin hexamer. Biochemistry 33:1512-1517.
231. Schlichtkrull, J. 1958. Insulin Crystals: chemical and biological studies on insulin crystals and insulin zinc suspensions. Copenhagen, Munksgaard: Kobenhavns universitet. 139.
232. Roy, M., Brader, M.L., Lee, R.W., Kaarsholm, N.C., Hansen, J.F., and Dunn, M.F. 1989. Spectroscopic signatures of the T to R conformational transition in the insulin hexamer. J. Biol. Chem. 264:19081-19085.
233. Wollmer, A., Rannefeld, B., Johansen, B.R., Hejnaes, K.R., Balschmidt, P., and Hansen, F.B. 1987. Phenol-promoted structural transformation of insulin in solution. Biol. Chem. Hoppe Seyler 368:903-911.
234. Coffman, F.D., and Dunn, M.F. 1988. Insulin-metal ion interactions: the binding of divalent cations to insulin hexamers and tetramers and the assembly of insulin hexamers. Biochemistry 27:6179-6187.
235. Wood, S.P., Blundell, T.L., Wollmer, A., Lazarus, N.R., and Neville, R.W. 1975. The relation of conformation and association of insulin to receptor binding; a-ray and circular-dichroism studies on bovine and hystricomorph insulins. Eur. J. Biochem. 55:531-542.
236. Renscheidt, H., Strassburger, W., Glatter, U., Wollmer, A., Dodson, G.G., and Mercola, D.A. 1984. A solution equivalent of the 2Zn→4Zn transformation of insulin in the crystal. Eur. J. Biochem. 142:7-14.
237. Williamson, K.L., and Williams, R.J. 1979. Conformational analysis by nuclear magnetic resonance: insulin. Biochemistry 18:5966-5972.
238. Ramesh, V., and Bradbury, J.H. 1986. 1 H-NMR studies of insulin. Reversible transformation of 2Zn→4Zn insulin hexamer. Int. J. Pept. Protein Res. 28:146-153.
239. Brange, J., editor. 1987. Galenics of Insulin: The Physico-chemical and Pharmaceutical Aspects of Insulin and Insulin Preparations . Berlin: Springer Berlin Heidelberg. 1-103 pp.
240. Brange, J., and Langkjoer, L. 1993. Insulin structure and stability. Pharm. Biotechnol. 5:315-350.
241. Weiss, M.A. 2013. Design of ultra-stable insulin analogues for the developing world. Journal of Health Specialties 1:59-70.
242. Ciszak, E., Beals, J.M., Frank, B.H., Baker, J.C., Carter, N.D., and Smith, G.D. 1995. Role of C-terminal B-chain residues in insulin assembly: the structure of hexameric Lys B28 Pro B29 -human insulin. Structure (Lond.) 3:615-622.
243. Howell, S.L., Young, D.A., and Lacy, P.E. 1969. Isolation and properties of secretory granules from rat islets of Langerhans. 3. Studies of the stability of the isolated b granules. J. Cell Biol. 41:167-176.
244. Howell, S.L., Kostianovsky, M., and Lacy, P.E. 1969. b granule formation in isolated islets of Langerhans: a study by electron microscopic radioautography. J. Cell Biol. 42:695-705.
245. Katsoyannis, P.G. 1966. Synthesis of insulin. Science 154:1509-1514.
246. De Meyts, P., van Obberghen, E., and Roth, J. 1978. Mapping of the residues responsible for the negative cooperativity of the receptor-binding region of insulin. Nature 273:504-509.
247. Kristensen, C., Kjeldsen, T., Wiberg, F.C., Schaffer, L., Hach, M., Havelund, S., Bass, J., Steiner, D.F., and Andersen, A.S. 1997. Alanine scanning mutagenesis of insulin. J. Biol. Chem. 272:12978-12983.
248. Liang, D.C., Chang, W.R., Zhang, J.P., and Wan, Z.L. 1992. The possible mechanism of binding interaction of insulin molecule with its receptor. Sci. China B 35:547-557.
249. Shoelson, S.E., Lee, J., Lynch, C.S., Backer, J.M., and Pilch, P.F. 1993. Bpa B25 insulins. Photoactivatable analogues that quantitatively cross-link, radiolabel, and activate the insulin receptor. J. Biol. Chem. 268:4085-4091.
250. Kurose, T., Pashmforoush, M., Yoshimasa, Y., Carroll, R., Schwartz, G.P., Burke, G.T., Katsoyannis, P.G., and Steiner, D.F. 1994. Cross-linking of a B25 azidophenylalanine insulin derivative to the carboxyl-terminal region of the α-subunit of the insulin receptor. Identification of a new insulin-binding domain in the insulin receptor. J. Biol. Chem. 269:29190-29197.
251. Xu, B., Hu, S.Q., Chu, Y.C., Wang, S., Wang, R.Y., Nakagawa, S.H., Katsoyannis, P.G., and Weiss, M.A. 2004. Diabetes-associated mutations in insulin identify invariant receptor contacts. Diabetes 53:1599-1602.
252. Xu, B., Huang, K., Chu, Y.C., Hu, S.Q., Nakagawa, S., Wang, S., Wang, R.Y., Whittaker, J., Katsoyannis, P.G., and Weiss, M.A. 2009. Decoding the cryptic active conformation of a protein by synthetic photoscanning: Insulin inserts a detachable arm between receptor domains. J. Biol. Chem. 284:14597-14608.
253. Tager, H.S. 1995. Diabetes: Clinical Science in Practice . Cambridge: Cambridge University Press. 492 pp.
254. Pullen, R.A., Lindsay, D.G., Wood, S.P., Tickle, I.J., Blundell, T.L., Wollmer, A., Krail, G., Brandenburg, D., Zahn, H., Gliemann, J., et al. 1976. Receptor-binding region of insulin. Nature 259:369-373.
255. Kwok, S.C., Steiner, D.F., Rubenstein, A.H., and Tager, H.S. 1983. Identification of a point mutation in the human insulin gene giving rise to a structurally abnormal insulin (insulin Chicago). Diabetes 32:872-875.
256. Shoelson, S., Haneda, M., Blix, P., Nanjo, A., Sanke, T., Inouye, K., Steiner, D., Rubenstein, A., and Tager, H. 1983. Three mutant insulins in man. Nature 302:540-543.
257. Shoelson, S., Fickova, M., Haneda, M., Nahum, A., Musso, G., Kaiser, E.T., Rubenstein, A., and Tager, H. 1983. Indentification of a mutant human insulin predicetd to contain a serine-for -phenylalanine substitition. Proc. Natl. Acad. Sci. U. S. A. 80:7390-7394.
258. Kobayashi, M., Ohgaku, S., Iwasaki, M., Maegawa, H., Shigeta, Y., and Inouye, K. 1982. Characterization of [Leu B24 ]- and [Leu B25 ]-insulin analogues. Receptor binding and biological activity. Biochem. J. 206:597-603.
259. Kobayashi, M., Ohgaku, S., Iwasaki, M., Maegawa, H., Shigeta, Y., and Inouye, K. 1982. Supernormal insulin: [D-Phe B24 ]-insulin with increased affinity for insulin receptors. Biochem. Biophys. Res. Commun. 107:329-336.
260. Nanjo, K., Sanke, T., Miyano, M., Okai, K., Sowa, R., Kondo, M., Nishimura, S., Iwo, K., Miyamura, K., Given, B.D., et al. 1986. Diabetes due to secretion of a structurally abnormal insulin (insulin Wakayama). Clinical and functional characteristics of [Leu A3 ] insulin. J. Clin. Invest. 77:514-519.
261. Hua, Q.X., Shoelson, S.E., Inouye, K., and Weiss, M.A. 1993. Paradoxical structure and function in a mutant human insulin associated with diabetes mellitus. Proc. Natl. Acad. Sci. U. S. A. 90:582-586.
262. Liu, M., Haataja, L., Wright, J., Wickramasinghe, D.N., Hua, Q.X., Phillips, N.B., Barbetti, F., Weiss, M.A., and Arvan, P. 2010. Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on nonmutant proinsulin transport. PLos-One 5:e13333.
263. Huang, K., Xu, B., Hu, S.Q., Chu, Y.C., Hua, Q.X., Qu, Y., Li, B., Wang, S., Wang, R.Y., Nakagawa, S.H., et al. 2004. How insulin binds: the B-chain α-helix contacts the L1 β-helix of the insulin receptor. J. Mol. Biol. 341:529-550.
264. Schäffer, L. 1994. A model for insulin binding to the insulin receptor. Eur. J. Biochem. 221:1127-1132.
265. De Meyts, P. 1994. The structural basis of insulin and insulin-like growth factor-I receptor binding and negative co-operativity, and its relevance to mitogenic versus metabolic signaling. Diabetologia 37:S135-S148.
266. De Meyts, P., Palsgaard, J., Sajid, W., Theede, A.M., and Aladdin, H. 2004. Structural biology of insulin and IGF-1 receptors. Novartis Found. Symp. 262:160-171.
267. De Meyts, P., and Shymko, R.M. 2000. Timing-dependent modulation of insulin mitogenic versus metabolic signalling. Novartis Found. Symp. 227:46-57.
268. Hansen, B.F., Kurtzhals, P., Jensen, A.B., Dejgaard, A., and Russell-Jones, D. 2011. Insulin X10 revisited: a super-mitogenic insulin analogue. Diabetologia 54:2226-2231.
269. Hemkens, L.G., Bender, R., Grouven, U., and Sawicki, P.T. 2009. Insulin glargine and cancer. Lancet 374:1743-1744.
270. Weinstein, D., Simon, M., Yehezkel, E., Laron, Z., and Werner, H. 2009. Insulin analogues display IGF-I mitogenic and anti-apoptotic activities in cultured cancer cells. Diabetes Metab. Res. Rev. 25:41-49.
271. Sciacca, L., Cassarino, M.F., Genua, M., Pandini, G., LeMoli, R., Squatrito, S., and Vigneri, R. 2010. Insulin analogues differently activate insulin receptor isoforms and post-receptor signaling. Diabetologia 53:1743-1753.
272. Sciacca, L., Le Moli, R., and Vigneri, R. 2012. Insulin analogs and cancer. Front. Endocrinol. (Lausanne) 3:21.
273. Cosmatos, A., Cheng, K., Okada, Y., and Katsoyannis, P.G. 1978. The chemical synthesis and biological evaluation of [1-L-alanine-A]-and [1-D-alanine-A]insulins. J. Biol. Chem. 253:6586-6590.
274. Geiger, R., Geisen, K., Summ, H.D., and Langer, D. 1975. (A1-D-alanine) insulin. Hoppe Seylers Z Physiol. Chem. 356:1635-1649.
275. Geiger, R., Geisen, K., Regitz, G., Summ, H.D., and Langner, D. 1980. Insulin analogues with substitution of A1-glycine by D-amino acids and omega-amino acids (author's transl). Hoppe Seylers Z Physiol. Chem. 361:563-570.
276. Geiger, R., Geisen, K., and Summ, H.D. 1982. Astausch von A1-Glycin in Rinderinsulin gegen L-und D-Tryptophan. Hoppe Seylers Z Physiol. Chem. 363:S1231-1239.
277. Nakagawa, S.H., and Tager, H.S. 1989. Perturbation of insulin-receptor interactions by intramolecular hormone cross-linking. Analysis of relative movement among residues A1, B1, and B29. J. Biol. Chem. 264:272-279.
278. Kitagawa, K., Ogawa, H., Burke, G.T., Chanley, J.D., and Katsoyannis, P.G. 1984. Critical role of the A2 amino acid residue in the biological activity of insulin: [2-glycine-A]- and [2-alanine-A]insulins. Biochemistry 23:1405-1413.
279. Nakagawa, S.H., and Tager, H.S. 1992. Importance of aliphatic side-chain structure at positions 2 and 3 of the insulin A chain in insulin-receptor interactions. Biochemistry 31:3204-3214.
280. Kitagawa, K., Ogawa, H., Burke, G.T., Chanley, J.D., and Katsoyannis, P.G. 1984. Interaction between the A2 and A19 amino acid residues is of critical importance for high biological activity in insulin: [19-leucine-A]insulin. Biochemistry 23:4444-4448.
281. Wieneke, H.J., Danho, W., Bullesbach, E.E., Gattner, H.G., and Zahn, H. 1983. The synthesis of [A 19-3-iodotyrosine] and [A 19-3,5-diiodotyrosine]insulin (porcine). Hoppe Seylers Z Physiol. Chem. 364:537-550.
282. Ohta, N., Burke, G.T., and Katsoyannis, P.G. 1988. Synthesis of an insulin analogue embodying a strongly fluorescent moiety, [19-tryptophan-A]insulin. J. Protein Chem. 7:55-65.
283. Du, X., and Tang, J.G. 1998. Hydroxyl group of insulin A19Tyr is essential for receptor binding: studies on (A19Phe)insulin. Biochem. Mol. Biol. Int. 45:255-260.
284. Sonne, O., Linde, S., Larsen, T.R., and Gliemann, J. 1983. Monoiodoinsulin labelled in tyrosine residue 16 or 26 of the B-chain or 19 of the A-chain. II. Characterization of the kinetic binding constants and determination of the biological potency. Hoppe Seylers Z Physiol. Chem. 364:101-110.
285. Hamlin, J.L., and Arquilla, E.R. 1974. Monoiodoinsulin. Preparation, purification, and characterization of a biologically active derivative substituted predominantly on tyrosine A14. J. Biol. Chem. 249:21-32.
286. Linde, S., Welinder, B.S., Hansen, B., and Sonne, O. 1986. Preparative reversed-phase high-performance liquid chromatography of iodinated insulin retaining full biological activity. J. Chromatogr. 369:327-339.
287. Chu, Y.C., Zong, L., Burke, G.T., and Katsoyannis, P.G. 1992. The A14 position of insulin tolerates considerable structural alterations with modest effects on the biological behavior of the hormone. J. Protein Chem. 11:571-577.
288. Slobin, L.I., and Carpenter, F.H. 1963. Action of carboxypeptidase- A on bovine insulin: preparation of desalanine-desasparagine-insulin. Biochemistry 2:16-22.
289. Chu, Y.C., Wang, R.Y., Burke, G.T., Chanley, J.D., and Katsoyannis, P.G. 1987. Possible involvement of the A20-A21 peptide bond in the biological activity of insulin. 1. [21-Desasparagine,20-cysteinamide-A]insulin and [21-desasparagine,20-cysteine isopropylamide-A]insulin. Biochemistry 26:6966-6971.
290. Chu, Y.C., Wang, R.Y., Burke, G.T., Chanley, J.D., and Katsoyannis, P.G. 1987. Possible involvement of the A20-A21 peptide bond in the expression of the biological activity of insulin. 3. [21- Des asparagine,20-cysteine ethylamide-A]insulin and [21- des asparagine,20-cysteine 2,2,2-trifluoroethylamide-A]insulin. Biochemistry 26:6975-6979.
291. Chu, Y.C., Burke, G.T., Chanley, J.D., and Katsoyannis, P.G. 1987. Possible involvement of the A20-A21 peptide bond in the expression of the biological activity of insulin. 2. [21-Asparagine diethylamide-A]insulin. Biochemistry 26:6972-6975.
292. Thomas, B., and Wollmer, A. 1989. Cobalt probing of structural alternatives for insulin in solution. Biol. Chem. Hoppe Seyler 370:1235-1244.
293. Kaarsholm, N.C., Ko, H.C., and Dunn, M.F. 1989. Comparison of solution structural flexibility and zinc binding domains for insulin, proinsulin, and miniproinsulin. Biochemistry 28:4427-4435.
294. Wollmer, A., Rannefeld, B., Stahl, J., and Melberg, S.G. 1989. Structural transition in the metal-free hexamer of protein-engineered [B13 Gln]insulin. Biol. Chem. Hoppe Seyler 370:1045-1053.
295. Kruger, P., Gilge, G., Cabuk, Y., and Wollmer, A. 1990. Cooperativity and intermediate states in the T----R-structural transformation of insulin. Biol. Chem. Hoppe Seyler 371:669-673.
296. Roy, M., Lee, R.W., Brange, J., and Dunn, M.F. 1990. 1 H NMR spectrum of the native human insulin monomer. Evidence for conformational differences between the monomer and aggregated forms. J. Biol. Chem. 265:5448-5452.
297. Mirmira, R.G., and Tager, H.S. 1989. Role of the phenylalanine B24 side chain in directing insulin interaction with its receptor: Importance of main chain conformation. J. Biol. Chem. 264:6349-6354.
298. Mirmira, R.G., Nakagawa, S.H., and Tager, H.S. 1991. Importance of the character and configuration of residues B24, B25, and B26 in insulin-receptor interactions. J. Biol. Chem. 266:1428-1436.
299. Mirmira, R.G., and Tager, H.S. 1991. Disposition of the phenylalanine B25 side chain during insulin-receptor and insulin-insulin interactions. Biochemistry 30:8222-8229.
300. Cao, Q.P., Geiger, R., Langner, D., and Geisen, K. 1986. Biological activity in vivo of insulin analogues modified in the N-terminal region of the B-chain. Biol. Chem. Hoppe Seyler 367:135-140.
301. Schwartz, G., and Katsoyannis, P.G. 1978. Synthesis of des(tetrapeptide B(1-4)) and des(pentapeptide B(1-5) human insulins. Two biologically active analogues. Biochemistry 17:4550-4556.
302. Geiger, R. 1977. In Molecular Endocrinology . I. MacIntyre, and M. Szelke, editors. Amsterdam: Holland Biomedical Press. 27-41.
303. Nakagawa, S.H., and Tager, H.S. 1991. Implications of invariant residue LeuB6 in insulin-receptor interactions. J. Biol. Chem. 266:11502-11509.
304. Hua, Q.X., Liu, M., Hu, S.Q., Jia, W., Arvan, P., and Weiss, M.A. 2006. A conserved histidine in insulin is required for the foldability of human proinsulin. Structure and function of an Ala B5 analog. J. Biol. Chem. 281:24889-24899.
305. Cosmatos, A., Ferderigos, N., and Katsoyannis, P.G. 1979. Chemical synthesis of [ des (tetrapeptide B27-30), Tyr(NH2)26-B] and [ des (pentapeptide B26-30), Phe(NH2)25-B] bovine insulins. Int. J. Protein Res. 14:457-471.
306. Fischer, W.H., Saunders, D., Brandenburg, D., Wollmer, A., and Zahn, H. 1985. A shortened insulin with full in vitro potency. Biol. Chem. Hoppe Seyler 366:521-525.
307. Zakova, L., Brynda, J., Au-Alvarez, O., Dodson, E.J., Dodson, G.G., Whittingham, J.L., and Brzozowski, A.M. 2004. Toward the insulin-IGF-I intermediate structures: functional and structural properties of the [TyrB25NMePheB26] insulin mutant. Biochemistry 43:16293-16300.
308. Žáková, L., Kazdová, L., Hančlová, I., Protivínská, E., Šanda, M., Buděšínský, M., and Jiráček, J. 2008. Insulin analogues with modifications at position B26. Divergence of binding affinity and biological activity. Biochemistry 47:5858-5868.
309. Dai, J.B., Lou, M.Z., You, J.M., and Liang, D.C. 1987. Refinement of the structure of despentapetide (B26-B30) insulin at 1.5 Å resolution. Sci. Sin. 30:55-65.
310. Casaretto, M., Spoden, M., Diaconescu, C., Gattner, H.G., Zahn, H., Brandenburg, D., and Wollmer, A. 1987. Shortened insulin with enhanced in vitro potency. Biol. Chem. Hoppe Seyler 368:709-716.
311. Nakagawa, S.H., and Tager, H.S. 1993. Importance of main-chain flexibility and the insulin fold in insulin-receptor interactions. Biochemistry 32:7237-7243.
312. Boelens, R., Ganadu, M.L., Verheyden, P., and Kaptein, R. 1990. Two-dimensional NMR studies on des-pentapeptide-insulin. Proton resonance assignments and secondary structure analysis. Eur. J. Biochem. 191:147-153.
313. Nakagawa, S.H., and Tager, H.S. 1986. Role of the phenylalanine B25 side chain in directing insulin interaction with its receptor. Steric and conformational effects. J. Biol. Chem. 261:7332-7341.
314. Xu, B., Hu, S.Q., Chu, Y.C., Huang, K., Nakagawa, S.H., Whittaker, J., Katsoyannis, P.G., and Weiss, M.A. 2004. Diabetes-associated mutations in insulin: consecutive residues in the B chain contact distinct domains of the insulin receptor. Biochemistry 43:8356-8372.
315. Shoelson, S.E., Lu, Z.X., Parlautan, L., Lynch, C.S., and Weiss, M.A. 1992. Mutations at the dimer, hexamer, and receptor-binding, surfaces of insulin independently affect insulin-insulin and insulin-receptor interactions. Biochemistry 31:1757-1767.
316. Pittman, I., Nakagawa, S.H., Tager, H.S., and Steiner, D.F. 1997. Maintenance of the B-chain β-turn in [Gly B24 ] insulin mutants: a steady-state fluorescence anisotropy study. Biochemistry 36:3430-3437.
317. De Meyts, P. 2004. Insulin and its receptor: structure, function and evolution. Bioessays 26:1351-1362.
318. Kiselyov, V.V., Versteyhe, S., Gaugin, L., and De Meyts, P. 2009. Harmonic oscillator model of the insulin and IGF1 receptors' allosteric binding and activation. Mol. Syst. Biol. 5:243.
319. Whittaker, J., Whittaker, L.J., Roberts, C.T., Jr., Phillips, N.B., Ismail-Beigi, F., Lawrence, M.C., and Weiss, M.A. 2012. α-Helical element at the hormone-binding surface of the insulin receptor functions as a signaling element to activate its tyrosine kinase. Proc. Natl. Acad. Sci. U. S. A. 109:11166-11171.
320. Ward, C.W., and Lawrence, M.C. 2009. Ligand-induced activation of the insulin receptor: a multi-step process involving structural changes in both the ligand and the receptor. Bioessays 31:422-434.
321. Henzen, C. 2012. Monogenic diabetes mellitus due to defects in insulin secretion. Swiss Med. Wkly. 142:w13690.
322. Chan, S.J., Seino, S., Gruppuso, P.A., Schwartz, R., and Steiner, D.F. 1987. A mutation in the B chain coding region is associated with impaired proinsulin conversion in a family with hyperproinsulinemia. Proc. Natl. Acad. Sci. U. S. A. 84:2194-2197.
323. Gruppuso, P.A., Gorden, P., Kahn, C.R., Cornblath, M., Zeller, W.P., and Schwartz, R. 1984. Familial hyperproinsulinemia due to a proposed defect in conversion of proinsulin to insulin. N. Engl. J. Med. 311:629-634.
324. Schwartz, G.P., Burke, G.T., and Katsoyannis, P.G. 1987. A superactive insulin: [B10-aspartic acid]insulin(human). Proc. Natl. Acad. Sci. U. S. A. 84:6408-6411.
325. Burgess, T.L., and Kelly, R.B. 1987. Constitutive and regulated secretion of proteins. Annu. Rev. Cell Biol. 3:243-293.
326. Carroll, R.J., Hammer, R.E., Chan, S.J., Swift, H.H., Rubenstein, A.H., and Steiner, D.F. 1988. A mutant human proinsulin is secreted from islets of Langerhans in increased amounts via an unregulated pathway. Proc. Natl. Acad. Sci. U. S. A. 85:8943-8947.
327. Robbins, D.C., Shoelson, S., Rubenstein, A., and Tager, H. 1984. Familial hyperproinsulinemia. Two cohorts secreting indistinguishable type II intermediates of proinsulin conversion. J. Clin. Invest. 73:714-719.
328. Gabbay, K.H., DeLuca, K., Fisher, J.N., Jr., Mako, M.E., and Rubenstein, A.H. 1976. Familial hyperproinsulinemia. An autosomal dominant defect. N. Engl. J. Med. 294:911-915.
329. Edghill, E.L., Flanagan, S.E., Patch, A.M., Boustred, C., Parrish, A., Shields, B., Shepherd, M.H., Hussain, K., Kapoor, R.R., Malecki, M., et al. 2008. Insulin mutation screening in 1044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 57:1034-1042.
330. Colombo, C., Porzio, O., Liu, M., Massa, O., Vasta, M., Salardi, S., Beccaria, L., Monciotti, C., Toni, S., Pedersen, O., et al. 2008. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J. Clin. Invest. 118:2148-2156.
331. Molven, A., Ringdal, M., Nordbo, A.M., Raeder, H., Stoy, J., Lipkind, G.M., Steiner, D.F., Philipson, L.H., Bergmann, I., Aarskog, D., et al. 2008. Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes 57:1131-1135.
332. Slingerland, A.S., and Hattersley, A.T. 2005. Mutations in the Kir6.2 subunit of the KATP channel and permanent neonatal diabetes: new insights and new treatment. Ann. Med. 37:186-195.
333. Babenko, A.P., Polak, M., Cave, H., Busiah, K., Czernichow, P., Scharfmann, R., Bryan, J., Aguilar-Bryan, L., Vaxillaire, M., and Froguel, P. 2006. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N. Engl. J. Med. 355:456-466.
334. Park, S.Y., Ye, H., Steiner, D.F., and Bell, G.I. 2010. Mutant proinsulin proteins associated with neonatal diabetes are retained in the endoplasmic reticulum and not efficiently secreted. Biochem. Biophys. Res. Commun. 391:1449-1454.
335. Meur, G., Simon, A., Harun, N., Virally, M., Dechaume, A., Bonnefond, A., Fetita, S., Tarasov, A.I., Guillausseau, P.J., Boesgaard, T.W., et al. 2010. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes 59:653-661.
336. Yoshioka, M., Kayo, T., Ikeda, T., and Koizumi, A. 1997. A novel locus, Mody4 , distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 46:887-894.
337. Wang, J., Takeuchi, T., Tanaka, S., Kubo, S.K., Kayo, T., Lu, D., Takata, K., Koizumi, A., and Izumi, T. 1999. A mutation in the insulin 2 gene induces diabetes with severe pancreatic b -cell dysfunction in the Mody mouse. J. Clin. Invest. 103:27-37.
338. Oyadomari, S., Koizumi, A., Takeda, K., Gotoh, T., Akira, S., Araki, E., and Mori, M. 2002. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Invest. 109:525-532.
339. Izumi, T., Yokota-Hashimoto, H., Zhao, S., Wang, J., Halban, P.A., and Takeuchi, T. 2003. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes 52:409-416.
340. Zuber, C., Fan, J.Y., Guhl, B., and Roth, J. 2004. Misfolded proinsulin accumulates in expanded pre-Golgi intermediates and endoplasmic reticulum subdomains in pancreatic β cells of Akita mice. FASEB J. 18:917-919.
341. Liu, M., Hodish, I., Rhodes, C.J., and Arvan, P. 2007. Proinsulin maturation, misfolding, and proteotoxicity. Proc. Natl. Acad. Sci. U. S. A. 104:15841-15846.
342. Yoshinaga, T., Nakatome, K., Nozaki, J., Naitoh, M., Hoseki, J., Kubota, H., Nagata, K., and Koizumi, A. 2005. Proinsulin lacking the A7-B7 disulfide bond, Ins2Akita, tends to aggregate due to the exposed hydrophobic surface. Biol. Chem. 386:1077-1085.
343. Hua, Q.X., Nakagawa, S.H., Jia, W., Hu, S.Q., Chu, Y.C., Katsoyannis, P.G., and Weiss, M.A. 2001. Hierarchical protein folding: asymmetric unfolding of an insulin analogue lacking the A7-B7 interchain disulfide bridge. Biochemistry 40:12299-12311.
344. Jia, X.Y., Guo, Z.Y., Wang, Y., Xu, Y., Duan, S.S., and Feng, Y.M. 2003. Peptide models of four possible insulin folding intermediates with two disulfides. Protein Sci. 12:2412-2419.
345. Herbach, N., Rahtkolb, B., Kemter, E., Pichl, L., Klaften, M., De Angelis, M.H., Halban, P., Wolf, E., Aigner, B., and Wanker, R. 2007. Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe β-cell loss in Munich Ins2 C95S mutant mice. Diabetes 56:1268-1276.
346. Hodish, I., Liu, M., Rajpal, G., Larkin, D., Holz, R.W., Adams, A., Liu, L., and Arvan, P. 2010. Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J. Biol. Chem. 285:685-694.
347. Hodish, I., Absood, A., Liu, L., Liu, M., Haataja, L., Larkin, D., Al-Khafaji, A., Zaki, A., and Arvan, P. 2011. In vivo misfolding of proinsulin below the threshold of frank diabetes. Diabetes 60:2092-2101.
348. Haataja, L., Snapp, E., Wright, J., Liu, M., Hardy, A.B., Wheeler, M.B., Markwardt, M.L., Rizzo, M., and Arvan, P. 2013. Proinsulin intermolecular interactions during secretory trafficking in pancreatic β cells. J. Biol. Chem. 288:1896-1906.
349. Absood, A., Gandomani, B., Zaki, A., Nasta, V., Michail, A., Habib, P.M., and Hodish, I. 2013. Insulin therapy for pre-hyperglycemic β-cell endoplasmic reticulum crowding. PLoS One 8:e54351.
350. Shoelson, S.E., Polonsky, K.S., Zeidler, A., Rubenstein, A.H., and Tager, H.S. 1984. Human insullin B24 (Phe ® Ser), secretion and metabolic clearance of the abnormal insulin in man and in a dog model. J. Clin. Invest. 73:1351-1358.
351. Fonseca, S.G., Gromada, J., and Urano, F. 2011. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 22:266-274.
352. Thomas, S.E., Dalton, L., Malzer, E., and Marciniak, S.J. 2011. Unravelling the story of protein misfolding in diabetes mellitus. World J. Diabetes 2:114-118.
353. Ron, D. 2002. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J. Clin. Invest. 109:443-445.
354. Yuan, Q., Tang, W., Zhang, X., Hinson, J.A., Liu, C., Osei, K., and Wang, J. 2012. Proinsulin atypical maturation and disposal induces extensive defects in mouse Ins2 +/Akita β-cells. PLoS One 7:e35098.
355. Despa, F. 2010. Endoplasmic reticulum overcrowding as a mechanism of β-cell dysfunction in diabetes. Biophys. J. 98:1641-1648.
- TAKE-HOME POINTS
- REGULATION OF PLASMA GLUCOSE BY INSULIN
- INSULIN BIOGENESIS AND MECHANISM OF RELEASE
- BIOCHEMISTRY AND ELECTROPHYSIOLOGY OF INSULIN SECRETION
- INSULIN STRUCTURE
- THE INSULIN MONOMER
- INSULIN SELF-ASSEMBLY
- INSULIN STRUCTURE-FUNCTION RELATIONSHIPS
- STRUCTURE OF THE INSULIN RECEPTOR
- MUTATIONS IN THE INSULIN GENE
- REFERENCES
- Review Proinsulin and the genetics of diabetes mellitus.[J Biol Chem. 2009]Review Proinsulin and the genetics of diabetes mellitus.Weiss MA. J Biol Chem. 2009 Jul 17; 284(29):19159-63. Epub 2009 Apr 24.
- Review Structural Lessons From the Mutant Proinsulin Syndrome.[Front Endocrinol (Lausanne). 2...]Review Structural Lessons From the Mutant Proinsulin Syndrome.Dhayalan B, Chatterjee D, Chen YS, Weiss MA. Front Endocrinol (Lausanne). 2021; 12:754693. Epub 2021 Sep 30.
- Review Diabetes mellitus due to toxic misfolding of proinsulin variants.[Mol Metab. 2021]Review Diabetes mellitus due to toxic misfolding of proinsulin variants.Dhayalan B, Chatterjee D, Chen YS, Weiss MA. Mol Metab. 2021 Apr 3; :101229. Epub 2021 Apr 3.
- Review Diabetes-Associated Mutations in Proinsulin Provide a "Molecular Rheostat" of Nascent Foldability.[Curr Diab Rep. 2022]Review Diabetes-Associated Mutations in Proinsulin Provide a "Molecular Rheostat" of Nascent Foldability.Dhayalan B, Weiss MA. Curr Diab Rep. 2022 Feb; 22(2):85-94. Epub 2022 Feb 4.
- Peptide Model of the Mutant Proinsulin Syndrome. I. Design and Clinical Correlation.[Front Endocrinol (Lausanne). 2...]Peptide Model of the Mutant Proinsulin Syndrome. I. Design and Clinical Correlation.Dhayalan B, Glidden MD, Zaykov AN, Chen YS, Yang Y, Phillips NB, Ismail-Beigi F, Jarosinski MA, DiMarchi RD, Weiss MA. Front Endocrinol (Lausanne). 2022; 13:821069. Epub 2022 Mar 1.
- Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships...Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...