

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Stress constitutes a state of threatened homeostasis triggered by intrinsic or extrinsic adverse forces (stressors) and is counteracted by an intricate repertoire of physiologic and behavioral responses aiming to maintain/reestablish the optimal body equilibrium (eustasis). The adaptive stress response depends upon a highly interconnected neuroendocrine, cellular, and molecular infrastructure, i.e. the stress system. Key components of the stress system are the hypothalamic-pituitary-adrenal (HPA) axis and the autonomic nervous system (ANS), which interact with other vital centers in the central nervous system (CNS) and tissues/organs in the periphery to mobilize a successful adaptive response against the imposed stressor(s). Dysregulation of the stress system (hyper- or hypo-activation) in association with potent and/or chronic stress can markedly disrupt the body homeostasis leading to a state of cacostasis or allostasis, with a spectrum of clinical manifestations. This chapter describes the organization and physiology of the stress system, focusing on its interactions with other CNS centers and endocrine axes, and reviews the existing evidence linking stress to pathophysiologic mechanisms implicated in the development of stress-related diseases affecting the endocrine, metabolic, gastrointestinal, and immune systems. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
STRESS AND STRESS SYNDROME- DEFINITIONS AND PHENOMENOLOGY
All vital physiologic systems of the body are inherently programmed, through rigorous fine-tuning achieved during evolution, to preserve a predefined steady state (homeostasis or eustasis), which is essential for life and well-being [1-3]. This optimal equilibrium is constantly challenged by adverse forces which are intrinsic or extrinsic, real or even perceived, and are described as stressors [1]. Thus, stress is defined as a state of disharmony (cacostasis or allostasis) and is counteracted by an intricate repertoire of physiologic and behavioral responses which aim to maintain/reestablish the threatened homeostasis (adaptive stress response) [1]. This adaptive stress response is mediated by a complex and interconnected neuroendocrine, cellular, and molecular infrastructure which constituents the stress system and is located in both the central nervous system (CNS) and the periphery [1, 2]. The adaptive response of each individual to stress is determined by a multiplicity of genetic, environmental, and developmental factors. Changes in the ability to effectively respond to stressors (e.g. inadequate, excessive and/or prolonged reactions) may lead to disease. Moreover, highly potent and/or chronic stressors can have detrimental effects on a variety of physiologic functions, including growth, metabolism, reproduction, and immune competence, as well as on behavior and personality development. Of note, prenatal life, infancy, childhood, and adolescence are critical periods in the process of forming the matrix of the adaptive stress response, characterized by high plasticity of the stress system and increased vulnerability to stressors.
The stress system receives and integrates a great diversity of neurosensory (i.e. visual, auditory, somatosensory, nociceptive, and visceral), blood-borne, and limbic signals which arrive at the various stress system centers/stations through distinct pathways. Acute stress system activation triggers a cluster of time-limited changes, both behavioral and physical, which are rather consistent in their qualitative presentation and are collectively defined as the stress syndrome [1-4]. Under normal conditions these changes are adaptive and improve the chances of survival. Initially, the stimulation of the stress system components follows a stressor-specific mode; however, as the potency of the stressor(s) increases the specificity of the adaptive response decreases in order to eventually present the relatively nonspecific stress syndrome phenomenology which follows exposure to potent stressors.
Behavioral adaptation includes enhanced arousal, alertness, vigilance, cognition, focused attention, and analgesia, whilst there is concurrent inhibition of vegetative functions, such as feeding and reproduction. In parallel, physical adaptation mediates an adaptive redirection of energy and body resources. As such, increases in the cardiovascular tone, respiratory rate and intermediate metabolism (gluconeogenesis and lipolysis) work in concert to promote this redirection of vital substrates, while energy consuming functions (e.g. digestion, reproduction, growth, and immunity) are temporally suppressed. Thus, oxygen and nutrients are primarily shunted to the CNS and to stressed body site(s) where they are needed the most.
In addition to the adaptive stress response, restraining forces are also activated during stress to prevent a potential excessive response of the various stress system components [1-4]. The ability to timely and precisely develop restraining forces is equally essential for a successful outcome against the imposed stressor(s), since prolonging the mobilized adaptive stress response can turn maladaptive and contribute to the development of disease.
Interestingly, the mobilization of the stress system is often of a magnitude and nature that allows the perception of control by the individual. Under such conditions, stress can be rewarding and pleasant, or even exciting, providing positive stimuli to the individual for emotional and intellectual growth and development [5]. Thus, it is not surprising that the stress system activation during feeding and sexual activity, both sine qua non functions for survival, is primarily linked to pleasure.
STRESS SYSTEM- PHYSIOLOGY AND INTERACTIONS
Neuroendocrine Effectors of the Stress Response- “The Stress System”
Although the entire CNS is directly or indirectly involved in preserving and fine-tuning the overall body homeostasis, specific areas of the brain have critical, distinct roles in orchestrating the stress response. As such, the central components of the stress system are located in the hypothalamus and the brainstem and include the parvocellular corticotropin-releasing hormone (CRH) and arginine-vasopressin (AVP) neurons of the paraventricular nuclei (PVN) of the hypothalamus, and the CRH neurons of the paragigantocellular and parabranchial nuclei of the medulla, as well as the locus coeruleus (LC) and other catecholaminergic, norepinephrine (NE)-synthesizing cell groups of the medulla and pons (central sympathetic nervous system) [1-4]. The peripheral limps of the hypothalamic-pituitary-adrenal (HPA) axis, together with the efferent sympathetic/adrenomedullary system, constitute the peripheral components of this interconnected system.
CENTRAL STRESS SYSTEM - CRH, AVP, & CATECHOLAMINERGIC NEURONS
The central neurochemical circuitry responsible for the stress system activation forms a highly complex physiological system within the CNS, consisting of both stimulatory and inhibitory networks with multiple sites of interaction which modulate and fine-tune the adaptive stress response [1-4]. The key components of these networks are the hypothalamic CRH and AVP neurons in combination with the central catecholaminergic (LC/NE) neurons (Figure 1). The central stress system activation is based on reciprocal reverberatory neural connections between the PVN CRH and the catecholaminergic LC/NE neurons, with CRH and NE stimulating the secretion of each other through CRH receptor-1 (CRH-R1) and α1-noradrenergic receptors, respectively [6-8]. Of note, autoregulatory ultrashort negative feedback loops exist in both the PVN CRH and the brainstem catecholaminergic neurons [9, 10], with collateral fibers inhibiting CRH and catecholamine secretion respectively, via inhibition of the corresponding presynaptic CRH- and α2-noradrenergic receptors [11]. In addition, multiple other regulatory central pathways exist, since both CRH and catecholaminergic neurons receive stimulatory innervation from the serotoninergic and cholinergic systems [12, 13], and inhibitory input from the gamma-aminobutyric acid (GABA)/benzodiazepine (BZD) and the opioid neuronal systems of the brain [14, 15], as well as by glucocorticoids (the end-product of the HPA axis) [16]. Interestingly, both α2-adrenoceptor and opiate agonists act through separate receptors on neurons in the LC, albeit sharing common post-receptor effector signaling mediated through Gi proteins [17].

Figure 1.
A simplified representation of the central and peripheral components of the stress system, their functional interrelations and their relationships to other central nervous system (CNS) pathways involved in the stress response. CRH: corticotropin-releasing hormone; LC/NE sympathetic system: locus coeruleus/norepinephrine-sympathetic system; POMC: proopiomelanocortin; AVP: arginine vasopressin; GABA: γ-aminobutyric acid; BZD: benzodiazepine; ACTH: adrenocorticotropic hormone (corticotrophin); NPY: neuropeptide Y; SP: substance P. Activation is represented by solid green lines and inhibition by dashed red lines.
CRH, a 41-amino acid peptide, was first isolated as the principal hypothalamic stimulus to the pituitary-adrenal axis by Vale et al. in 1981 [18]. The subsequent availability of synthetic CRH and of inhibitory analogues opened huge vistas for stress research. Thus, CRH and CRH-receptors were identified in numerous extra-hypothalamic sites of the brain, including parts of the limbic system, the basal forebrain, the anterior pituitary and the central arousal-sympathetic systems (LC-sympathetic systems) in the brainstem and spinal cord [19, 20]. Moreover, central administration of CRH was shown to set in motion a coordinated series of physiologic and behavioral responses which included activation of the pituitary-adrenal axis and the sympathetic nervous system (SNS), as well as characteristic stress-related behaviors [21]. Hence, it became evident that CRH plays a broader role in coordinating the stress response than had been previously suspected [3, 4]. In fact, this neuropeptide appears to reproduce the stress response phenomenology, as summarized in Table 1.
Table 1.
Behavioral and Physical Adaptation During Stress
| Behavioral Adaptation |
| Adaptive redirection of behavior |
| Increased arousal and alertness |
| Increased cognition, vigilance and focused attention |
| Suppression of feeding behavior |
| Suppression of reproductive behavior |
| Inhibition of gastric motility; stimulation of colonic motility |
| Containment of the stress response |
| Physical Adaptation |
| Adaptive redirection of energy |
| Oxygen and nutrients directed to the central nervous system and stressed body site(s) |
| Altered cardiovascular tone; increased blood pressure and heart rate |
| Increased respiratory rate |
| Increased gluconeogenesis and lipolysis |
| Detoxification from toxic products |
| Inhibition of reproductive and growth axes |
| Containment of the stress response |
| Containment of the inflammatory/immune response |
Adapted from Chrousos G.P. and Gold P.W., JAMA, 1992; 267,1244.
CRH binds to specific receptors which belong to the class II seven-transmembrane G-protein-coupled receptor superfamily of receptors (GPCRs) [22]. In addition to their wide expression throughout the brain, CRH receptors are found in a number of peripheral sites, including the adrenal medulla, prostate, gut, spleen, liver, kidney and testis. Distinct CRH receptor subtypes have been identified in humans, i.e. CRH-R1 and CRH-R2, which are encoded by distinct genes on chromosomes 17 and 7, respectively (Figure 2) [23, 24]. CRH-R1 and CRH-R2 share a 70% homology of their amino acid sequence, but exhibit unique pharmacologic profiles and are differentially expressed, hence they appear to mediate selective actions of CRH at different target organs/tissues. CRH-R1 is widely distributed in the brain, mainly in the anterior pituitary, neocortex and cerebellum, whilst is also expressed in the adrenal gland, gastrointestinal tract, skin, ovary and testis [25]. On the other hand, CRH-R2 receptors are mainly expressed in the peripheral vasculature, skeletal muscles, gastrointestinal tract and heart, while they also exhibit a widespread distribution in subcortical structures of the brain (e.g. in the lateral septum, amygdala, hypothalamus and brain stem) [26]. Importantly, CRH-R1 is considered the only CRH-R type present in the LC, cerebellar cortex, thalamus and striatum, whereas exclusive CRH-R2 expression has been reported in the bed nucleus of the stria terminalis [27-29]. Of note, both CRH receptor genes have the ability of variant splicing, producing different isoforms for each subtype. As such, the CRH-R1 gene appears to have several splice variants (R1b, R1c, R1d, R1e, R1f, R1g and R1h) which encode proteins with altered N-terminal (CRH-R1c, CRH-R1e, CRH-R1h), intracellular (CRH-R1b, CRH-R1f) and transmembrane (CRH-R1g, CRH-R1d) segments compared to the prototypic CRH-R1a; however, their ligand-binding affinity is low and their expression in native tissues has not been fully characterized [30]. Similarly, the CRH-R2 gene has three splice variants, respectively, encoding the CRH-R2a, CRH-R2b, CRH-R2c isoforms which differ only in the extracellular N-terminus and have distinct tissue distributions. Indeed, CRH-R2a is localized in subcortical regions, including the lateral septum and the hypothalamic paraventricular and ventromedial nuclei. Conversely, CRH-R2b in rodents is primarily localized in the heart, gastrointestinal tract, skeletal muscles and in non-neural brain tissues (e.g. in cerebral arterioles and the choroid plexus), whilst CRH-R2c expression has been detected in human limbic regions [26]. This diversity of CRH receptor subtype and isoform expression is considered to play an important role in modulating the stress response by implicating locally different ligands (CRH and CRH-related peptides) and different intracellular second messengers (e.g. CRH-R2 is now recognized to play a significant role in the physiology and pathophysiology of the cardiovascular system) [22, 31].

Figure 2.
Corticotropin-releasing hormone (CRH) receptor subtypes, splice variants and tissue distribution. CRH is considered the specific endogenous ligand for CRH-R1, while Urocortin 2 and Urocortin 3 are considered the specific endogenous ligands of CRH-R2. Urocortin 1 is considered an endogenous ligand for both CRH-R subtypes. CRH binds to CRH-R2 with an affinity that is 100-fold lower compared to the binding affinity of urocortins. CRH-R: Corticotropin-releasing hormone receptor, TM: transmembrane.
AVP is a nonapeptide produced by PVN parvocellular neurons and by the magnocellular neurons of the neurohypophysis [32]. While the AVP from the posterior pituitary is secreted into the circulation and modulates fluid and electrolyte homeostasis, AVP of PVN origin, like CRH, is secreted into the hypophyseal portal system and holds a key role in the stress response, representing the second most important modulator of pituitary ACTH secretion [32, 33]. Notably, whilst CRH appears to directly stimulate ACTH secretion, AVP and other factors (e.g. angiotensin II) have primarily synergistic or additive effects [32-35]. Indeed, AVP exhibits synergy with CRH in vivo, when these peptides are co-administered in humans [36], by acting on a V1-type receptor (V1β, also referred as V3) and exerting its effects through calcium/phospholipid-dependent mechanisms [37]. This synergistic effect on pituitary ACTH secretion offers an alternate pathway to influence the consequent HPA axis activation at the hypothalamic level, since the secretion of CRH and AVP is further regulated by a variety of different neuropeptides, including catecholamines which stimulate CRH secretion, and ghrelin (a GH-secretagogue factor) which appears to stimulate predominantly AVP secretion [38, 39]. Similarly, leptin which is expressed in the central branch of the HPA axis can regulate both CRH and ACTH secretion acting in an autocrine/paracrine manner with most evidence indicating that it exerts an inhibitory effect on the HPA axis, although depending on the species, it may also stimulate the HPA activity [40]. Furthermore, endocannabinoids appear to negatively regulate basal and stimulated ACTH release at multiple levels of the HPA axis [41].
Interestingly, a subset of parvocellular neurons synthesize and secrete both CRH and AVP and the relative proportion of this subset is increased significantly by stress conditions [42-44]. Moreover, the terminals of the parvocellular PVN CRH and AVP neurons project to different CNS sites, including noradrenergic neurons of the brainstem and the hypophyseal portal system in the median eminence. PVN CRH and AVP neurons also send projections to and activate pro-opiomelanocortin (POMC)-containing neurons in the arcuate nucleus of the hypothalamus. In turn, these POMC-containing neurons project reciprocally to the PVN CRH and AVP neurons, innervate LC/NE-sympathetic neurons of the central stress system in the brainstem and terminate on pain control neurons of the hind brain and spinal cord. Thus, stress system activation, via CRH and catecholamines, stimulates the hypothalamic secretion of β-endorphin and other POMC-peptides which reciprocally inhibit the stress system activity, induce analgesia ("stress-induced" analgesia) and may also influence the emotional tone (Figure 1).
It is also noteworthy that, among the multiple regulatory central pathways which influence the central stress system activity, neuropeptide Y (NPY) stimulates CRH neurons, whereas it inhibits the central SNS [45, 46]. This may be of particular relevance to changes in stress system activity in states of dysregulated food intake and obesity. Interestingly, glucocorticoids, which stimulate appetite, have been also shown to stimulate the hypothalamic NPY gene expression, while they inhibit the PVN CRH and LC/NE-sympathetic systems [47]. Of note, in addition to its appetite stimulating and anxiolytic activities, NPY can also act peripherally exerting detrimental actions on the cardiovascular system and metabolism, related to adaptation to stress [48]. On the other hand, substance P (SP) has reciprocal actions to those of NPY, since it inhibits CRH neurons [49], whereas it activates the central catecholaminergic system [50]. It is considered that, SP release is increased centrally by peripheral activation of somatic afferent fibers and, hence, may be relevant to stress system activity changes induced by chronic inflammatory and/or painful states [51]. Along with NPY and SP, a number of other neuropeptides, including the Tyr-MIF-1 family of peptides, teneurin C-terminal associated peptides (TCAP), oxytocin, cholecystokinin (CCK) and galanin, appear to be implicated in the regulation of stress-like behavior [52].
HYPOTHALAMIC-PITUITARY-ADRENAL (HPA) AXIS
The HPA axis is a vital component of both the central and the peripheral limb of the stress system [1, 4]. As such, HPA axis integrity and precise regulation of its function are essential characteristics of the successful adaptive response to any stressor. At the level of the hypothalamic-pituitary unit, CRH is released into the hypophyseal portal system and acts as the principal regulator of the anterior pituitary ACTH secretion [4]. As aforementioned, CRH binding on CRH-R1 of the corticotrophs is permissive for ACTH secretion, whilst AVP acts as a potent synergistic factor to CRH with little ACTH secretagogue activity by itself [32-34, 53]. Under non-stressful conditions, both CRH and AVP are secreted into the portal system in a circadian and highly concordant pulsatile fashion [54, 55]. Indeed, the HPA axis activity is characterized not only by a typical circadian rhythm, but also by an ultradian pattern of discrete pulsatile release of glucocorticoids, with a pulse of production every 1-2 hours [56]. The amplitude of the CRH and AVP pulses increases in the early morning hours, consequently resulting in increased amplitude and frequency of ACTH and cortisol secretory bursts in the systemic circulation [57, 58]. Of note, recent data indicate that various factors including age, body mass index (BMI), and gender, are individually and in some cases jointly associated with endogenous ACTH-induced stimulation of overnight pulsatile cortisol secretion [59].
The circadian release of CRH/AVP/ACTH/cortisol in their characteristic pulsatile manner appears to be controlled by one or more CNS pace makers, as will be more precisely described in the following section on the “CLOCK system” [60, 61]. These diurnal variations are perturbed by changes in lighting, feeding and physical activity patterns, whilst they are disrupted when a stressor is imposed. During acute stress, the amplitude and synchronization of both CRH and AVP secretory pulses increases, with additional recruitment of PVN CRH and AVP secretion. Furthermore, angiotensin II, various cytokines and lipid mediators of inflammation are also secreted, depending on the stressor, and act on various levels of the HPA axis to mainly stimulate its activity. Interestingly, nicotine can also induce the HPA axis via both CRH-R and AVP V(1b) receptors; hence, when CRH-R is blocked, nicotine may utilize the AVP V(1b) receptor to induce its action and increase the secretion of ACTH and glucocorticoids [62].
The adrenal cortex constitutes the principal target organ of the pituitary-derived circulating ACTH. The latter is the key regulator of glucocorticoid and adrenal androgen secretion by the zona fasciculata and zona reticularis, respectively, whilst it is also implicated in the regulation of aldosterone secretion by the zona glomerulosa [63]. Notably, existing evidence suggests that the adrenal cortisol secretion is further regulated by other hormones and/or cytokines coming from the adrenal medulla or the systemic circulation, and by neuronal signals via the autonomic innervation of the adrenal cortex (Figure 1).
Glucocorticoids are the final hormonal effectors of the HPA axis, exerting their pleiotropic effects via their ubiquitously distributed intracellular receptors (GRα and GRβ; both members of the nuclear receptor superfamily) [64]. The non-activated glucocorticoid receptor resides in the cytosol as a hetero-oligomer with heat shock proteins and immunophilin [65]. Upon ligand binding, glucocorticoid receptors dissociate from the rest of this hetero-oligomer, and subsequently homodimerize and translocate into the nucleus, where they interact with specific glucocorticoid response elements (GREs) of the DNA to transactivate or transrepress appropriate hormone-responsive genes [66]. Transactivation has been suggested as mediating most of the adverse effects of glucocorticoids, while transrepression is considered to mediate mostly anti-inflammatory glucocorticoid effects by inhibiting several inflammatory mediators/pathways (e.g. AP-1, NF-κB). Post-translational modifications of glucocorticoid receptors (e.g. phosphorylation, acetylation, ubiquitination and sumoylation) regulate the receptor stability and nuclear localization, as well as its interaction with other proteins [67-69]. Furthermore, glucocorticoid receptor activation causes changes in the stability of other mRNAs and, thus, the translation rates of several glucocorticoid-responsive proteins. Notably, glucocorticoids influence the secretion rates of specific proteins and alter the electrical potential of neuronal cells, through mechanisms that remain to be elucidated. Glucocorticoids can further induce rapid non-genomic effects, via mechanisms which are also not fully clarified yet [70]. Moreover, there are also data indicating that glucocorticoids have the ability to regulate mitochondrial functions and energy metabolism. Indeed, the presence of both GRα and GRβ in mitochondria of animal and human cells has been associated with modulation of mitochondrial functions indicating that the cross-talk of glucocorticoid receptors with mitochondria may be involved in cell survival [71, 72].
Glucocorticoids play a crucial role in the regulation of the basal HPA axis activity and in the termination of the stress response by acting at multiple levels, including extra-hypothalamic regulatory centers, the hypothalamus and the pituitary (Figure 1) [73]. As such, the inhibitory glucocorticoid feedback on the ACTH secretory response limits the duration of the total tissue exposure to glucocorticoids, thus minimizing the catabolic, anti-reproductive and immunosuppressive effects of these hormones. Interestingly, a dual glucocorticoid receptor system exists in the CNS, including both type I glucocorticoid receptors (mineralocorticoid receptor) which respond to low levels of glucocorticoids and primarily act to induce activation; and the classic glucocorticoid receptor (type II) which responds to higher levels of glucocorticoids, stress-related or not, and can either dampen some systems or activate other. The negative feedback control of the CRH and ACTH secretion is mediated through type II glucocorticoid receptors.
Finally, it must be highlighted that, the glucocorticoid secretion pulsatility is among the main factors determining the HPA axis responsiveness to stress and the transcriptional responses of glucocorticoid responsive genes [74, 75]. Data on the downstream effects of short-term fluctuations in serum glucocorticoid concentrations indicate that ultradian cortisol pulsatility can impact on the gene expression and phenotype of target cells. Importantly, pulsatile cortisol has been shown to significantly reduce cell survival due to increased apoptosis compared to continuous exposure to the same cumulative dose [76].
SYMPATHETIC/ADRENOMEDULLARY AND PARASYMPATHETIC SYSTEMS
The autonomic nervous system (ANS) provides a rapidly responsive mechanism to control a wide range of physiologic functions. As such, the cardiovascular, respiratory, gastrointestinal, renal, endocrine, and other vital systems are tightly regulated by either the SNS or the parasympathetic system or the combined activity of both [77]. Indeed, the ANS activity is typically regulated through a dual reaction mechanism, since the parasympathetic system can equally assist or antagonize most of the SNS functions by withdrawing or increasing its activity, respectively.
Sympathetic innervation of peripheral organs is derived from the efferent preganglionic fibers whose cell bodies lie in the intermediolateral column of the spinal cord. These nerves synapse in the bilateral chain of sympathetic ganglia with postganglionic sympathetic neurons, which innervate the smooth muscle cells of the vasculature, skeletal muscles, heart, kidneys, gut, adipose tissue and many other organs [78]. The preganglionic neurons are primarily cholinergic, whereas the postganglionic neurons release mostly noradrenaline. The SNS activity has an additional humoral contribution consisting of circulating epinephrine and, to a lesser extent, norepinephrine released by the adrenal medulla which can be considered as a modified sympathetic ganglion.
Moreover, a plethora of additional neurotransmitters is implicated in the regulation of the ANS activity, complementing the effects of acetylcholine and norepinephrine. Both the sympathetic and parasympathetic system contain several subpopulations of target-selective and neurochemically coded neurons which express a variety of neuropeptides and, in some cases, adenosine triphosphate (ATP), nitric oxide (NO), or lipid mediators of inflammation [79]. Interestingly, CRH, NPY, somatostatin, and galanin are colocalized in noradrenergic vasoconstrictive neurons, whereas vasoactive intestinal polypeptide (VIP) and, to a lesser extent, SP and calcitonin gene-related peptide (CGRP) are colocalized in cholinergic neurons. In addition, the signal transmission in sympathetic ganglia is further modulated by neuropeptides released from preganglionic fibers and short interneurons (e.g. enkephalin and neurotensin), as well as by primary afferent (e.g. VIP and SP) nerve collaterals [80]. Thus, the particular combination of neurotransmitters in sympathetic neurons is markedly influenced by central and local factors which may trigger or suppress specific genes.
Interactions with Other CNS Components
The stress system not only sets the arousal level and regulates vital signs, but further interacts with other crucial CNS components, including the mesocorticolimbic dopaminergic system (“reward” system), the amygdala/hippocampus complex and the arcuate nucleus POMC neuronal system [81-83]. In turn, following activation by stress, these CNS systems act via specific neuronal pathways to modify the stress system activity, hence forming a complex reciprocal mechanism which fine-tunes the adaptive response. Of note, well-established interactions exist between the stress system and distinct CNS centers which are essential for survival, such as the thermoregulatory and appetite-satiety centers [84].
MESOCORTICOLIMBIC DOPAMINERGIC SYSTEM
The mesocortical and mesolimbic components of the dopaminergic system are highly innervated by PNV CRH neurons and the LC/NE-sympathetic noradrenergic system and, thus, are activated by CRH, catecholamines and glucocorticoids during stress. The mesocortical system contains dopaminergic neurons of the ventral tegmentum which send projections to the prefrontal cortex. Activation of these neurons appears to centrally suppress the stress system response and is implicated in anticipatory phenomena and cognitive functions [82]. Similarly, the mesolimbic system also consists of dopaminergic neurons of the ventral tegmentum. These neurons innervate the nucleus accumbens and are considered to play a pivotal role in motivational/reinforcement/reward phenomena and in forming the central dopaminergic “reward” system [84]. Hence, euphoria and dysphoria are likely to be mediated by the mesocorticolimbic system which is considered the central target of several addictive substances (e.g. cocaine).
AMYGDALA/HIPPOCAMPUS
The amygdala/hippocampus complex is activated during stress primarily by ascending catecholaminergic neurons originating in the brain stem or by inner emotional stressors (e.g. conditioned fear) possibly from cortical association areas [83]. The amygdala nuclei constitute the principal CNS center for fear-related behaviors and their activation is important for both retrieval and emotional analysis of all relevant stored information for any given stressor. In response to emotional stressors, the amygdala can directly stimulate central stress system components and the mesocorticolimbic dopaminergic system. Interestingly, there are CRH peptidergic neurons in the amygdala which respond positively to glucocorticoids and whose activation leads to stress system stimulation and anxiety. Of note, CRH neurons in the central nucleus of the amygdala send projections to the PVN parvocellular regions and the parabrachial nucleus of the brain stem which are considered crucial for CRH-induced neuroendocrine, autonomic and behavioral effects. Moreover, CRH fibers also interconnect the amygdala with the bed nucleus of the stria terminalis and the hypothalamus [85, 86]. Conversely to the stimulatory CRH and norepinephrine effect, the hippocampus exerts a tonic and stimulated inhibitory effect on the amygdala activity and the PVN CRH and LC/NE-sympathetic systems. Indeed, the hippocampus plays an important role in shutting off the HPA stress response; hence, hippocampal atrophy or damage impairs this shut off function and can lead to prolonged HPA responses to psychological stressors [87]. These findings led to the "glucocorticoid cascade hypothesis" of stress and aging. Accordingly, Lupien et al. have shown that progressively increased salivary cortisol levels during annual exams over a 5-year period can predict reduced hippocampal volume and decreased performance on hippocampal-dependent learning and memory tasks [88]. Moreover, Refojo et al. have demonstrated, through specific CRH-R1 deletions in glutamatergic, GABAergic, dopaminergic and serotonergic cells, that CRH-R1 absence in forebrain glutamatergic circuits reduces anxiety and impairs neurotransmission in the amygdala and hippocampus, whilst elective CRH-R1 deletion in midbrain dopaminergic neurons results in increased anxiety-like behavior, suggesting a bidirectional model for the CRH-R1 role in anxiety [89].
ARCUATE NUCLEUS PROOPIOMELANOCORTIN (POMC) NEURONAL SYSTEM
Reciprocal innervation exists between opioid peptide (POMC-producing) neurons of the hypothalamic arcuate nucleus and both the CRH/AVP-producing and LC/NE-noradrenergic neurons [6, 81]. Stress system activation stimulates hypothalamic release of POMC-derived peptides, including α-melanocyte-stimulating hormone (α-MSH) and β-endorphin, which reciprocally inhibit the activity of both the central stress system components. Moreover, through projections of these neurons to the hind brain and the spinal cord, "stress- induced analgesia” is achieved by inhibition of the ascending pain pathways (Figure 1).
THERMOREGULATORY CENTER- TEMPERATURE REGULATION
It is well-established that the activation of the LC/NE-noradrenergic and PVN CRH systems by stressors increases the body core temperature. Intracerebroventricular administration of both norepinephrine and CRH can cause temperature elevation, possibly through prostanoid-mediated actions on the septal and hypothalamic temperature-regulating center. CRH has also been shown to partly mediate the pyrogenic effects of the three major inflammatory cytokines, i.e. tumor necrosis factor-α (TNF-α), interleukin 1 (IL-1), and interleukin-6 (IL-6), following stimulation by lipopolysaccharide (LPS; endotoxin, a potent exogenous pyrogen) [84].
Interestingly, psychological stress appears to also significantly affect the central thermoregulatory system, inducing a rise in body core temperature through activation of thermoregulatory sympathetic premotor neurons in the medullary raphe region [90]. This psychogenic fever can last for as long as the underlying psychological stressor(s) exist(s) [91], whilst in animal stress models it can be reduced by systemic injection of an antagonist of the β3-adrenoreceptor [90-92]. Of note, the latter is the adrenoreceptor subtype which is abundantly expressed in brown adipose tissue (BAT) and mediates BAT thermogenesis [93].
APPETITE/SATIETY CENTERS- APPETITE REGULATION
Stress is directly implicated in the regulation of appetite by influencing the central appetite/satiety centers in the hypothalamus. As such, CRH can acutely cause anorexia, whilst NPY (a potent orexigenic neuropeptide) also stimulates CRH secretion via Y1 receptors, probably to counter-regulate its own actions. In parallel, NPY also inhibits the LC/NE-sympathetic system and activates the parasympathetic system, with both effects decreasing thermogenesis and facilitating digestion and storage of nutrients [45, 46]. On the other hand, leptin (an adipocyte-derived satiety hormone/adipokine), inhibits the secretion of hypothalamic NPY, whilst it also stimulates arcuate nucleus POMC neurons which secrete α-MSH (a potent anorexigenic and thermogenic peptide, acting through specific melanocortin receptors type 4; MC4) (Figure 3). Importantly, apart from its appetite enhancing effects, NPY appears to be critical for maintaining stress responses, although its range of actions in the rest of the body and its exact role as a stress mediator remain to be fully clarified [48]. Of note, existing evidence indicates that stress-induced eating behavior in obese women with binge-eating disorders is characterized both by stronger motivation to eat (as manifested by a fast-initial eating rate) and by absence of satiety perception (as manifested by a lower deceleration of the eating rate) [94]. Finally, recent data support the direct implication of glucocorticoids in appetite regulation [95].

Figure 3.
Schematic representation of interactions between the hypothalamic-pituitary-adrenal (HPA) axis, adipose tissue and hypothalamic appetite-satiety centers. ARC: arcuate nucleus; PVN: paraventricular nucleus; LHA: lateral hypothalamic area; CRH: corticotropin-releasing hormone; ACTH: adrenocorticotropic hormone (corticotrophin); POMC: proopiomelanocortin; NPY: neuropeptide Y; AgRP: agouti related peptide; α-MSH: α-melanocyte-stimulating hormone; Y1: neuropeptide Y receptor type 1; MC4R: melanocortin receptor type 4; TRH: thyrotropin-releasing hormone; MCH: melanin concentrating hormone; OXY: oxytocin. Activation is represented by solid green lines and inhibition by dashed red lines.
CLOCK System
More recently, it became evident that the stress system is interconnected and communicates at multiple levels with an additional vital system, defined as the CLOCK system, which generates the body circadian rhythms and regulates a wide range of physiologic functions [60]. This system is comprised by a main central hypothalamic component and numerous associated extra-hypothalamic, peripheral components [60, 96].
The central CLOCK system component is located in the suprachiasmatic nuclei (SCN) of the hypothalamus and acts as a “master” CLOCK under the influence of light/dark input through the eyes (Figure 4) [96]. Indeed, light/dark information can travel through the retinohypothalamic tract (RHT; a photic neural input pathway implicated in the regulation of circadian rhythms in mammals). As such, this information travels from the retina, and specifically from the photosensitive retina ganglion cells, to the SCN. Subsequently, SCN neurons of the central CLOCK system send efferent projections: (i) to the other CNS sites [e.g. to the PVN, medial preoptic area (MPA) and dorsomedial nucleus (DMH) of the hypothalamus and to the pineal gland] to transfer timing information, regulate melatonin and pituitary hormone secretion and control sleep, food intake and body temperature; and (ii) to ANS centers (sympathetic and parasympathetic) [96, 97]. As a result, all these basic physiologic functions of the body follow circadian rhythm patterns under the control of the central CLOCK system which facilitates the entrainment of these circadian rhythms to the daily light/dark cycle and essentially to the rotation of earth (Figure 4) [98, 99]. Notably, an important intracellular signaling pathway which couples light to entrainment of the mammalian “master” CLOCK is mediated via the p42/44 mitogen-activated protein kinase (MAPK) pathway and mitogen- and stress-activated protein kinase 1 (MSK1; a downstream target of the MAPK cascade) [100].
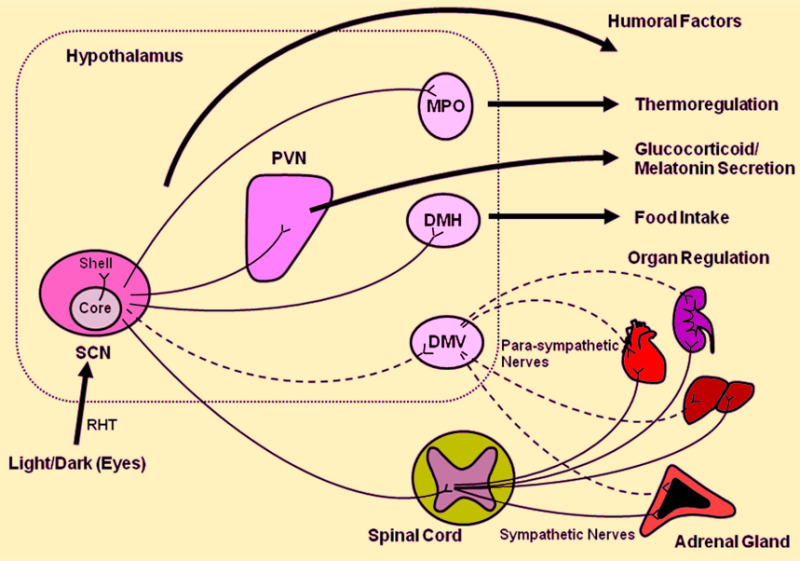
Figure 4.
Central CLOCK synchronizes the peripheral CLOCKs and regulates peripheral organ activities via neural and humoral interactions. Light/dark information travels via the retinohypothalamic tract (RHT) from the retina (specifically from the retina ganglion cells which are intrinsically photosensitive) to the suprachiasmatic nucleus (SCN) where efferent neurons: (i) transfer timing information to other parts of the CNS, such as the paraventricular nucleus (PVN), medial preoptic area (MPO) and dorsomedial nucleus (DMH) of the hypothalamus and the pineal gland; and (ii) affect the autonomic nervous system (sympathetic and parasympathetic); in order to regulate the secretion of pituitary hormones and melatonin, which in turn control basic physiologic functions, including regulation of sleep, food intake and body temperature. DMV: dorsal motor nucleus of vagus. [Adapted from Nader, N, Chrousos, GP, Kino T. Trends Endocrinol Metab 2010;21:277].
The extra-hypothalamic, peripheral components of the CLOCK system are located in all other organs/tissues, including brain centers beyond the SCN [98, 101]. Interestingly, in order to generate intrinsic circadian rhythms, the central and peripheral CLOCKs utilize almost the same transcriptional regulatory machinery [98, 102]. A central role in this machinery is played by two specific transcription factors, i.e. the circadian locomotor output cycles kaput (Clock; a histone acetyltransferase) and the brain-muscle-arnt-like protein 1 (Bmal1; the heterodimer partner of Clock) transcription factor, which both belong to the basic helix-loop-helix (bHLH) PER-ARNT-SIM (PAS) superfamily of transcription factors [96].
During the day, the Clock/Bmal1 interaction leads to transcriptional activation of two principal clock genes, i.e. the Per (Period 1,2,3) and Cry (Cryptochrome 1,2) gene, resulting in high levels of these transcripts. The Per and Cry proteins, after heterodimerization, translocate to the nucleus and interact with the Clock/Bmal1 complex, thus inhibiting their own transcription. During the night, the Per/Cry repressor complex is degraded and the Clock/Bmal1 complex can then activate a new cycle of transcription [103]. This entire cycle lasts approximately 24 hours and results from a combination of transcriptional and post-translational negative feedback loops, where Per and Cry proteins periodically suppress their own expression. Notably, post-translational modification and degradation of circadian clock proteins appear to play crucial roles in determining the circadian periodicity of the CLOCK [104], whilst rhythmic alterations in 3',5'-cyclic adenosine monophosphate (cAMP) signaling can determine central CLOCK properties, including amplitude, phase and period [105]. In addition, a number of other candidate CLOCK mediators, such as Timeless, Dec1, Dec2, Rev-erbα, retinoic acid receptor-related orphan receptor α (RORα) and E4bp4, appear to play further roles in this system which are not fully explored yet [106, 107].
Importantly, the central (master) CLOCK can synchronize the circadian rhythm of peripheral CLOCKs via both humoral and neural connections which remain to be further clarified [99]. Thus, destruction of the central CLOCK can revoke the synchronization of peripheral CLOCKs in different organs/tissues, while the circadian rhythm of each peripheral CLOCK is still retained. The latter suggests that peripheral CLOCKs exhibit a relative autonomy from the central CLOCK.
The circadian rhythm which characterizes the fluctuation of circulating glucocorticoid levels is well-established, with peak levels in the early morning and a nadir in the late evening in humans [108]. It is now evident that the light-activated central CLOCK system is orchestrating the daily rhythmic release of glucocorticoids by regulating the HPA axis activity via efferent connections from the SCN to the PVN CRH/AVP-neurons (Figure 5) [99, 109]. In addition, splanchnic innervation to the adrenal medulla via the aforementioned SCN-ANS axis also contributes to the circadian glucocorticoid secretion and resets the local adrenal clock via modulating adrenal sensitivity to ACTH through effects of epinephrine and other secretory products of the adrenal medulla, such as NPY (Figure 5) [110].

Figure 5.
The light-activated central CLOCK located in the suprachiasmatic nucleus (SCN) is orchestrating the daily rhythmic release of glucocorticoids by influencing the activity of the hypothalamic-pituitary-adrenal (HPA) axis through efferent connections from the SCN to the CRH/AVP-containing neurons of the PVN. Additionally, splanchnic nerve innervation to the adrenal medulla via the SCN-ANS axis also contributes to circadian glucocorticoid secretion and resets the adrenal local clock through modulating the adrenal sensitivity to ACTH by the action of epinephrine. In turn, secreted glucocorticoids reset and phase-delay circadian rhythm of the peripheral CLOCKs by stimulating the expression of several CLOCK-related genes; this is particularly important for temporal adjustment of the body’s activity against stress. The peripheral CLOCKs also regulate the effects of glucocorticoids in local tissues through interactions between Clock/Bmal1 and glucocorticoid receptors, providing a local counter regulatory feedback loop to the effect of central CLOCK on the HPA axis. CRH: corticotropin-releasing hormone; AVP: arginine vasopressin; PVN: paraventricular nucleus; Clock: circadian locomotor output cycles kaput transcription factor; Bmal1: brain-muscle-arnt-like protein 1 transcription factor (the heterodimer partner of Clock). [Adapted from Nader, N, Chrousos, GP, Kino T. Trends Endocrinol Metab 2010;21:277].
Along with these central mechanisms, experimental evidence supports the existence of a peripheral clock machinery which is intrinsic to the adrenal gland and may also underlie the circadian regulation of the glucocorticoid rhythm [111-114]. As such, glucocorticoid biosynthesis is also closely linked with the local adrenal oscillator by clock-controlled expression of steroidogenic acute regulatory protein (StAR; the rate-limiting step of steroidogenesis). In turn, rhythmic StAR expression promotes a daily oscillation in adrenal steroidogenesis, thus contributing to the generation of a robust circadian glucocorticoid rhythm in the systemic circulation [115].
Reciprocally, the HPA axis affects the circadian rhythm of the CLOCK system through glucocorticoids. Glucocorticoids are considered to exert their effects on peripheral CLOCKs in almost all organs/tissues, but not on the central CLOCK in the SCN. In support of this, glucocorticoid receptors are not expressed in the SCN [116]. Glucocorticoids reset the peripheral CLOCKs via influencing the expression of several clock-related genes (e.g. Per1 and Per2) in both peripheral tissues (e.g. in the liver, kidney and heart) and in certain CNS sites (e.g. in the amygdala) in a GRE-dependent manner (Figure 5) [117-119]. Interestingly, acetylation of the glucocorticoid receptors at multiple lysine residues in their hinge region can lead to repression of their transcriptional effects on several glucocorticoid responsive genes either through reducing binding of glucocorticoid receptors to GREs, or by altering the translocation of the receptor into the nucleus, or both. Of note, Clock/Bmail1 acetylates glucocorticoid receptors at these lysine residues, hence regulating the transcription of glucocorticoid responsive genes [69].
Overall, strong evidence indicates that there is bidirectional crosstalk between the CLOCK system and the HPA axis at the level of peripheral target organs/tissues, whereas the master CLOCK in the SCN retains its intrinsic circadian rhythm independently of HPA axis activation by external or internal stimuli. The aforementioned findings suggest that the CLOCK system acts as a reverse-phase negative regulator of glucocorticoid action in target organ/tissues, potentially by antagonizing the biological glucocorticoid effects through synchronizing the peak glucocorticoid concentrations to coincide with the peak glucocorticoid resistance at the target organs/tissues [96]. Importantly, this protective feedback loop which acts as an intrinsic safety valve against over-exposure to glucocorticoids becomes decoupled when glucocorticoid secretion is stimulated by stress. Over a prolonged period of time, such a disruption in the synchronization/coupling between the HPA axis activity and the circadian glucocorticoid receptor acetylation could create a sustained/chronic stress-related hypercortisolism (mild or even functional hypercortisolism) which promotes the development of various pathologic conditions, including metabolic and cardiovascular disorders [60, 120-123].
Stress System- Endocrine Axes Interactions
The stress system is tightly interconnected with all the major endocrine axes, including the reproductive, growth and thyroid axis. This ensures that the activity of the endocrine system is rapidly regulated in a coordinated and precise way in order to serve the adaptive stress response and maximize the chances of survival against the imposed stressor(s).
REPRODUCTIVE AXIS
Although the observation that stress can impact negatively on the reproductive function traces back to antiquity, the exact pathophysiologic and molecular mechanisms which mediate this effect still pose a research challenge [124-126]. The reproductive system, both in females and males, is inhibited at all levels by various components of the HPA axis (Figure 6). As such, CRH suppresses the gonadotropin-releasing hormone (GnRH) neurons directly and indirectly via enhancing β-endorphin secretion by the arcuate POMC neurons. Recent data indicate that CRH-R1 mediates, at least in part, the effects of restraint acute-stress on the reproductive axis, whilst antalarmin (a selective CRH-R1 antagonist) can abolish these effects [127]. In addition, glucocorticoids exert inhibitory effects on GnRH neurons, pituitary gonadotrophs and directly on the gonads, whilst also rendering target organs/tissues resistant to sex steroids [128, 129]. Thus, steroidogenesis is directly inhibited at both the ovaries and testes, with concomitant inhibition of the pulsatile GnRH secretion from the hypothalamus. Notably, certain pro-inflammatory circulating cytokines (e.g. IL-6) can also suppress the reproductive function at multiple levels, providing a link between inflammatory stress and reproductive dysfunction [130].

Figure 6.
Schematic representation of the interactions between the hypothalamic-pituitary-adrenal (HPA) axis and the reproductive and growth axes. Chronic hyperactivation of the stress system may lead to both osteoporosis and metabolic syndrome. CRH: corticotropin-releasing hormone; GnRH: gonadotropin-releasing hormone; ACTH: adrenocorticotropic hormone (corticotrophin); LH: luteinizing hormone; FSH: follicle-stimulating hormone; GHRH: growth hormone releasing hormone; STS: somatostatin; GH: growth hormone; SmC: somatomedin C. Activation is represented by solid green lines and inhibition by dashed red lines.
In women these suppressing effects HPA axis on reproduction are responsible for the hypothalamic amenorrhea of stress which is manifested under various conditions of prolonged/chronic stress, including anxiety, depression, eating disorders, and chronic excessive exercise [131]. Similarly, in men these HPA axis effects result in decreased libido and hypo-fertility [132]. Of note, in addition to stress-induced testosterone decrease, direct effects of stress on the seminiferous epithelium have also been reported [133].
Moreover, the presence of CRH and its receptors in the female and male reproductive system suggests the presence of a local reproductive CRH system [134, 135]. Existing evidence supports the role of this local CRH system in the physiology and pathophysiology of reproduction, highlighting its implication in several reproductive functions as an additional autocrine/paracrine modulator. Ovarian CRH is primarily localized in thecal cells and in luteinized cells of the stroma, mediating ovulation and luteolysis processes [136]. Furthermore, ovarian CRH is also potentially implicated in the premature ovarian failure observed in women exposed to high psychosocial stress [137]. In addition, intrauterine CRH appears to play a critical role in mechanisms responsible for embryo implantation and maintenance of pregnancy by killing activated T-cells and regulating the expression of carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM1), respectively [133, 138].
Both epidemiologic and experimental data indicate that adverse intrauterine stressors (e.g. abnormal trophoblast invasion, deficient remodeling of spiral arteries with high-resistance placental vessels and subsequent placental dysfunction) may lead to preterm labor, fetal growth restriction and pre-eclampsia. Notably, all these conditions are characterized by increased CRH levels both in the maternal circulation and in the fetus; although it is still unclear whether this CRH increase is causally related to or only a consequence of the underlying pathophysiology. Importantly, elevated CRH levels and abnormally increased cortisol in the fetus are recognized as predisposing risk factors of adult disease, including insulin resistance, cardio-metabolic complications and psychiatric disorders [139]. Indeed, adverse intrauterine stressors, as well as maternal prenatal stress, anxiety and depression can impact on the fetal programming and lead to development of chronic disease later in life (e.g. type 2 diabetes, cardiovascular disease, and neurodevelopmental disorders) [140-142]. Interestingly, maternal gestational stress may also lead to low birth weight which, in turn, appears associated with increased plasma cortisol levels in adult life and risk for developing metabolic syndrome [121, 143].
It is noteworthy that, the third trimester of pregnancy by itself constitutes a condition characterized by hypercortisolism of a degree similar to that observed in severe depression, anorexia nervosa, and mild Cushing’s syndrome, whilst it is the only known physiological state in humans which exhibits increased CRH levels in the circulation that are high enough to directly cause HPA axis activation [144-146]. This circulating CRH has a placental origin and, although it is bound with high affinity to CRH-binding protein, its circulating free fraction is sufficient to explain the observed escalating hypercortisolism when the CRH-binding protein plasma levels start to gradually decrease after the 35th week of pregnancy [147, 148].
A model for fetal programming by altered placental function and/or glucocorticoid overexposure has been proposed. According to this model, prenatal maternal stress reduces the activity of the placental 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD 2; an enzyme which metabolizes cortisol to its inactive form, i.e. cortisone), hence allowing the high circulating levels of maternal glucocorticoids to enter the fetal circulation [149]. Additional molecular mechanisms which are implicated in the programming effects of fetal stress and exposure to increased glucocorticoid levels include the epigenetic changes in target chromatin, affecting the tissue-specific expression of glucocorticoid receptors [139]. As such, excess glucocorticoid exposure in early life can alter tissue glucocorticoid signaling in a permanent way, which, although it may confer short-term adaptive benefits, in the long-term increases the risk of later life disease [139].
Finally, it must be noted that the interaction between CRH and the gonadal axis appears to be bidirectional [132]. Indeed, studies have documented both the presence of estrogen response elements in the promoter area of the CRH gene and direct stimulatory estrogen effects on CRH gene expression [150]. This implicates the CRH gene and, hence, the HPA axis, as a target of ovarian steroids and a potential mediator of gender-related differences in the HPA axis activity and the overall stress response [151]. On the other hand, the activated estrogen receptor interacts with and, on occasion, potentiates the c-jun/c-fos heterodimer which mediates several cytokine effects. Furthermore, estrogen appears to stimulate adhesion molecules and their receptors in immune and immune accessory cells, thus offering a possible explanation as to why autoimmune diseases afflict more frequently females than males.
GROWTH AXIS
The growth axis is also inhibited at various levels during stress (Figure 6). Prolonged activation of the HPA axis leads to suppression of growth hormone (GH) secretion and inhibition of somatomedin C (SmC) and other growth factor effects on their target tissues by glucocorticoids [152, 153], presumably via inhibition of the c-jun/c-fos heterodimer. However, acute transient elevations of GH concentrations in plasma may occur at the onset of the stress response, as well as after acute administration of glucocorticoids, potentially mediated through GRE-stimulated GH expression [154]. In addition to the direct effects of glucocorticoids which play a key role in the suppression of growth observed under prolonged stress, increased somatostatin (STS) secretion caused by CRH which results in inhibition of GH secretion, appears to also contribute to the stress-related suppression of the growth axis (Figure 6) [155]. Redirection of oxygen, nutrients and vital substrates to the brain and other stressed organ/tissues where they are needed most in the context of the adaptive stress response is the apparent teleology for the suppressive effects of stress on growth.
Interestingly, psychosocial dwarfism is a term that has been used to describe severe childhood/adolescent growth arrest and/or delayed puberty due to emotional deprivation and/or psychologic harassment [156-158]. Decreased GH secretion that is reversible after separation of the child from the responsible environment is a characteristic finding in this condition, which is further associated with a spectrum of behavioral abnormalities, including depression and disturbed eating behaviors. This form of growth arrest was first studied in infants housed in foundling homes or orphanages, who exhibited decreased growth and high mortality rates. Although deficient nutrition may contribute to this failure to thrive, it has been shown that in these infants’ weight gain is also independent of food intake, whilst a caring and attentive environment improved both their growth rate and psychological profile. Little is known about the HPA axis activity in infants/children with this condition; however, it is suggested that chronic activation of the HPA axis is implicated, thus explaining the other endocrine abnormalities observed in these children.
It must be also noted that, premature infants are at increased risk for delayed growth and development, particularly after prolonged hospitalization in the intensive care nursery. This is known as reactive attachment disorder of infancy and exhibits similarities to psychosocial dwarfism. The key role that the quality of parental care plays on later growth, development and behavior has been also shown in nonhuman primates which are socially organized in extended families, such as the common marmoset (a small primate species) [159]. Finally, infantile malnutrition is characterized by hypercortisolism, decreased responsiveness to CRH, incomplete dexamethasone suppression, growth arrest and thyroid function test changes reminiscent of the euthyroid sick syndrome as will be discussed in the following section [2, 160]. These abnormalities can be restored following nutritional rehabilitation [2, 160].
THYROID AXIS
Stress-related inhibition of thyroid axis activity has also been documented (Figure 7). Chronic HPA axis activation is associated with decreased production of thyroid stimulating hormone (TSH) and inhibited conversion of the relatively inactive thyroxine (T4) to the more biologically active triiodothyronine (T3) in peripheral tissues (a condition described as the "euthyroid sick" syndrome) [161-163]. Although the exact mechanism(s) underlying these effects have not been fully clarified, increased circulating glucocorticoid levels are considered to mediate the stress-induced suppression of the thyroid axis which serves a desired energy conservation during the adaptive stress response. Indeed, existing evidence suggests decreased efficacy of TRH in stimulating TSH release in patients with hypercortisolism and in healthy subjects after glucocorticoid administration, which is dose-dependent [161, 164]. Interestingly, even a single dose of glucocorticoids (1-2 mg of dexamethasone) can cause an acute decrease in pulsatile TSH production in healthy men [165], whilst mildly elevated cortisol plasma levels induced by timed cortisol infusions can also decrease the pulsatile TSH secretion by 50% [166]. Of note, in Cushing’s syndrome patient’s cortisol excess decreases TSH secretion by diminishing its pulsatile release, while surgically cured patients exhibit elevated non-pulsatile TSH release [163]. Moreover, in cases of hypercortisolism-induced TSH-decrease, the circulating free-T4 levels can remain within normal limits, suggesting that the biological activity of TSH may be increased potentially through altered posttranslational processing of the oligosaccharide chains of the TSH molecule [161, 163, 167]. Finally, in the case of inflammatory stress inhibition of TSH secretion and enhanced somatostatin production may be mediated, at least in part, by effects of cytokines on the hypothalamus and/or the pituitary [168, 169].

Figure 7.
Schematic representation of the interactions between the hypothalamic-pituitary-adrenal (HPA) axis and the thyroid and immune function. CRH: corticotropin-releasing hormone; STS: somatostatin; TRH: thyrotropin releasing hormone; TSH: thyroid stimulating hormone; T4: thyroxine; T3: triiodothyronine; TNF-α: tumor necrosis factor-α; IL-1: interleukin-1; IL-6: interleukin-6. Activation is represented by solid green lines and inhibition by dashed red lines.
Stress System- Metabolism
In the context of the adaptive stress response, glucocorticoids exert primarily catabolic effects as part of a generalized effort to utilize every available energy resource against the imposed stressor(s). Thus, glucocorticoids increase hepatic gluconeogenesis and glucose plasma levels, induce lipolysis (although they favor abdominal and dorsocervical fat accumulation) and cause protein degradation at multiple tissues (e.g. in skeletal muscles, bone and skin) to provide amino acids which can be utilized as an additional substrate for oxidative pathways [1, 170, 171]. In parallel to their direct catabolic actions, glucocorticoids also antagonize the anabolic actions of GH, insulin and sex steroids on their target organs/tissues [1, 170, 171]. This shift of the metabolism to a catabolic state by the activated HPA axis normally reverses upon retraction of the imposed stressor(s). However, chronic HPA axis activation can have a range of detrimental effects, including increased visceral adiposity, suppressed osteoblastic activity, decreased lean body mass (decreased muscle and bone mass causing sarcopenia and osteopenia) and insulin resistance (Figure 8) [1, 170, 171].
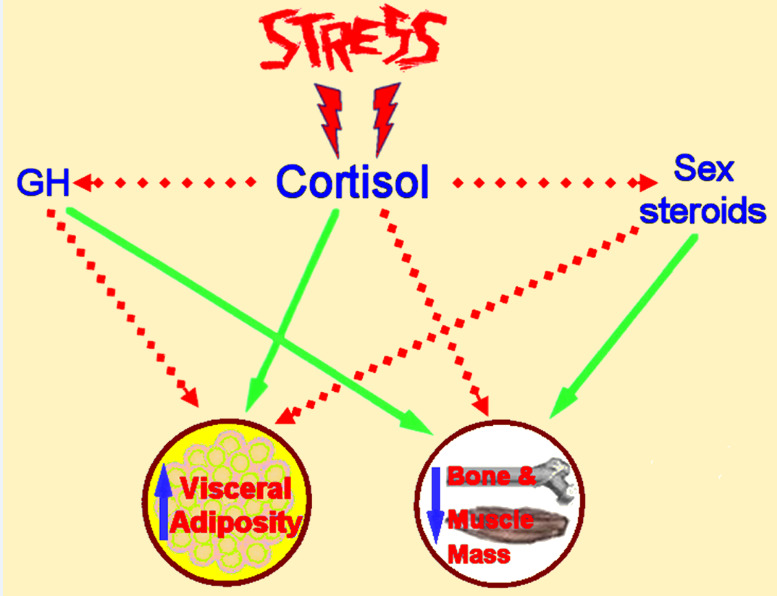
Figure 8.
Schematic representation of the detrimental effects of chronic stress on adipose tissue, bone and muscle metabolism. GH: growth hormone. Activation is represented by solid green lines and inhibition by dashed red lines.
In addition, metabolic homeostasis is also centrally affected by the neuroendocrine crosstalk between the central stress system components, HPA axis and the CNS centers which control appetite/satiety and energy expenditure (Figure 3) [172]. It is a common observation that acute stressful situations are frequently associated with anorexia and marked suppression of food intake. Indeed, CRH stimulates the POMC neurons of the arcuate nucleus which, via α-MSH release, elicit anorexigenic signals and increase thermogenesis [173]. The anorexigenic effects of CRH appear to involve the lateral septum or the bed nucleus of the stria terminalis and are probably mediated through CRH-R2 receptors [174]. Anorexia nervosa represents an interesting example of the implication of the stress system in the regulation of appetite and energy intake. As such, anorexia nervosa can be regarded as a complex condition of chronic stress which is associated with HPA axis dysregulation and suppression of multiple other endocrine axes (e.g. gonadal, growth and thyroid axis), whilst it is characterized by low levels of insulin and leptin and high levels of ghrelin and NPY [175-177]. Interestingly, high cortisol and NPY levels have been shown to exhibit an association with disordered eating psychopathology, independently of BMI [178]. Moreover, existing evidence also indicates that insulin and leptin play important roles in the regulation of central pathways related to food reward [179]. However, it should also be noted that, under normal conditions glucocorticoids enhance the intake of carbohydrates and fat and inhibit energy expenditure by stimulating the secretion of NPY at the hypothalamus. NPY additionally inhibits the LC-norepinephrine system and activates the parasympathetic system, facilitating digestion and storage of nutrients [180-182].
The association between chronic, experimentally induced psychosocial stress, hypercortisolism and the development of a metabolic syndrome-like state with increased incidence of atherosclerosis, has been documented in cynomolgus monkeys. In such animal studies, HPA axis activation induced by chronic stress and the consequent hypercortisolism has been shown to result in visceral obesity, insulin resistance and suppression of GH secretion, hence promoting the development of the metabolic syndrome phenotype (physical and biochemical) [170]. Similar findings have been documented in humans where epidemiological data suggest strong associations between chronic stress exposure and metabolic disease [170, 183-185]. Indeed, chronic HPA hyperactivation in individuals with a genetic predisposition exposed to a permissive environment may lead to visceral fat accumulation and decreased lean body mass (muscle and bone mass) as a result of chronic hypercortisolism and stress-induced low GH secretion and hypogonadism [1, 120, 184, 185]. Moreover, hypercortisolism can directly cause insulin resistance in peripheral target organs/tissues which appears to be proportional to both the glucocorticoid levels and to glucocorticoid sensitivity of the target organs/tissues, as suggested by studies on polymorphisms of the glucocorticoid receptor gene [186]. This can cause reactive compensatory insulin hypersecretion and further increased visceral obesity and sarcopenia, resulting in type 2 diabetes, dyslipidemia and hypertension [1, 120].
More recently, chronic stress has been also associated with a low-grade inflammatory state which follows fat accumulation, especially visceral [187-189]. Thus, obese patients typically exhibit increased circulating levels of pro-inflammatory adipokines and cytokines (e.g. leptin, resistin, TNF-a and IL-6) and decreased levels of anti-inflammatory adipokines (e.g. adiponectin and omentin), creating an adverse adipokine profile which strongly correlates to the metabolic syndrome manifestations [187-189]. Indeed, this obesity related chronic inflammatory stress can cause a range of detrimental effects on peripheral tissues/organs (e.g. on the liver, skeletal muscles and cardiovascular system), promoting enhanced secretion of acute-phase reactants (e.g. fibrinogen and C-reactive protein), insulin resistance, hypertension, atherosclerosis, hypercoagulability, thrombosis and cardiac dysfunction [170, 187-189]. Of note, glucocorticoids have also been shown to induce both insulin and leptin secretion, thus further contributing to the leptin-resistant state which characterizes obesity.
Interestingly, because intracellular glucocorticoid levels are regulated by 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1; an enzyme which converts inactive cortisone to cortisol), research has focused on tissue specific changes in 11β-HSD1 expression and activity in obesity and insulin resistance. As such, it has been shown that the global 11β-HSD1 activity, as measured by urinary corticosteroid metabolite analysis, is impaired in obesity [190, 191], whilst selective 11β-HSD1 inhibitors are in development as novel therapeutic approaches for obesity and metabolic syndrome [192].
It is also noteworthy that, obstructive sleep apnea (OSA) appears also associated with an adverse metabolic profile consisting of increased visceral adiposity and insulin resistance, as well as elevated levels of circulating stress hormones and pro-inflammatory adipokines/cytokines [193-197]. Indeed, obesity, particularly central/visceral, and insulin resistance may contribute to OSA development, whilst, in turn, OSA may promote fat accumulation and reduce insulin sensitivity, potentially through progressive elevation of stress hormones and cytokines (e.g. increased cortisol, noradrenaline, TNFα and IL-6 plasma levels) [196]. Thus, a vicious cycle appears to fuel the association between OSA, chronic stress and metabolic dysregulation.
Increased LC/NE sympathoadrenal system activity, including the central LC/NE neurons, is also an important pathophysiologic component of chronic stress which appears to contribute to the development of impaired glucose tolerance and to the particularly increased risk of acute cardiovascular events (e.g. myocardial infraction and stroke) [198-200]. Finally, chronic stress disorders exhibit a strong positive correlation to a number of behavioral changes with an adverse effect on physical activity (e.g. sedentary lifestyle and increased hours of sleep) and dietary habits (e.g. increased portion size, binge eating and alcohol consumption); hence leading to further weight gain and potentially to dysregulation of glucose and lipid metabolism (Figure 9) [187, 201].

Figure 9.
Schematic representation of the proposed links between stress and dysregulation of metabolic homeostasis. Chronic stress induces hyperactivation of both the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic nervous system (SNS) which together with distinct changes in certain health behaviors can progressively lead to the development of obesity (particularly central/visceral) and metabolic syndrome manifestations.
Moreover, the circadian CLOCK system is also implicated in the pathophysiologic mechanisms linking stress and metabolic syndrome [60, 120]. Notably, most of the metabolic phenotypes associated with dysregulation of the CLOCK system and the HPA axis overlap [96]. As aforementioned, the Clock-mediated repression of the glucocorticoid receptor transcriptional activity oscillates during the day in inverse phase to the normal diurnal rhythm of the HPA axis, whilst stress disrupts this synchronization/coupling. As such, even mild elevations of circulating cortisol levels during the evening, as frequently observed in chronic stress conditions, can cause a type of functional hypercortisolism with disproportionately more potent glucocorticoid-induced effects due to the concurrently increased glucocorticoid sensitivity of the target organs/tissues. This stress-related functional hypercortisolism further promotes the development of metabolic syndrome manifestations [69, 96, 122, 202].
It must be highlighted that, the links between stress and metabolic dysregulation are also particularly significant during fetal life, childhood and adolescence which constitute periods of heightened vulnerability to intense acute and/or chronic stress. As aforementioned, both early nutritional stress (even during fetal or early infant life) and low birth weight are associated with higher risk for obesity and obesity-related cardio-metabolic disease later in life, highlighting the impact on fetal programming of adiposity and its consequences [203, 204]. Moreover, most of the children who experienced chronic stress, anxiety, depression or post-traumatic stress disorder (PTSD) exhibit higher cortisol and catecholamine plasma levels than in the resting state, especially during evening hours [205]. These children are also at higher risk to develop obesity, hypertension and other related comorbidities later in adulthood [206, 207]. As observed in adults, both biological and behavioral pathways mediated the links between chronic stress and obesity in children (Figure 9) [204].
Finally, prolonged stress can also have a significant negative impact on bone metabolism (Figure 8). Indeed, chronic stress can shift the balance of bone remodeling in favor of bone resorption due to both direct effects of increased glucocorticoid and IL-6 plasma levels on bones and indirect effects resulting from the suppression of the growth, gonadal and thyroid axes, thus leading over time to osteopenia and potentially manifestations of osteoporosis [1, 187].
Stress System- Immune System Interactions
EFFECTS OF THE STRESS SYSTEM ON THE IMMUNE/INFLAMMATORY CASCADE
HPA axis activation exerts primarily suppressing effects on the inflammatory/immune response, since both the innate and adaptive immunity are modulated by glucocorticoids with cortisol suppressing the immune system at multiple levels (Figure 7) [1, 208, 209]. At the cellular level, the main anti-inflammatory effects of glucocorticoids include changes in leukocyte trafficking and function, decreased production of cytokines and other mediators of inflammation, and inhibition of pro-inflammatory signaling pathways in target organs/tissues [63, 208, 209]. For example, glucocorticoid-mediated suppression of TNF-α and IL-1β production appears to be the basis for their efficacy in relieving symptoms of rheumatoid arthritis, inflammatory bowel disease, and psoriasis. Indeed, cytokine signaling is affected by glucocorticoids through multiple mechanisms, including direct transcriptional repression of cytokine gene expression by activated glucocorticoid receptors [209-211]. Of note, transcriptional interference between activated glucocorticoid receptors and other transcription factors, such as the nuclear factor-κB (NF-κB) and activator protein-1 (AP-1; a key transcription factor mediating inflammatory responses and pro-inflammatory cytokine production), at various cytokine promoters is a typical example of repression through protein-protein interactions [209]. However, not all cytokines are suppressed, since anti-inflammatory cytokines (e.g. IL-10) are up-regulated by glucocorticoids in accord to the immunosuppressive activities of these hormones [211].
A large infrastructure of anatomical, chemical and molecular connections further facilitates the close communication between the neuroendocrine and immune systems. As such, the efferent sympathetic/adrenomedullary system is also considered too closely participate in interactions between the HPA axis and the immune/inflammatory cascade since: (i) it is reciprocally connected with the CRH system; (ii) it receives and transmits humoral and nervous immune signals from the periphery; (iii) it densely innervates both primary and secondary lymphoid organs; and (iv) it reaches all sites of inflammation via the postganglionic sympathetic neurons [212, 213]. The innate immune system constitutes one of the SNS targets with adrenergic signaling directly affecting pro-inflammatory pathways [214, 215]. Furthermore, similar to what is noted for the HPA axis, a neuroendocrine immune feedback loop appears to exist in order to allow the peripheral immune activation to signal to the CNS and activate the central stress system, thus allowing the CNS to sense and regulate inflammation in the periphery [216-218]. Hence, when activated during stress, the ANS exerts its own direct effects on immune organs/cells which can be immunosuppressive (e.g. inhibition of natural killer cell activity) or both immunopotentiating and immunosuppressive by inducing secretion of IL-6 in the systemic circulation [219, 220]. Indeed, the SNS can exert both pro- and anti-inflammatory effects with various factors determining which of these effects will prevail, including the underlying state of the respective target tissue. For example, it has been shown that an already activated inflammatory pathway can be downregulated by adrenergic signaling, whereas in non-activated immune cells adrenergic signals can activate the pro-inflammatory cascade [220]. In light of these findings, the stress system effects on the immune system can be more accurately characterized as immunomodulating, rather than immunosuppressing.
In addition to affecting antigen presentation, cytokine secretion and leukocyte proliferation and trafficking, the principal stress hormones, i.e. glucocorticoids and catecholamines, further modulate the balance between T helper-1 (Th1) versus Th2 responses (Figure 10). It is now established that, both glucocorticoids and catecholamines directly inhibit the production of type 1 cytokines (e.g. IL-12, IL-2, TNF-α and INF-γ) which enhance cellular immunity and Th1 formation, whilst conversely favor the production of type 2 cytokines (e.g. IL-10, IL-4, IL-13) which induce humoral immunity and Th2 activity [221]. Interestingly, glucocorticoids may inhibit Th1 cell activity also indirectly through manipulating dendritic cell subsets by regulating the expression of Toll-like receptor 2 (TLR2) [211, 222]. In accord with these effects, during immune challenges stress causes an adaptive Th1 to Th2 shift in order to protect the organs/tissues against the potentially destructive actions of pro-inflammatory type 1 cytokines and other products of activated macrophages. Of note, this potentially protective role of the stress-induced Th2 shift against overshooting of cellular immunity often complicates pathologic conditions in which either cellular immunity is beneficial (e.g. carcinogenesis and infections) or humoral immunity is deleterious (e.g. allergy and autoimmune diseases) [223]. Indeed, HPA axis hyperactivation has been associated with increased susceptibility to both infectious agents and tumors. Thus, relapse of mycobacterial infections, progression of HIV infection and infections following major traumatic injuries or burns have been associated to excessive HPA axis responses and a sustained/prolonged Th2 shift. Similarly, several studies have documented a higher incidence of tumor growth and metastases in relation to chronic stress, highlighting the role of cellular immunity in surveillance and eradication of tumor cells [224].
More recent evidence indicates that, stress can influence the immune response in an even more complicated way. Indeed, although stress hormones systemically inhibit Th1/pro-inflammatory responses and induce a Th2 shift, in certain local responses these hormones can induce pro-inflammatory cytokine production and activation of the peripheral CRH-mast cell-histamine axis [223]. This constitutes an additional mechanism via which the stress system may be implicated in the pathogenesis of chronic inflammation and immune-related disease [223]. Adding to the complexity of the interactions between stress and the immune system, there are also data indicating that glucocorticoids may impact on Th17 differentiation and function through molecular mechanisms which have not been fully clarified [225, 226]. Th17 cells constitute a newer effector T-cell subset which secrete IL-17 and appear to play an important role in autoimmune processes, thus providing another potential link between stress and autoimmune disease [210].
Finally, the CLOCK system induces circadian fluctuations of several cytokines (e.g. IFN-γ, ΙL-1β, IL-6 and TNFα) and of natural killer cell and T- and B-lymphocyte populations [227, 228]. Hence, it appears that the central CLOCK system, by regulating glucocorticoid secretion, can also influence the peripheral CLOCK system of immune cells. Moreover, the CLOCK system can also modulate immune functions by regulating the actions of endogenous glucocorticoids on various components of the immune system via interactions between the Clock/Bmal1 transcription factors and glucocorticoid receptors.
EFFECTS OF THE IMMUNE SYSTEM ON THE STRESS SYSTEM
The immune system exerts its surveillance/defense function constantly and mostly unconsciously for the individual. It has been well-established that immune/inflammatory stimuli/insults (e.g. infectious diseases, accidental or operative trauma and active autoimmune processes) are associated with concurrent HPA axis activation. More recently, it also became evident that immune cytokines and other humoral mediators of inflammation are potent activators of the central stress-responsive neurotransmitter systems, constituting the afferent limb of the feedback loop via which the immune/inflammatory system and the CNS communicate (Figure 10). Indeed, through this pathway, the peripheral immune apparatus signals the brain to participate in maintaining immunological and behavioral homeostasis [108, 229].

Figure 10.
Schematic representation of interactions between the stress and immune system. LC/NE: locus coeruleus/norepinephrine-sympathetic system; SPGN: sympathetic postganglionic neurons; CRH: corticotropin-releasing hormone; AVP: arginine vasopressin; ACTH: adrenocorticotropic hormone (corticotrophin); PAF: platelet activating factor; NE/E: norepinephrine/epinephrine; Th1: T-helper lymphocyte 1; Th2: T-helper lymphocyte 2. Activation is represented by solid green lines and inhibition by dashed red lines.
The three main pro-inflammatory cytokines, i.e. TNF-α, IL-1 and IL-6, are produced in this order and in a cascade-like fashion at inflammatory sites, whilst by entering the systemic circulation they can cause HPA axis stimulation in vivo, alone or in synergy with each other [230-232]. These effects can be significantly blocked with CRH-neutralizing antibodies, prostanoid synthesis inhibitors and glucocorticoids. In addition, all of these three cytokines can directly stimulate hypothalamic CRH secretion in vitro, an action which may also be suppressed by glucocorticoids and prostanoid synthesis inhibitors [233-235]. Similarly, IL-2 can stimulate ACTH secretion indirectly and potentially directly [236-238]. Of note, IL-1β also increases the hypothalamic and anterior pituitary expression of leukemia-inhibitory factor (LIF) which is a member of the IL-6 family and a potent ACTH secretagogue [239, 240]. The specific effects mediated by CRH and LIF in HPA axis regulation during inflammation remain under investigation. Existing evidence suggests that central CRH appears to be more critical in mediating ACTH release in response to shock or alcohol rather than to LPS [241]. This is further supported by data showing that CRH knockout animals have nearly normal HPA axis reaction to inflammatory challenges [242]. Furthermore, LIF deficient animals exhibit markedly lower POMC and ACTH responses to inflammation induced by high LPS doses [243], whilst exogenous LIF injection restores the pituitary POMC expression in LIF knockout animals [244]. In vitro studies have also shown that LIF greatly potentiates the CRH effects on POMC transcription. Therefore, although CRH is required for rapid increase of ACTH synthesis and secretion in response to any nonspecific stressful challenge, it appears that LIF is important for maintaining a sustained HPA axis activation during inflammatory stress. However, mice deficient in both CRH and LIF still demonstrate robust ACTH and corticosterone responses to inflammation, probably due to abundant TNFα, IL-1β and IL-6 activation observed in the hypothalamus and pituitary of these animals [245].
A body of evidence also suggests that IL-6, which constitutes the main endocrine/circulating cytokine, plays the primary role in immune stimulation of the human HPA axis, particularly in the long-term. Indeed, IL-6 has been shown to be an extremely potent activator of the HPA axis in humans [168, 220, 246]. Notably, the ACTH and cortisol elevations attained by IL-6 are well above those observed with maximal stimulatory doses of CRH, suggesting that parvocellular AVP and other ACTH secretagogues are additionally stimulated by this cytokine. Moreover, high doses of IL-6 have been shown to further stimulate peripheral elevations of AVP, presumably as a result of a stimulatory effect on magnocellular AVP-secreting neurons [247]. This suggests that IL-6 may be involved in the pathogenesis of the syndrome of inappropriate antidiuretic hormone secretion (SIADH) which is observed during the course of infectious/inflammatory disease or during trauma.
Some of the activating effects of inflammation on the HPA axis may be exerted indirectly through stimulation of the central catecholaminergic pathways by pro-inflammatory cytokines and other humoral mediators of inflammation. Furthermore, activation of peripheral nociceptive, somatosensory and visceral afferent fibers could lead to stimulation of both the catecholaminergic and CRH neuronal systems via ascending spinal pathways. Of note, in chronic inflammatory states which may be characterized by chronic central elevations of SP, impaired HPA axis responsiveness to stimuli or stress may be observed, potentially due to the suppressive effect of SP on CRH neurons [49, 84]. This impairment has been documented in AIDS, African trypanosomiasis and extensive burns in humans and also in animal models of chronic inflammation [84, 248].
In addition to the three main pro-inflammatory cytokines, other mediators of inflammation may also participate in the HPA axis activation. Indeed, several eicosanoids, platelet activating factor (PAF) and serotonin show potent CRH-releasing properties [249, 250]. However, it is still unclear exactly which of these effects are endocrine and which are paracrine. Although delayed, direct effects on pituitary ACTH secretion have been documented by most of the above cytokines and mediators of inflammation [168, 251, 252], whilst direct effects of these substances on adrenal glucocorticoid secretion appear to be also present [253]. Notably, both prostaglandins and nitric oxide (NO), which are key mediators of inflammation and immunity, have been found to impact on ACTH and cortisol secretion. Experimental evidence suggests opposite actions of prostaglandins generated by cyclooxygenase (COX) and NO synthesized by the inducible NO synthase (iNOS; a pro-inflammatory enzyme which dysregulates NO production) in the LPS-induced HPA axis response, with prostaglandins stimulating the ACTH response to endotoxin, while NO inhibits it [254, 255]. Further data on the role of prostaglandins in the HPA axis response to LPS indicate that induced prostaglandin synthesis, mediated via Cox-2, contributes to the delayed HPA axis activation, whereas constitutive prostaglandin synthesis, mediated preferentially via Cox-1, is involved in the early HPA response [256].
An intriguing aspect of the immune response is that CRH is also secreted peripherally at inflammatory sites (peripheral or immune CRH) by postganglionic sympathetic neurons and by cells of the immune system (e.g. macrophages and tissue fibroblasts) [257]. The secretion of immune CRH has been studied both in experimental animal models of inflammation [257], and in patients with rheumatoid arthritis [258], Hashimoto thyroiditis and other inflammatory illnesses [259]. Glucocorticoids and somatostatin have been shown to suppress this immune CRH secretion [257]. Of note, mast cells are considered as the primary target of immune CRH where, along with SP, it acts via CRH-R1 receptors causing degranulation. As a result, histamine is released causing vasodilation, increased vascular permeability and other manifestations of local inflammation. Hence, locally secreted CRH triggers a peripheral CRH-mast cell-histamine axis, which has potent pro-inflammatory properties, whereas central CRH alleviates the immune response [108, 260].
Another interesting topic relating to the interactions between the immune and stress system is the study of critically ill patients with systemic inflammation. To date, the results of studies investigating the adrenal response to critical illness have been conflicting. As such, the initial phase of critical illness is characterized by excessive release of ACTH and cortisol as a result of increased CRH/AVP secretion and cytokine production. Although the magnitude of the increase of cortisol plasma levels may not correlate linearly with the illness severity, some studies have documented that patients with the highest circulating cortisol levels had also the highest mortality [261]. However, the ACTH and cortisol responses may diverge during prolonged critical illness, with high cortisol plasma levels persisting despite ACTH suppression, thus suggesting that cortisol secretion is further stimulated by alternative pathways, other than hypothalamic CRH, potentially involving factors such as AVP, atrial natriuretic peptide (ANP), endothelin and a variety of cytokines (especially IL-6) [261, 262]. Notably, in severe critical illness a relative corticosteroid insufficiency may also develop, characterized as critical illness-related corticosteroid insufficiency (CIRCI) [263]. The exact mechanisms of adrenal suppression in critical illness remain largely unclear [264-266]. Cytokines and adipokines derived from the adipose tissue may influence the normal synthesis and release of ACTH and cortisol, as well as the activity of glucocorticoid receptors. Indeed, TNF-α and peptides derived from immune cells, such as the corticostatins, may compete with ACTH on its receptor, negatively influencing adrenal cortisol secretion and inducing tissue resistance to glucocorticoids [264-266].
Stress System- Gastrointestinal Function
The stress system activity is implicated in the regulation of gastrointestinal function and exhibits a strong association with gastrointestinal illness [267-269]. Interestingly, data from a study in patients with chronic painful gastrointestinal disorders revealed a high incidence of physically and sexually abused women in this patient population [270]. It has been also shown that sexually abused girls exhibit chronic HPA axis activation, similarly to patients with melancholic depression [271]. Thus, it has been proposed that CRH hypersecretion may constitute a hidden link between the symptomatology of chronic painful gastrointestinal disorders and a history of physical and/or psychological abuse [270, 272].
Indeed, increasing evidence suggests that CRH is involved in the mechanisms by which stress affects the gastrointestinal function. Of note, several studies have identified immunoreactive CRH and urocortin, as well as CRH-R1 and CRH-R2 in the human colonic mucosa [273, 274]. During acute stress, PVN CRH, independently of the associated HPA axis stimulation, induces both inhibition of gastric emptying and stimulation of colonic motor function by alterations in the ANS activity (Figure 11) [275]. It is considered that inhibition of the vagus nerve activity at the dorsal vagal complex results in selective inhibition of gastric motility, while stimulation of the sacral parasympathetic system activity results in selective stimulation of colonic motility, with the latter possibly mediated through CRH projections of the Barrington nucleus which is part of the LC complex [276, 277]. At the receptor level, it appears that stress-induced delayed gastric emptying involves the central medullary CRH-R2 receptors and also the peripheral CRH-R2 receptors in the gastrointestinal tract, whereas the CRH-R1 subtype seems to mediate the colonic motor responses (e.g. stimulation of distal colonic transit) [273]. Hence, CRH may play a role in mediating the gastric stasis which is associated with the stress of surgery and the increased IL-1 levels during surgery and the immediate postoperative period, whilst it is also implicated in the stress-induced colonic hyper-motility of the irritable bowel syndrome (IBS) [274, 275, 278]. Moreover, the colonic contraction in IBS patients can activate the LC-noradrenergic system, thus, creating a vicious cycle which may explain the chronicity of this condition [275]. Importantly, CRH can also modulate the visceral pain hypersensitivity in IBS [274, 279]. Existing evidence suggests contrasting roles of the two CRH-R subtypes in visceral nociception, with CRH-R1 being involved in the pro-nociceptive effects of visceral pain, while CRH-R2 mediates an anti-nociceptive response [274].

Figure 11.
Schematic representation of stress system effects on gastrointestinal function. CRH: corticotropin-releasing hormone; ACTH: adrenocorticotropic hormone (corticotrophin); PVN: paraventricular nucleus; LC: locus coeruleus. Activation is represented by solid green lines and inhibition by dashed red lines.
Additional studies exploring the underlying mechanisms mediating the stress-induced stimulation of colonic motility further revealed that restraint stress in conscious rats can stimulate vagal efferent nerves innervating the proximal colon via central CRH-R1 receptors, resulting in 5-hydroxytryptamine (5-HT) release from the proximal colon. This released 5-HT activates 5-HT3 receptors located at the vagal afferent fibers, whilst, in turn, this 5-HT3 receptor activation stimulates colonic motility via the vagovagal reflex [280]. Thus, it appears that the primary target of restraint stress may be the enterochromaffin cells of the proximal colon [280].
Although acute stress stimulates colonic motor function via a central CRH, it seems that colonic motility is decreased following chronic stress through an adaptation mechanism which does not implicate reduced sensitivity to central CRH [281]. Apart from the colonic motility, the delayed gastric emptying induced by acute stress can be also completely restored following chronic homotypic stress in rats [282]. The latter appears mediated by a mechanism involving oxytocin expression upregulation in the hypothalamus which, in turn, attenuates CRH expression [282]. More recently, additional mechanisms have been proposed as potential mediators of stress-induced changes in the gastrointestinal motility, implicating changes in circulating ghrelin and ghrelin-O-acyltransferase levels, as well as the neuropeptide S (NPS; a neuropeptide mainly produced by neurons in the amygdala and between the Barrington nucleus and the LC) [283, 284].
In addition to altering gastrointestinal motility patterns, stressors can further exert profound effects in several other aspects of the gastrointestinal function. As such, it has been shown that stress-induced activation of central and peripheral CRH receptors can cause dysfunction of the intestinal barrier, increase the gastrointestinal permeability and promote inflammatory bowel disease (IBD) relapse [267-269]. Finally, chronic activation of the HPA axis and/or the LC/NE-sympathetic system may induce depletion or tachyphylaxis of the opioid-peptide system responsible for stress-induced analgesia, which may account for the observed lower pain threshold for visceral sensation in patients with functional gastrointestinal disorders [267-269].
STRESS: ENDOCRINE PATHOPHYSIOLOGY
In the context of the adaptive stress response, the mobilization of different stress system components must be of intensity that correlates to the presented threat by the imposed stressor(s) and of duration that allows a timely return to the desired steady state (homeostasis) [1-4]. Thus, a successful stress system response should be both of magnitude to overpower the homeostatic threat posed by the stressor(s) without overshooting and of time-limited duration which would render its accompanying catabolic, anti-reproductive, antigrowth and immunosuppressive adaptive effects transient and temporarily beneficial rather than sustained and detrimental [1-4]. The dose-response relationship between the stress system response activity and the potency of any given stressor can be depicted in a simplified way by a sigmoidal curve which starts from the basal stress system activity levels at rest and plateaus at a maximum activity level when all available adaptive forces have been mobilized (Figure 12A). Of note, this sigmoidal response curve varies in each individual; however, there is a relatively limited, narrow range between basal and maximum activity which characterizes the normal reactive individuals. Therefore, dose-response curves of stress activity extending outside the two extremes of this normal range denote pathologic stress responses, with higher and lower-shifted curves denoting excessive and defective reactions, respectively (Figure 12A). Similarly, the dose-response relationship between the sense of well-being or performance ability of each individual and the stress system activity can be represented by an inverted U-shaped curve which covers the normal range of the stress system activity. Shifts to the left or the right of this range result in hypoarousal or hyperarousal (anxiety) states, respectively, with a suboptimal sense of well-being and/or diminished performance (Figure 12B). The following sections present briefly the principles which characterize the pathophysiology of chronic hyper- and hypo-activation of the stress system in relation to the aforementioned stress system organization and physiology.

Figure 12.
A. The dose-response curve between the potency of an imposed stressor and the activity of the stress system components responding to this specific stressor. Curve 1 (green): the normal dose-response curve; Curve 2 (red): the dose-response curve which defines the upper physiologic level of stress system activity; Curve 3 (purple): the dose-response curve which defines the lower physiologic level of stress system activity. Any curve higher than Curve 2 represents stress system hyperactivity, while any curve lower than Curve 3 represents stress system hypoactivity. B. The inverted U-shaped dose-response curve between the sense of well-being or performance ability and the stress system activity. Curve 1 (green): optimal stress system activity curve; Curve 2 (red): excessive stress system activity curve; Curve 3 (purple): defective stress system activity curve. Curves 2 and 3 curtail the top of the optimal curve and represent shifts to the right (hyperarousal/anxiety) and to the left (hypoarousal), respectively, whilst both are associated with suboptimal sense of well-being or diminished performance. [Adapted from Chrousos G.P. and Gold P.W., JAMA, 1992, 267,1244].
Chronic Hyperactivation of the Stress System - Pathophysiology
Chronic stress system hyperactivation leads to the syndromal state which Selye first described in 1936 [1, 2, 285]. As previously discussed, CRH coordinates the neuroendocrine, autonomic, immune and behavioral adaptation during stress, hence increased and prolonged CRH production is regarded to play a pivotal role in the pathogenesis of the chronic stress syndrome and its clinical manifestations, including endocrine, cardio-metabolic, immune and psychiatric complications [1, 2].
In this context, the syndrome of adult melancholic depression represents a typical example of dysregulation of the generalized stress response, leading to chronic, dysphoric hyperarousal, with hyperactivation of both the HPA axis and the SNS and relative immunosuppression [286, 287]. Indeed, these patients exhibit increased cortisol excretion, decreased plasma ACTH response to exogenous CRH and elevated CRH levels in the cerebrospinal fluid (CSF) [288, 289]. These findings suggest that melancholic depression correlates with distinct hypersecretion of CRH which may participate in the initiation and/or perpetuation of a vicious pathophysiologic cycle. As such, patients with depression history were found on autopsy to have a significantly increased number of CRH neurons in the PVN [290], whilst imaging studies have also documented marked hippocampal atrophy and a small and hypo-functioning section of the medial frontal lobe (Figure 13) [291, 292]. Whether and to what extent this pathology is genetically determined, or environmentally induced, or both is still the subject of intense research.
Figure 13.
Schematic representation of the central neurocircuitry and its altered activity implicated in acute stress and melancholic depression (chronic stress system hyperactivation). Hyperfunctioning amygdala, hypofunctioning hippocampus and/or hypofunctioning mesocorticolimbic system (MCLS) could be associated with chronic hyperactivation of the PVN CRH-AVP system and predispose to melancholic depression. PVN: paraventricular nucleus; CRH: corticotropin-releasing hormone; AVP: arginine vasopressin. Activation is represented by solid green lines and inhibition by dashed red lines.
Similarly to the prototypic example of melancholic depression, a broad spectrum of other clinical conditions have been associated with various degrees of increased and prolonged stress system activation (Table 2), including panic anxiety disorders [293]; obsessive-compulsive disorder [294]; childhood physical/sexual abuse [295]; chronic alcohol abuse [296]; alcohol/narcotic withdrawal [297, 298]; anorexia nervosa [175, 299, 300]; central (visceral) obesity [170, 301]; diabetes (especially when complicated by diabetic neuropathy) [302, 303].
Table 2.
Clinical Conditions Associated with Altered Hypothalamic-Pituitary-Adrenal (HPA) Axis Activity and Dysregulation of the Adaptive Stress Response
| Increased HPA axis activity | Decreased HPA axis activity |
|---|---|
| Chronic stress | Adrenal insufficiency |
| Melancholic depression | Atypical/Seasonal depression |
| Anorexia nervosa | Chronic fatigue syndrome |
| Obsessive-compulsive disorder | Fibromyalgia |
| Panic disorder | Hypothyroidism |
| Excessive exercise (obligate athleticism) | Nicotine withdrawal |
| Chronic active alcoholism | Post glucocorticoid therapy |
| Alchohol and narcotic withdrawal | Post Cushing's syndrome cure |
| Diabetes mellitus | Postpartum period |
| Central obesity (Pseudo-Cushing syndrome) | Post chronic stress |
| Childhood sexual abuse | Rheumatoid arthritis |
| Hyperthyroidism | Premenstrual tension syndrome |
| Cushing's syndrome | Climacteric depression |
| Pregnancy |
Adapted from Chrousos G.P. and Gold P.W., JAMA, 1992; 267,1244.
Chronic Hypoactivation of the Stress System - Pathophysiology
Chronic stress system hypoactivation with reduced CRH secretion may result in pathologic hypoarousal which characterizes another group of pathophysiologic states (Table 2). Patients with atypical and seasonal depression or chronic fatigue syndrome appear to belong in this category [304, 305]. As such, periods of depression (winter period) of the former and periods of marked fatigue in the latter are characterized by prolonged HPA axis hypoactivity. Similarly, patients with fibromyalgia exhibit decreased urinary free cortisol excretion and frequently complain of fatigue [306]. Interestingly, amongst the clinical manifestations of hypothyroidism is atypical depression, with hypothyroid patients exhibiting evidence of CRH hyposecretion [307].
Moreover, smoking withdrawal has been documented as a state associated with decreased cortisol and catecholamine secretion [308, 309]. Decreased CRH secretion in the early period of nicotine abstinence in habitual smokers could explain the hyperphagia, low metabolic rate and weight gain frequently observed during attempts to stop smoking. It should be also noted that, the clinical presentation of Cushing’s syndrome with atypical depression, hyperphagia, weight gain, fatigue and anergia is consistent with suppression of CRH neurons by the associated hypercortisolism. Periods following cure of hypercortisolism or cessation of chronic stress, as well as the postpartum period are also associated with suppressed PVN CRH secretion and decreased HPA axis activity (Table 2) [1, 2, 142, 310, 311].
Finally, a defective HPA axis response to inflammatory stimuli can reproduce the glucocorticoid-deficient state and may lead to relative resistance to infections and neoplastic disease, but increased susceptibility to autoimmune/inflammatory diseases [108, 224, 312, 313]. Indeed, such findings were documented in studies utilizing an interesting pair of near-histocompatible, highly inbred rat strains, i.e. the Fischer and Lewis rats which were genetically selected out of Sprague-Dawley rats for their resistance or susceptibility, respectively, to inflammatory disease [314, 315]. In accord with the findings of animal studies in these models, an increasing body of clinical evidence indicates that rheumatoid arthritis patients may exhibit a mild form of central hypocortisolism, with reduced 24-hour cortisol excretion, less pronounced diurnal rhythm of cortisol secretion and blunted adrenal responses to surgical stress [316, 317]. Taken together these findings suggest that HPA axis dysfunction can play a role in the development and/or perpetuation of autoimmune disease, rather than being an epiphenomenon. This rationale may also explain the high incidence of autoimmune disease in the period after cure of hypercortisolism and during the postpartum period, as well as in untreated or under-replaced adrenal insufficiency [224].
Potential Role of CRH Antagonists in Clinical Practice
Based on the implication of stress system dysregulation in clinical manifestations of a wide spectrum of diseases, research has focused on identifying novel therapeutic approaches targeting this underlying pathophysiologic link. As such, small molecular weight antagonists of CRH-R1 and CRH-R2 have been developed which can be absorbed orally and cross the blood brain barrier, thus exhibiting a therapeutic potential in the treatment of disorders linked to disturbances of CRH-regulated pathways [30, 318].
Antalarmin is a non-peptidic prototype CRH antagonist which binds with high affinity to CRH-R1. This small lipophilic pyrrolopyrimidine agent decreases the activity of both the HPA axis and the LC/NE-sympathetic system, blocking a variety of manifestations associated with anxiety and the development/expression of conditioned fear [319]. In addition, antalarmin can suppress neurogenic inflammation, stress-induced peptic ulcers and colonic hyperfunction, whilst it also blocks CRH-induced skin mast cell degranulation [30, 320-324]. Importantly, chronic administration of antalarmin is not associated with glucocorticoid or catecholamine deficiency and permits adequate HPA axis and LC/NE responses to severe stress [325]. Overall, data from several studies that tested the efficacy of such CRH-R1 antagonists indicate a potential therapeutic role in various disorders, including melancholic depression, chronic anxiety, narcotic withdrawal, IBS, allergic reactions and autoimmune/inflammatory disease. Indeed, clinical studies with the CRH-R1 antagonists NBI-30775/R121919 and NBI-34041 have reported promising outcomes in depression and anxiety [326].
In addition, it is considered that CRH-R2 antagonists could potentially play a role in the treatment of atypical depression, chronic fatigue syndrome, fibromyalgia and stress-induced anorexia [30, 327, 328]. However, the available data on effects of selective CRH-R2 antagonists are relatively limited [30]. Identifying specific CRH-R2 neuronal pathways which are implicated in various disease states in humans and better understanding of the role of CRH-related peptides, such as urocortin (Ucn) I, UcnII, UcnIII and urotensin I, is expected to shed more light on the overall therapeutic potential of CRH-R2 antagonists. Notably, urocortin appears to participate in the regulation of anxiety levels, learning, memory and body temperature, whilst it may also exhibit neuro- and cardio-protective properties [329-331]. Hence, there is also increasing research interest on the role of these agents and the effects of CRH-R2 inhibition in neuro-inflammation and cardiovascular function [330-336].
Finally, an additional study showed that astressin-2B (a selective CRH-R2 antagonist) attenuated stress-induced bacterial growth and prevented severe sepsis in an animal model [337], indicating that CRH-R2 inhibition may be protective against stress-induced pneumococcal disease and suggesting that the therapeutic potential of such agents may be even broader.
CONCLUSION
Despite the multiple challenges in studying the stress system and identifying the exact mechanisms underlying stress-induced pathology, stress research represents an important field of biomedical research with high translational value for targeted prevention and/or management of a broad spectrum of clinical conditions. It is now recognized that strong interdependent links exist between neurobehavioral/psychoemotional states relating to stress and certain “classic” disease states relating to autoimmunity, inflammation, malignancy, as well as metabolic, reproductive and growth disorders. Understanding the organization and integration of specific stress system pathways and neurochemical networks which facilitate these links constitutes a significant step forward in exploring the pathogenesis of stress-related complications. To date, a compelling body of experimental, epidemiologic and clinical evidence strongly supports the significant impact of acute and chronic stress on both physical health and emotional well-being, highlighting the need for further ongoing research in this field.
REFERENCES
- 1.
- Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5(7):374–81. [PubMed: 19488073]
- 2.
- Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA. 1992;267:1244–1252. [PubMed: 1538563]
- 3.
- Chrousos GP. Regulation and dysregulation of the hypothalamic-pituitary-adrenal axis. The corticotropin-releasing hormone perspective. Endocrinol Metab Clin North Am. 1992;21:833–858. [PubMed: 1486878]
- 4.
- Tsigos C, Chrousos GP. Physiology of the hypothalamic-pituitary-adrenal axis in health and dysregulation in psychiatric and autoimmune disorders. Endocrinol Metab Clin North Am. 1994;23:451–466. [PubMed: 7805648]
- 5.
- Dorn LD, Chrousos GP. The endocrinology of stress and stress system disorders in adolescence. Endocrinol Metab Clin North Am. 1993;22:685–700. [PubMed: 8243455]
- 6.
- Calogero AE, Gallucci WT, Gold PW, Chrousos GP. Multiple feedback regulatory loops upon rat hypothalamic corticotropin-releasing hormone secretion. Potential clinical implications. J Clin Invest. 1988;82:767–774. [PMC free article: PMC303581] [PubMed: 2843570]
- 7.
- Valentino RJ, Foote SL, Aston-Jones G. Corticotropin-releasing factor activates noradrenergic neurons of the locus coeruleus. Brain Res. 1983;270:363–367. [PubMed: 6603889]
- 8.
- Kiss A, Aguilera G. Participation of alpha 1-adrenergic receptors in the secretion of hypothalamic corticotropin-releasing hormone during stress. Neuroendocrinology. 1992;56:153–160. [PubMed: 1328915]
- 9.
- Calogero AE, Gallucci WT, Chrousos GP, Gold PW. Catecholamine effects upon rat hypothalamic corticotropin-releasing hormone secretion in vitro. J Clin Invest. 1988;82:839–846. [PMC free article: PMC303591] [PubMed: 2901433]
- 10.
- Silverman AJ, Hou-Yu A, Chen WP. Corticotropin-releasing factor synapses within the paraventricular nucleus of the hypothalamus. Neuroendocrinology. 1989;49:291–299. [PubMed: 2785663]
- 11.
- Aghajanian GK, VanderMaelen CP. alpha 2-adrenoceptor-mediated hyperpolarization of locus coeruleus neurons: intracellular studies in vivo. Science. 1982;215:1394–1396. [PubMed: 6278591]
- 12.
- Calogero AE, Bagdy G, Szemeredi K, Tartaglia ME, Gold PW, Chrousos GP. Mechanisms of serotonin receptor agonist-induced activation of the hypothalamic-pituitary-adrenal axis in the rat. Endocrinology. 1990;126:1888–1894. [PubMed: 2156670]
- 13.
- Fuller RW. The involvement of serotonin in regulation of pituitary-adrenocortical function. Front Neuroendocrinol. 1992;13:250–270. [PubMed: 1334001]
- 14.
- Calogero AE, Gallucci WT, Chrousos GP, Gold PW. Interaction between GABAergic neurotransmission and rat hypothalamic corticotropin-releasing hormone secretion in vitro. Brain Res. 1988;463:28–36. [PubMed: 3264201]
- 15.
- Overton JM, Fisher LA. Modulation of central nervous system actions of corticotropin-releasing factor by dynorphin-related peptides. Brain Res. 1989;488:233–240. [PubMed: 2568150]
- 16.
- Keller-Wood ME, Dallman MF. Corticosteroid inhibition of ACTH secretion. Endocr Rev. 1984;5:1–24. [PubMed: 6323158]
- 17.
- Aghajanian GK, Wang YY. Common alpha 2- and opiate effector mechanisms in the locus coeruleus: intracellular studies in brain slices. Neuropharmacology. 1987;26:793–799. [PubMed: 2443865]
- 18.
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. [PubMed: 6267699]
- 19.
- Aguilera G, Millan MA, Hauger RL, Catt KJ. Corticotropin-releasing factor receptors: distribution and regulation in brain, pituitary, and peripheral tissues. Ann N Y Acad Sci. 1987;512:48–66. [PubMed: 2831785]
- 20.
- De Souza EB, Insel TR, Perrin MH, Rivier J, Vale WW, Kuhar MJ. Corticotropin-releasing factor receptors are widely distributed within the rat central nervous system: an autoradiographic study. J Neurosci. 1985;5:3189–3203. [PMC free article: PMC6565229] [PubMed: 3001239]
- 21.
- Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotropin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Brain Res Rev. 1990;15:71–100. [PubMed: 1980834]
- 22.
- Slater PG, Yarur HE, Gysling K. Corticotropin-Releasing Factor Receptors and Their Interacting Proteins: Functional Consequences. Mol Pharmacol. 2016 Nov;90(5):627–632. [PubMed: 27612874] [CrossRef]
- 23.
- Polymeropoulos MH, Torres R, Yanovski JA, Chandrasekharappa SC, Ledbetter DH. The human corticotropin-releasing factor receptor (CRHR) gene maps to chromosome 17q12-q22. Genomics. 1995;28:123–124. [PubMed: 7590738]
- 24.
- Meyer AH, Ullmer C, Schmuck K, Morel C, Wishart W, Lubbert H, Engels P. Localization of the human CRF2 receptor to 7p21-p15 by radiation hybrid mapping and FISH analysis. Genomics. 1997;40:189–190. [PubMed: 9070940]
- 25.
- Chen R, Lewis KA, Perrin MH, Vale WW. Expression cloning of a human corticotropin-releasing-factor receptor. Proc Natl Acad Sci U S A. 1993;90:8967–8971. [PMC free article: PMC47482] [PubMed: 7692441]
- 26.
- Lovenberg TW, Chalmers DT, Liu C, De Souza EB. CRF2 alpha and CRF2 beta receptor mRNAs are differentially distributed between the rat central nervous system and peripheral tissues. Endocrinology. 1995;136:4139–4142. [PubMed: 7544278]
- 27.
- Chalmers DT, Lovenberg TW, De Souza EB. Localization of novel corticotropin-releasing factor receptor (CRF2) mRNA expression to specific subcortical nuclei in rat brain: comparison with CRF1 receptor mRNA expression. J Neurosci. 1995;15:6340–6350. [PMC free article: PMC6577987] [PubMed: 7472399]
- 28.
- Sanchez MM, Young LJ, Plotsky PM, Insel TR. Autoradiographic and in situ hybridization localization of corticotropin-releasing factor 1 and 2 receptors in nonhuman primate brain. J Comp Neurol. 1999;408:365–377. [PubMed: 10340512]
- 29.
- Wong ML, Licinio J, Pasternak KI, Gold PW. Localization of corticotropin-releasing hormone (CRH) receptor mRNA in adult rat brain by in situ hybridization histochemistry. Endocrinology. 1994;135:2275–2278. [PubMed: 7956950]
- 30.
- Grammatopoulos DK, Chrousos GP. Functional characteristics of CRH receptors and potential clinical applications of CRH-receptor antagonists. Trends Endocrinol Metab. 2002;13:436–444. [PubMed: 12431840]
- 31.
- Takefuji M, Murohara T. Corticotropin-Releasing Hormone Family and Their Receptors in the Cardiovascular System. Circ J. 2019 Jan 25;83(2):261–266. [PubMed: 30584229]
- 32.
- Rotondo F, Butz H, Syro LV, Yousef GM, Di Ieva A, Restrepo LM, Quintanar-Stephano A, Berczi I, Kovacs K. Arginine vasopressin (AVP): a review of its historical perspectives, current research and multifunctional role in the hypothalamo-hypophysial system. Pituitary. 2016 Aug;19(4):345–55. [PubMed: 26762848]
- 33.
- Antoni FA. Vasopressinergic control of pituitary adrenocorticotropin secretion comes of age. Front Neuroendocrinol. 1993;14:76–122. [PubMed: 8387436]
- 34.
- Gillies GE, Linton EA, Lowry PJ. Corticotropin releasing activity of the new CRF is potentiated several times by vasopressin. Nature. 1982;299:355–357. [PubMed: 6287293]
- 35.
- Vale W, Vaughan J, Smith M, Yamamoto G, Rivier J, Rivier C. Effects of synthetic ovine corticotropin-releasing factor, glucocorticoids, catecholamines, neurohypophysial peptides, and other substances on cultured corticotropic cells. Endocrinology. 1983;113:1121–1131. [PubMed: 6307665]
- 36.
- Lamberts SW, Verleun T, Oosterom R. de JF, Hackeng WH. Corticotropin-releasing factor (ovine) and vasopressin exert a synergistic effect on adrenocorticotropin release in man. J Clin Endocrinol Metab. 1984;58:298–303. [PubMed: 6319446]
- 37.
- Antoni FA. Receptors mediating the CRH effects of vasopressin and oxytocin. Ann N Y Acad Sci. 1987;512:195–204. [PubMed: 2831775]
- 38.
- Al-Damluji S, Cunnah D, Grossman A, Perry L, Ross G, Coy D, Rees LH, Besser GM. Effect of adrenaline on basal and ovine corticotrophin-releasing factor-stimulated ACTH secretion in man. J Endocrinol. 1987;112:145–150. [PubMed: 3029258]
- 39.
- Korbonits M, Kaltsas G, Perry LA, Putignano P, Grossman AB, Besser GM, Trainer PJ. The growth hormone secretagogue hexarelin stimulates the hypothalamo-pituitary-adrenal axis via arginine vasopressin. J Clin Endocrinol Metab. 1999;84:2489–2495. [PubMed: 10404825]
- 40.
- Malendowicz LK, Rucinski M, Belloni AS, Ziolkowska A, Nussdorfer GG. Leptin and the regulation of the hypothalamic-pituitary-adrenal axis. Int Rev Cytol. 2007;263:63–102. [PubMed: 17725965]
- 41.
- Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grubler Y, Stalla J, Pasquali R, Lutz B, Stalla GK, Pagotto U. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology. 2007;148:1574–1581. [PubMed: 17194743]
- 42.
- Aguilera G, Rabadan-Diehl C. Vasopressinergic regulation of the hypothalamic-pituitary-adrenal axis: implications for stress adaptation. Regul Pept. 2000 Dec 22;96(1-2):23–9. [PubMed: 11102648]
- 43.
- Aguilera G. Regulation of pituitary ACTH secretion during chronic stress. Front Neuroendocrinol. 1994 Dec;15(4):321–50. [PubMed: 7895891]
- 44.
- Aguilera G, Subburaju S, Young S, Chen J. The parvocellular vasopressinergic system and responsiveness of the hypothalamic pituitary adrenal axis during chronic stress. Prog Brain Res. 2008;170:29–39. [PMC free article: PMC2536760] [PubMed: 18655869]
- 45.
- Egawa M, Yoshimatsu H, Bray GA. Neuropeptide Y suppresses sympathetic activity to interscapular brown adipose tissue in rats. Am J Physiol. 1991;260:R328–R334. [PubMed: 1996720]
- 46.
- Oellerich WF, Schwartz DD, Malik KU. Neuropeptide Y inhibits adrenergic transmitter release in cultured rat superior cervical ganglion cells by restricting the availability of calcium through a pertussis toxin-sensitive mechanism. Neuroscience. 1994;60:495–502. [PubMed: 8072693]
- 47.
- White BD, Dean RG, Edwards GL, Martin RJ. Type II corticosteroid receptor stimulation increases NPY gene expression in basomedial hypothalamus of rats. Am J Physiol. 1994;266:R1523–R1529. [PubMed: 8203629]
- 48.
- Hirsch D, Zukowska Z. NPY and Stress 30 Years Later: The Peripheral View. Cell Mol Neurobiol. 2012 [PMC free article: PMC3492947] [PubMed: 22271177]
- 49.
- Larsen PJ, Jessop D, Patel H, Lightman SL, Chowdrey HS. Substance P inhibits the release of anterior pituitary adrenocorticotrophin via a central mechanism involving corticotrophin-releasing factor-containing neurons in the hypothalamic paraventricular nucleus. J Neuroendocrinol. 1993;5:99–105. [PubMed: 7683556]
- 50.
- Culman J, Tschope C, Jost N, Itoi K, Unger T. Substance P and neurokinin A induced desensitization to cardiovascular and behavioral effects: evidence for the involvement of different tachykinin receptors. Brain Res. 1993;625:75–83. [PubMed: 7694777]
- 51.
- Jessop DS, Chowdrey HS, Larsen PJ, Lightman SL. Substance P: multifunctional peptide in the hypothalamo-pituitary system? J Endocrinol. 1992;132:331–337. [PubMed: 1373432]
- 52.
- Rotzinger S, Lovejoy DA, Tan LA. Behavioral effects of neuropeptides in rodent models of depression and anxiety. Peptides. 2010;31:736–756. [PubMed: 20026211]
- 53.
- Abou-Samra AB, Harwood JP, Catt KJ, Aguilera G. Mechanisms of action of CRF and other regulators of ACTH release in pituitary corticotrophs. Ann N Y Acad Sci. 1987;512:67–84. [PubMed: 2831786]
- 54.
- Engler D, Pham T, Fullerton MJ, Ooi G, Funder JW, Clarke IJ. Studies of the secretion of corticotropin-releasing factor and arginine vasopressin into the hypophysial-portal circulation of the conscious sheep. I. Effect of an audiovisual stimulus and insulin-induced hypoglycemia. Neuroendocrinology. 1989;49:367–381. [PubMed: 2541360]
- 55.
- Redekopp C, Irvine CH, Donald RA, Livesey JH, Sadler W, Nicholls MG, Alexander SL, Evans MJ. Spontaneous and stimulated adrenocorticotropin and vasopressin pulsatile secretion in the pituitary venous effluent of the horse. Endocrinology. 1986;118:1410–1416. [PubMed: 3004914]
- 56.
- Young EA, Abelson J, Lightman SL. Cortisol pulsatility and its role in stress regulation and health. Front Neuroendocrinol. 2004;25:69–76. [PubMed: 15571755]
- 57.
- Horrocks PM, Jones AF, Ratcliffe WA, Holder G, White A, Holder R, Ratcliffe JG, London DR. Patterns of ACTH and cortisol pulsatility over twenty-four hours in normal males and females. Clin Endocrinol (Oxf). 1990;32:127–134. [PubMed: 2158868]
- 58.
- Iranmanesh A, Lizarralde G, Short D, Veldhuis JD. Intensive venous sampling paradigms disclose high frequency adrenocorticotropin release episodes in normal men. J Clin Endocrinol Metab. 1990;71:1276–1283. [PubMed: 2172277]
- 59.
- Veldhuis JD, Iranmanesh A, Roelfsema F, Aoun P, Takahashi P, Miles JM, Keenan DM. Tripartite control of dynamic ACTH-cortisol dose responsiveness by age, body mass index, and gender in 111 healthy adults. J Clin Endocrinol Metab. 2011;96:2874–2881. [PMC free article: PMC3167672] [PubMed: 21752885]
- 60.
- Nicolaides NC, Charmandari E, Kino T, Chrousos GP. Stress-Related and Circadian Secretion and Target Tissue Actions of Glucocorticoids: Impact on Health. Front Endocrinol (Lausanne). 2017 Apr 28;8:70. [PMC free article: PMC5408025] [PubMed: 28503165] [CrossRef]
- 61.
- Veldhuis JD, Iranmanesh A, Johnson ML, Lizarralde G. Amplitude, but not frequency, modulation of adrenocorticotropin secretory bursts gives rise to the nyctohemeral rhythm of the corticotropic axis in man. J Clin Endocrinol Metab. 1990;71:452–463. [PubMed: 2166071]
- 62.
- Lutfy K, Aimiuwu O, Mangubat M, Shin CS, Nerio N, Gomez R, Liu Y, Friedman TC. Nicotine stimulates secretion of corticosterone via both CRH and AVP receptors. J Neurochem. 2011 [PMC free article: PMC3296864] [PubMed: 22191943]
- 63.
- Aguilera G. Factors controlling steroid biosynthesis in the zona glomerulosa of the adrenal. J Steroid Biochem Mol Biol. 1993;45:147–151. [PubMed: 8481339]
- 64.
- Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44. [PubMed: 6368214]
- 65.
- Smith DF, Toft DO. Steroid receptors and their associated proteins. Mol Endocrinol. 1993;7:4–11. [PubMed: 8446107]
- 66.
- Pratt WB. Glucocorticoid receptor structure and the initial events in signal transduction. Prog Clin Biol Res. 1990;322:119–132. [PubMed: 2406728]
- 67.
- Ismaili N, Garabedian MJ. Modulation of glucocorticoid receptor function via phosphorylation. Ann N Y Acad Sci. 2004;1024:86–101. [PubMed: 15265775]
- 68.
- Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, Rogatsky I, Logan SK, Garabedian MJ. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol. 2008;22:1754–1766. [PMC free article: PMC2725771] [PubMed: 18483179]
- 69.
- Nader N, Chrousos GP, Kino T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB J. 2009;23:1572–1583. [PMC free article: PMC2669420] [PubMed: 19141540]
- 70.
- Song IH, Buttgereit F. Non-genomic glucocorticoid effects to provide the basis for new drug developments. Mol Cell Endocrinol. 2006;246:142–146. [PubMed: 16388891]
- 71.
- Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, Khairova R, Zhou R, Yuan P, Machado-Vieira R, McEwen BS, Manji HK. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci U S A. 2009;106:3543–3548. [PMC free article: PMC2637276] [PubMed: 19202080]
- 72.
- Koufali MM, Moutsatsou P, Sekeris CE, Breen KC. The dynamic localization of the glucocorticoid receptor in rat C6 glioma cell mitochondria. Mol Cell Endocrinol. 2003;209:51–60. [PubMed: 14604816]
- 73.
- de Kloet ER. Stress in the brain. Eur J Pharmacol. 2000;405:187–198. [PubMed: 11033326]
- 74.
- Lightman SL, Wiles CC, Atkinson HC, Henley DE, Russell GM, Leendertz JA, McKenna MA, Spiga F, Wood SA, Conway-Campbell BL. The significance of glucocorticoid pulsatility. Eur J Pharmacol. 2008;583:255–262. [PubMed: 18339373]
- 75.
- Stavreva DA, Wiench M, John S, Conway-Campbell BL, McKenna MA, Pooley JR, Johnson TA, Voss TC, Lightman SL, Hager GL. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol. 2009;11:1093–1102. [PMC free article: PMC6711162] [PubMed: 19684579]
- 76.
- McMaster A, Jangani M, Sommer P, Han N, Brass A, Beesley S, Lu W, Berry A, Loudon A, Donn R, Ray DW. Ultradian cortisol pulsatility encodes a distinct, biologically important signal. PLoS One. 2011;6:e15766. [PMC free article: PMC3022879] [PubMed: 21267416]
- 77.
- Gilbey MP, Spyer KM. Essential organization of the sympathetic nervous system. Baillieres Clin Endocrinol Metab. 1993;7:259–278. [PubMed: 8098208]
- 78.
- Burnstock G, Miller P. Structural and chemical organization of the autonomic nervous system with special reference to noradrenergic non-cholinergic transmission, in Autonomic Failure. A Textbook of Clinical Disorders of the Autonomic Nervous System. Bannister R, Mathias CJ (eds), Oxford Medical Press, Oxford, 1989, pp107.
- 79.
- Benarroch EE. Neuropeptides in the sympathetic system: presence, plasticity, modulation, and implications. Ann Neurol. 1994;36:6–13. [PubMed: 8024263]
- 80.
- Elfvin LG, Lindh B, Hokfelt T. The chemical neuroanatomy of sympathetic ganglia. Annu Rev Neurosci. 1993;16:471–507. [PubMed: 8384808]
- 81.
- Nikolarakis KE, Almeida OF, Herz A. Stimulation of hypothalamic beta-endorphin and dynorphin release by corticotropin-releasing factor (in vitro). Brain Res. 1986;399:152–155. [PubMed: 2879612]
- 82.
- Roth RH, Tam SY, Ida Y, Yang JX, Deutch AY. Stress and the mesocorticolimbic dopamine systems. Ann N Y Acad Sci. 1988;537:138–147. [PubMed: 3059920]
- 83.
- Gray TS, Carney ME, Magnuson DJ. Direct projections from the central amygdaloid nucleus to the hypothalamic paraventricular nucleus: possible role in stress-induced adrenocorticotropin release. Neuroendocrinology. 1989;50:433–446. [PubMed: 2554178]
- 84.
- Chrousos GP. Stressors, stress, and neuroendocrine integration of the adaptive response. The 1997 Hans Selye Memorial Lecture. Ann N Y Acad Sci. 1998;851:311–335. [PubMed: 9668623]
- 85.
- Grigoriadis DE, Heroux JA, De Souza EB. Characterization and regulation of corticotropin-releasing factor receptors in the central nervous, endocrine and immune systems. Ciba Found Symp. 1993;172:85–101. [PubMed: 8387906]
- 86.
- Millan MJ. Stress and endogenous opioid peptides: a review. Mod Probl Pharmacopsychiatry. 1981;17:49–67. [PubMed: 6276731]
- 87.
- McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. [PubMed: 17615391]
- 88.
- Lupien SJ. de LM, de SS, Convit A, Tarshish C, Nair NP, Thakur M, McEwen BS, Hauger RL, Meaney MJ. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1:69–73. [PubMed: 10195112]
- 89.
- Refojo D, Schweizer M, Kuehne C, Ehrenberg S, Thoeringer C, Vogl AM, Dedic N, Schumacher M. von WG, Avrabos C, Touma C, Engblom D, Schutz G, Nave KA, Eder M, Wotjak CT, Sillaber I, Holsboer F, Wurst W, Deussing JM. Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science. 2011;333:1903–1907. [PubMed: 21885734]
- 90.
- Lkhagvasuren B, Nakamura Y, Oka T, Sudo N, Nakamura K. Social defeat stress induces hyperthermia through activation of thermoregulatory sympathetic premotor neurons in the medullary raphe region. Eur J Neurosci. 2011;34:1442–1452. [PubMed: 21978215]
- 91.
- Oka T, Oka K. Age and gender differences of psychogenic fever: a review of the Japanese literature. Biopsychosoc Med. 2007;1:11. [PMC free article: PMC1888691] [PubMed: 17511878]
- 92.
- Nakamura K. Central circuitries for body temperature regulation and fever. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1207–R1228. [PubMed: 21900642]
- 93.
- Mund RA, Frishman WH. Brown adipose tissue thermogenesis: β3-adrenoreceptors as a potential target for the treatment of obesity in humans. Cardiol Rev. 2013 Nov-Dec;21(6):265–9. [PubMed: 23707990]
- 94.
- Schulz S, Laessle RG. Stress-induced laboratory eating behavior in obese women with binge eating disorder. Appetite. 2012;58:457–461. [PubMed: 22200410]
- 95.
- Spencer SJ, Tilbrook A. The glucocorticoid contribution to obesity. Stress. 2011;14:233–246. [PubMed: 21294656]
- 96.
- Nader N, Chrousos GP, Kino T. Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol Metab. 2010;21:277–286. [PMC free article: PMC2862789] [PubMed: 20106676]
- 97.
- Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295:1070–1073. [PubMed: 11834835]
- 98.
- Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–775. [PMC free article: PMC3758473] [PubMed: 18802415]
- 99.
- Kalsbeek A, Palm IF, La Fleur SE, Scheer FA, Perreau-Lenz S, Ruiter M, Kreier F, Cailotto C, Buijs RM. SCN outputs and the hypothalamic balance of life. J Biol Rhythms. 2006;21:458–469. [PubMed: 17107936]
- 100.
- Butcher GQ, Lee B, Cheng HY, Obrietan K. Light stimulates MSK1 activation in the suprachiasmatic nucleus via a PACAP-ERK/MAP kinase-dependent mechanism. J Neurosci. 2005;25:5305–5313. [PMC free article: PMC6724997] [PubMed: 15930378]
- 101.
- Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Hum Mol Genet. 2006;15(Spec No 2):R271–R277. [PubMed: 16987893]
- 102.
- Hastings M, O'Neill JS, Maywood ES. Circadian clocks: regulators of endocrine and metabolic rhythms. J Endocrinol. 2007;195:187–198. [PubMed: 17951531]
- 103.
- Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. [PubMed: 11779462]
- 104.
- Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol. 2007;8:139–148. [PubMed: 17245414]
- 105.
- O'Neill JS, Maywood ES, Chesham JE, Takahashi JS, Hastings MH. cAMP-dependent signaling as a core component of the mammalian circadian pacemaker. Science. 2008;320:949–953. [PMC free article: PMC2735813] [PubMed: 18487196]
- 106.
- Lowrey PL, Takahashi JS. Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004;5:407–441. [PMC free article: PMC3770722] [PubMed: 15485355]
- 107.
- Ripperger JA, Schibler U. Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat Genet. 2006;38:369–374. [PubMed: 16474407]
- 108.
- Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–1362. [PubMed: 7715646]
- 109.
- Ishida A, Mutoh T, Ueyama T, Bando H, Masubuchi S, Nakahara D, Tsujimoto G, Okamura H. Light activates the adrenal gland: timing of gene expression and glucocorticoid release. Cell Metab. 2005;2:297–307. [PubMed: 16271530]
- 110.
- Ulrich-Lai YM, Arnhold MM, Engeland WC. Adrenal splanchnic innervation contributes to the diurnal rhythm of plasma corticosterone in rats by modulating adrenal sensitivity to ACTH. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1128–R1135. [PubMed: 16357102]
- 111.
- Yang S, Liu A, Weidenhammer A, Cooksey RC, McClain D, Kim MK, Aguilera G, Abel ED, Chung JH. The role of mPer2 clock gene in glucocorticoid and feeding rhythms. Endocrinology. 2009;150:2153–2160. [PMC free article: PMC2671901] [PubMed: 19179447]
- 112.
- Lemos DR, Downs JL, Urbanski HF. Twenty-four-hour rhythmic gene expression in the rhesus macaque adrenal gland. Mol Endocrinol. 2006;20:1164–1176. [PubMed: 16439464]
- 113.
- Oster H, Damerow S, Kiessling S, Jakubcakova V, Abraham D, Tian J, Hoffmann MW, Eichele G. The circadian rhythm of glucocorticoids is regulated by a gating mechanism residing in the adrenal cortical clock. Cell Metab. 2006;4:163–173. [PubMed: 16890544]
- 114.
- Chung S, Son GH, Kim K. Adrenal peripheral oscillator in generating the circadian glucocorticoid rhythm. Ann N Y Acad Sci. 2011;1220:71–81. [PubMed: 21388405]
- 115.
- Son GH, Chung S, Choe HK, Kim HD, Baik SM, Lee H, Lee HW, Choi S, Sun W, Kim H, Cho S, Lee KH, Kim K. Adrenal peripheral clock controls the autonomous circadian rhythm of glucocorticoid by causing rhythmic steroid production. Proc Natl Acad Sci U S A. 2008;105:20970–20975. [PMC free article: PMC2634940] [PubMed: 19091946]
- 116.
- Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, Schutz G, Schibler U. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science. 2000;289:2344–2347. [PubMed: 11009419]
- 117.
- Yamamoto T, Nakahata Y, Tanaka M, Yoshida M, Soma H, Shinohara K, Yasuda A, Mamine T, Takumi T. Acute physical stress elevates mouse period1 mRNA expression in mouse peripheral tissues via a glucocorticoid-responsive element. J Biol Chem. 2005;280:42036–42043. [PubMed: 16249183]
- 118.
- So AY, Bernal TU, Pillsbury ML, Yamamoto KR, Feldman BJ. Glucocorticoid regulation of the circadian clock modulates glucose homeostasis. Proc Natl Acad Sci U S A. 2009;106:17582–17587. [PMC free article: PMC2757402] [PubMed: 19805059]
- 119.
- Segall LA, Milet A, Tronche F, Amir S. Brain glucocorticoid receptors are necessary for the rhythmic expression of the clock protein, PERIOD2, in the central extended amygdala in mice. Neurosci Lett. 2009;457:58–60. [PubMed: 19429162]
- 120.
- Kassi EN, Chrousos GP. The central CLOCK system and the stress axis in health and disease. Hormones (Athens). 2013;12(2):172–191. [PubMed: 23933687]
- 121.
- Chrousos GP, Kino T. Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress. 2007;10:213–219. [PubMed: 17514590]
- 122.
- Kino T, Chrousos GP. Acetylation-mediated epigenetic regulation of glucocorticoid receptor activity: circadian rhythm-associated alterations of glucocorticoid actions in target tissues. Mol Cell Endocrinol. 2011;336:23–30. [PMC free article: PMC3057275] [PubMed: 21146585]
- 123.
- Kino T, Chrousos GP. Circadian CLOCK-mediated regulation of target-tissue sensitivity to glucocorticoids: implications for cardiometabolic diseases. Endocr Dev. 2011;20:116–126. [PMC free article: PMC3163295] [PubMed: 21164265]
- 124.
- Chand D, Lovejoy DA. Stress and reproduction: controversies and challenges. Gen Comp Endocrinol. 2011;171:253–257. [PubMed: 21362426]
- 125.
- Rincón-Cortés M, Herman JP, Lupien S, Maguire J, Shansky RM. Stress: Influence of sex, reproductive status and gender. Neurobiol Stress. 2019 Mar 9;10:100155. [PMC free article: PMC6430637] [PubMed: 30949564] [CrossRef]
- 126.
- Edwards KL, Edes AN, Brown JL. Stress, Well-Being and Reproductive Success. Adv Exp Med Biol. 2019;1200:91–162. [PubMed: 31471796]
- 127.
- Traslaviña GA, Franci CR. The CRH-R1 receptor mediates luteinizing hormone, prolactin, corticosterone and progesterone secretion induced by restraint stress in estrogen-primed rats. Brain Res. 2011 Nov 3;1421:11–9. [PubMed: 21959177]
- 128.
- Rabin DS, Schmidt PJ, Campbell G, Gold PW, Jensvold M, Rubinow DR, Chrousos GP. Hypothalamic-pituitary-adrenal function in patients with the premenstrual syndrome. J Clin Endocrinol Metab. 1990;71:1158–1162. [PubMed: 2172272]
- 129.
- Rivier C, Rivier J, Vale W. Stress-induced inhibition of reproductive functions: role of endogenous corticotropin-releasing factor. Science. 1986;231:607–609. [PubMed: 3003907]
- 130.
- Tsigos C, Papanicolaou DA, Kyrou I, Raptis SA, Chrousos GP. Dose-dependent effects of recombinant human interleukin-6 on the pituitary-testicular axis. J Interferon Cytokine Res. 1999;19:1271–1276. [PubMed: 10574620]
- 131.
- Valsamakis G, Chrousos G, Mastorakos G. Stress, female reproduction and pregnancy. Psychoneuroendocrinology. 2019;100:48–57. [PubMed: 30291988]
- 132.
- Kyrou I, Tsigos C. Chronic stress, visceral obesity and gonadal dysfunction. Hormones (Athens). 2008 Oct-Dec;7(4):287–93. [PubMed: 19121989]
- 133.
- Kalantaridou SN, Zoumakis E, Makrigiannakis A, Lavasidis LG, Vrekoussis T, Chrousos GP. Corticotropin-releasing hormone, stress and human reproduction: an update. J Reprod Immunol. 2010;85:33–39. [PubMed: 20412987]
- 134.
- Mastorakos G, Webster EL, Friedman TC, Chrousos GP. Immunoreactive corticotropin-releasing hormone and its binding sites in the rat ovary. J Clin Invest. 1993;92:961–968. [PMC free article: PMC294936] [PubMed: 8394389]
- 135.
- Mastorakos G, Scopa CD, Kao LC, Vryonidou A, Friedman TC, Kattis D, Phenekos C, Rabin D, Chrousos GP. Presence of immunoreactive corticotropin-releasing hormone in human endometrium. J Clin Endocrinol Metab. 1996;81:1046–1050. [PubMed: 8772574]
- 136.
- Wypior G, Jeschke U, Kurpisz M, Szekeres-Bartho J. Expression of CRH, CRH-related peptide and CRH receptor in the ovary and potential CRH signalling pathways. J Reprod Immunol. 2011;90:67–73. [PubMed: 21696829]
- 137.
- Bromberger JT, Matthews KA, Kuller LH, Wing RR, Meilahn EN, Plantinga P. Prospective study of the determinants of age at menopause. Am J Epidemiol. 1997;145:124–133. [PubMed: 9006309]
- 138.
- Vrekoussis T, Kalantaridou SN, Mastorakos G, Zoumakis E, Makrigiannakis A, Syrrou M, Lavasidis LG, Relakis K, Chrousos GP. The role of stress in female reproduction and pregnancy: an update. Ann N Y Acad Sci. 2010;1205:69–75. [PubMed: 20840255]
- 139.
- Cottrell EC, Seckl JR. Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci. 2009;3:19. [PMC free article: PMC2759372] [PubMed: 19826624]
- 140.
- Sandman CA, Davis EP, Buss C, Glynn LM. Prenatal programming of human neurological function. Int J Pept. 2011;2011:837596. [PMC free article: PMC3133795] [PubMed: 21760821]
- 141.
- Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet. 2011;378:169–181. [PubMed: 21705072]
- 142.
- Garcia-Caceres C, Lagunas N, Calmarza-Font I, Azcoitia I, Diz-Chaves Y, Garcia-Segura LM, Baquedano E, Frago LM, Argente J, Chowen JA. Gender differences in the long-term effects of chronic prenatal stress on the HPA axis and hypothalamic structure in rats. Psychoneuroendocrinology. 2010;35:1525–1535. [PubMed: 20558007]
- 143.
- Kinsella MT, Monk C. Impact of maternal stress, depression and anxiety on fetal neurobehavioral development. Clin Obstet Gynecol. 2009;52:425–440. [PMC free article: PMC3710585] [PubMed: 19661759]
- 144.
- Chrousos GP, Torpy DJ, Gold PW. Interactions between the hypothalamic-pituitary-adrenal axis and the female reproductive system: clinical implications. Ann Intern Med. 1998;129:229–240. [PubMed: 9696732]
- 145.
- Sasaki A, Sato S, Murakami O, Go M, Inoue M, Shimizu Y, Hanew K, Andoh N, Sato I, Sasano N, et al. Immunoreactive corticotropin-releasing hormone present in human plasma may be derived from both hypothalamic and extrahypothalamic sources. J Clin Endocrinol Metab. 1987;65:176–182. [PubMed: 3294879]
- 146.
- Behan DP, Linton EA, Lowry PJ. Isolation of the human plasma corticotrophin-releasing factor-binding protein. J Endocrinol. 1989;122:23–31. [PubMed: 2549150]
- 147.
- Linton EA, Perkins AV, Woods RJ, Eben F, Wolfe CD, Behan DP, Potter E, Vale WW, Lowry PJ. Corticotropin releasing hormone-binding protein (CRH-BP): plasma levels decrease during the third trimester of normal human pregnancy. J Clin Endocrinol Metab. 1993;76:260–262. [PubMed: 8421097]
- 148.
- Margioris AN, Grino M, Rabin D, Chrousos GP. Human placenta and the hypothalamic-pituitary-adrenal axis. Adv Exp Med Biol. 1988;245:389–398. [PubMed: 2852461]
- 149.
- Mairesse J, Lesage J, Breton C, Breant B, Hahn T, Darnaudery M, Dickson SL, Seckl J, Blondeau B, Vieau D, Maccari S, Viltart O. Maternal stress alters endocrine function of the feto-placental unit in rats. Am J Physiol Endocrinol Metab. 2007;292:E1526–E1533. [PubMed: 17264224]
- 150.
- Vamvakopoulos NC, Chrousos GP. Evidence of direct estrogenic regulation of human corticotropin-releasing hormone gene expression. Potential implications for the sexual dimophism of the stress response and immune/inflammatory reaction. J Clin Invest. 1993;92:1896–1902. [PMC free article: PMC288355] [PubMed: 8408641]
- 151.
- Vamvakopoulos NC, Chrousos GP. Hormonal regulation of human corticotropin-releasing hormone gene expression: implications for the stress response and immune/inflammatory reaction. Endocr Rev. 1994;15:409–420. [PubMed: 7988479]
- 152.
- Burguera B, Muruais C, Penalva A, Dieguez C, Casanueva FF. Dual and selective actions of glucocorticoids upon basal and stimulated growth hormone release in man. Neuroendocrinology. 1990;51:51–58. [PubMed: 2106087]
- 153.
- Dieguez C, Page MD, Scanlon MF. Growth hormone neuroregulation and its alterations in disease states. Clin Endocrinol (Oxf). 1988;28:109–143. [PubMed: 2901921]
- 154.
- Casanueva FF, Burguera B, Muruais C, Dieguez C. Acute administration of corticoids: a new and peculiar stimulus of growth hormone secretion in man. J Clin Endocrinol Metab. 1990;70:234–237. [PubMed: 2104624]
- 155.
- Rivier C, Vale W. Involvement of corticotropin-releasing factor and somatostatin in stress-induced inhibition of growth hormone secretion in the rat. Endocrinology. 1985;117:2478–2482. [PubMed: 2866087]
- 156.
- Money J. The syndrome of abuse dwarfism (psychosocial dwarfism or reversible hyposomatotropism). Am J Dis Child. 1977 May;131(5):508–13. [PubMed: 857651]
- 157.
- Bowden ML, Hopwood NJ. Psychosocial dwarfism: identification, intervention and planning. Soc Work Health Care. 1982 Spring;7(3):15–36. [PubMed: 7123443]
- 158.
- Green WH, Campbell M, David R. Psychosocial dwarfism: a critical review of the evidence. J Am Acad Child Psychiatry. 1984 Jan;23(1):39–48. [PubMed: 6319472]
- 159.
- Johnson EO, Kamilaris TC, Calogero AE, Gold PW, Chrousos GP. Effects of early parenting on growth and development in a small primate. Pediatr Res. 1996;39:999–1005. [PubMed: 8725261]
- 160.
- Malozowski S, Muzzo S, Burrows R, Leiva L, Loriaux L, Chrousos G, Winterer J, Cassorla F. The hypothalamic-pituitary-adrenal axis in infantile malnutrition. Clin Endocrinol (Oxf). 1990;32:461–465. [PubMed: 2112070]
- 161.
- Benker G, Raida M, Olbricht T, Wagner R, Reinhardt W, Reinwein D. TSH secretion in Cushing's syndrome: relation to glucocorticoid excess, diabetes, goitre, and the 'sick euthyroid syndrome'. Clin Endocrinol (Oxf). 1990;33:777–786. [PubMed: 2128925]
- 162.
- Duick DS, Wahner HW. Thyroid axis in patients with Cushing's syndrome. Arch Intern Med. 1979;139:767–772. [PubMed: 110278]
- 163.
- Roelfsema F, Pereira AM, Biermasz NR, Frolich M, Keenan DM, Veldhuis JD, Romijn JA. Diminished and irregular TSH secretion with delayed acrophase in patients with Cushing's syndrome. Eur J Endocrinol. 2009;161:695–703. [PubMed: 19687167]
- 164.
- Re RN, Kourides IA, Ridgway EC, Weintraub BD, Maloof F. The effect of glucocorticoid administration on human pituitary secretion of thyrotropin and prolactin. J Clin Endocrinol Metab. 1976;43:338–346. [PubMed: 820709]
- 165.
- Brabant G, Brabant A, Ranft U, Ocran K, Kohrle J, Hesch RD. von zur MA. Circadian and pulsatile thyrotropin secretion in euthyroid man under the influence of thyroid hormone and glucocorticoid administration. J Clin Endocrinol Metab. 1987;65:83–88. [PubMed: 3584402]
- 166.
- Samuels MH, McDaniel PA. Thyrotropin levels during hydrocortisone infusions that mimic fasting-induced cortisol elevations: a clinical research center study. J Clin Endocrinol Metab. 1997;82:3700–3704. [PubMed: 9360528]
- 167.
- Adriaanse R, Brabant G, Endert E, Wiersinga WM. Pulsatile thyrotropin secretion in patients with Cushing's syndrome. Metabolism. 1994;43:782–786. [PubMed: 8201971]
- 168.
- Tsigos C, Papanicolaou DA, Defensor R, Mitsiadis CS, Kyrou I, Chrousos GP. Dose effects of recombinant human interleukin-6 on pituitary hormone secretion and energy expenditure. Neuroendocrinology. 1997;66:54–62. [PubMed: 9258919]
- 169.
- Torpy DJ, Tsigos C, Lotsikas AJ, Defensor R, Chrousos GP, Papanicolaou DA. Acute and delayed effects of a single-dose injection of interleukin-6 on thyroid function in healthy humans. Metabolism. 1998;47:1289–1293. [PubMed: 9781636]
- 170.
- Chrousos GP. The role of stress and the hypothalamic-pituitary-adrenal axis in the pathogenesis of the metabolic syndrome: neuro-endocrine and target tissue-related causes. Int J Obes Relat Metab Disord. 2000;24 Suppl 2:S50–S55. [PubMed: 10997609]
- 171.
- Kyrou I, Tsigos C. Stress mechanisms and metabolic complications. Horm Metab Res. 2007 Jun;39(6):430–8. [PubMed: 17578760]
- 172.
- Kalra SP, Dube MG, Pu S, Xu B, Horvath TL, Kalra PS. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev. 1999;20:68–100. [PubMed: 10047974]
- 173.
- Richard D, Lin Q, Timofeeva E. The corticotropin-releasing factor family of peptides and CRF receptors: their roles in the regulation of energy balance. Eur J Pharmacol. 2002;440:189–197. [PubMed: 12007535]
- 174.
- Ohata H, Shibasaki T. Involvement of CRF2 receptor in the brain regions in restraint-induced anorexia. Neuroreport. 2011;22:494–498. [PubMed: 21666520]
- 175.
- Misra M, Klibanski A. Neuroendocrine consequences of anorexia nervosa in adolescents. Endocr Dev. 2010;17:197–214. [PMC free article: PMC3731628] [PubMed: 19955768]
- 176.
- Misra M, Klibanski A. The neuroendocrine basis of anorexia nervosa and its impact on bone metabolism. Neuroendocrinology. 2011;93:65–73. [PMC free article: PMC3214929] [PubMed: 21228564]
- 177.
- Chan JL, Mantzoros CS. Role of leptin in energy-deprivation states: normal human physiology and clinical implications for hypothalamic amenorrhoea and anorexia nervosa. Lancet. 2005;366:74–85. [PubMed: 15993236]
- 178.
- Lawson EA, Eddy KT, Donoho D, Misra M, Miller KK, Meenaghan E, Lydecker J, Herzog D, Klibanski A. Appetite-regulating hormones cortisol and peptide YY are associated with disordered eating psychopathology, independent of body mass index. Eur J Endocrinol. 2011;164:253–261. [PMC free article: PMC3677777] [PubMed: 21098684]
- 179.
- Figlewicz DP. Adiposity signals and food reward: expanding the CNS roles of insulin and leptin. Am J Physiol Regul Integr Comp Physiol. 2003;284:R882–R892. [PubMed: 12626355]
- 180.
- Cavagnini F, Croci M, Putignano P, Petroni ML, Invitti C. Glucocorticoids and neuroendocrine function. Int J Obes Relat Metab Disord. 2000;24 Suppl 2:S77–S79. [PubMed: 10997615]
- 181.
- Kaye WH, Berrettini W, Gwirtsman H, George DT. Altered cerebrospinal fluid neuropeptide Y and peptide YY immunoreactivity in anorexia and bulimia nervosa. Arch Gen Psychiatry. 1990;47:548–556. [PubMed: 2350207]
- 182.
- Munglani R, Hudspith MJ, Hunt SP. The therapeutic potential of neuropeptide Y. Analgesic, anxiolytic and antihypertensive. Drugs. 1996;52:371–389. [PubMed: 8875128]
- 183.
- Balsevich G, Abizaid A, Chen A, Karatsoreos IN, Schmidt MV. Stress and glucocorticoid modulation of feeding and metabolism. Neurobiol Stress. 2019 May 15;11:100171. [PMC free article: PMC6529856] [PubMed: 31193462]
- 184.
- Tamashiro KL, Sakai RR, Shively CA, Karatsoreos IN, Reagan LP. Chronic stress, metabolism, and metabolic syndrome. Stress. 2011;14:468–474. [PubMed: 21848434]
- 185.
- Charmandari E, Tsigos C, Chrousos G. Endocrinology of the stress response. Annu Rev Physiol. 2005;67:259–284. [PubMed: 15709959]
- 186.
- Chrousos GP, Kino T. Glucocorticoid signaling in the cell. Expanding clinical implications to complex human behavioral and somatic disorders. Ann N Y Acad Sci. 2009;1179:153–166. [PMC free article: PMC2791367] [PubMed: 19906238]
- 187.
- Kyrou I, Tsigos C. Stress hormones: physiological stress and regulation of metabolism. Curr Opin Pharmacol. 2009;9:787–793. [PubMed: 19758844]
- 188.
- Kyrou I, Mattu H, Chatha K, Randeva H. Fat hormones, adipokines. In Schisler JC, Lang CH, Willis M (eds); Endocrinology of the Heart in Health and Disease. Academic Press, 2017, 167-205; www
.sciencedirect.com /science/book/9780128031117. - 189.
- Su X, Peng D. Adipokines as novel biomarkers of cardio-metabolic disorders. Clin Chim Acta. 2020;507:31–38. [PubMed: 32283064]
- 190.
- Stewart PM, Boulton A, Kumar S, Clark PM, Shackleton CH. Cortisol metabolism in human obesity: impaired cortisone-cortisol conversion in subjects with central adiposity. J Clin Endocrinol Metab. 1999;84:1022–1027. [PubMed: 10084590]
- 191.
- Pereira CD, Azevedo I, Monteiro R, Martins MJ. 11β-Hydroxysteroid dehydrogenase type 1: relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes Metab. 2012 Oct;14(10):869–81. [PubMed: 22321826]
- 192.
- Anagnostis P, Katsiki N, Adamidou F, Athyros VG, Karagiannis A, Kita M, Mikhailidis DP. 11beta-Hydroxysteroid dehydrogenase type 1 inhibitors: novel agents for the treatment of metabolic syndrome and obesity-related disorders? Metabolism. 2013 Jan;62(1):21–33. [PubMed: 22652056]
- 193.
- Vgontzas AN, Chrousos GP. Sleep-disordered breathing, sleepiness, and insulin resistance: is the latter a consequence, a pathogenetic factor, or both? Sleep Med. 2002;3:389–391. [PubMed: 14592169]
- 194.
- Vgontzas AN, Chrousos GP. Sleep, the hypothalamic-pituitary-adrenal axis, and cytokines: multiple interactions and disturbances in sleep disorders. Endocrinol Metab Clin North Am. 2002;31:15–36. [PubMed: 12055986]
- 195.
- Vgontzas AN, Bixler EO, Chrousos GP. Obesity-related sleepiness and fatigue: the role of the stress system and cytokines. Ann N Y Acad Sci. 2006;1083:329–344. [PubMed: 17148748]
- 196.
- Vgontzas AN, Bixler EO, Chrousos GP. Sleep apnea is a manifestation of the metabolic syndrome. Sleep Med Rev. 2005;9:211–224. [PubMed: 15893251]
- 197.
- Vgontzas AN, Papanicolaou DA, Bixler EO, Hopper K, Lotsikas A, Lin HM, Kales A, Chrousos GP. Sleep apnea and daytime sleepiness and fatigue: relation to visceral obesity, insulin resistance, and hypercytokinemia. J Clin Endocrinol Metab. 2000;85:1151–1158. [PubMed: 10720054]
- 198.
- Grippo AJ, Johnson AK. Stress, depression and cardiovascular dysregulation: a review of neurobiological mechanisms and the integration of research from preclinical disease models. Stress. 2009;12:1–21. [PMC free article: PMC2613299] [PubMed: 19116888]
- 199.
- Dimsdale JE. Psychological stress and cardiovascular disease. J Am Coll Cardiol. 2008;51:1237–1246. [PMC free article: PMC2633295] [PubMed: 18371552]
- 200.
- Kyrou I, Kollia N, Panagiotakos D, Georgousopoulou E, Chrysohoou C, Tsigos C, Randeva HS, Yannakoulia M, Stefanadis C, Papageorgiou C, Pitsavos C., ATTICA Study investigators. Association of depression and anxiety status with 10-year cardiovascular disease incidence among apparently healthy Greek adults: The ATTICA Study. Eur J Prev Cardiol. 2017 Jan;24(2):145–152. [PubMed: 27671771]
- 201.
- Adam TC, Epel ES. Stress, eating and the reward system. Physiol Behav. 2007;91:449–458. [PubMed: 17543357]
- 202.
- Charmandari E, Chrousos GP, Lambrou GI, Pavlaki A, Koide H, Ng SS, Kino T. Peripheral CLOCK regulates target-tissue glucocorticoid receptor transcriptional activity in a circadian fashion in man. PLoS One. 2011;6:e25612. [PMC free article: PMC3182238] [PubMed: 21980503]
- 203.
- Pervanidou P, Chrousos GP. Metabolic consequences of stress during childhood and adolescence. Metabolism. 2011 [PubMed: 22146091]
- 204.
- Pervanidou P, Chrousos GP. Stress and obesity/metabolic syndrome in childhood and adolescence. Int J Pediatr Obes. 2011;6 Suppl 1:21–28. [PubMed: 21905812]
- 205.
- Pervanidou P. Biology of post-traumatic stress disorder in childhood and adolescence. J Neuroendocrinol. 2008;20:632–638. [PubMed: 18363804]
- 206.
- Noll JG, Zeller MH, Trickett PK, Putnam FW. Obesity risk for female victims of childhood sexual abuse: a prospective study. Pediatrics. 2007;120:e61–e67. [PubMed: 17606550]
- 207.
- Koupil I, Shestov DB, Sparen P, Plavinskaja S, Parfenova N, Vagero D. Blood pressure, hypertension and mortality from circulatory disease in men and women who survived the siege of Leningrad. Eur J Epidemiol. 2007;22:223–234. [PubMed: 17436055]
- 208.
- Chrousos GP. The stress response and immune function: clinical implications. The 1999 Novera H. Spector Lecture. Ann N Y Acad Sci. 2000;917:38–67. [PubMed: 11268364]
- 209.
- Charmandari E, Kino T, Chrousos GP. Glucocorticoids and their actions: an introduction. Ann N Y Acad Sci. 2004 Jun;1024:1–8. [PubMed: 15265770]
- 210.
- Flammer JR, Rogatsky I. Minireview: Glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol. 2011;25:1075–1086. [PMC free article: PMC5417249] [PubMed: 21511881]
- 211.
- Rogatsky I, Ivashkiv LB. Glucocorticoid modulation of cytokine signaling. Tissue Antigens. 2006;68:1–12. [PubMed: 16774534]
- 212.
- Ottaway CA, Husband AJ. Central nervous system influences on lymphocyte migration. Brain Behav Immun. 1992;6:97–116. [PubMed: 1504372]
- 213.
- Bellinger DL, Lorton D, Felten SY, Felten DL. Innervation of lymphoid organs and implications in development, aging, and autoimmunity. Int J Immunopharmacol. 1992;14:329–344. [PubMed: 1319962]
- 214.
- Besedovsky HO. del RA, Sorkin E, Da PM, Keller HH. Immunoregulation mediated by the sympathetic nervous system. Cell Immunol. 1979;48:346–355. [PubMed: 389444]
- 215.
- Downing JE, Miyan JA. Neural immunoregulation: emerging roles for nerves in immune homeostasis and disease. Immunol Today. 2000;21:281–289. [PubMed: 10825740]
- 216.
- Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52:595–638. [PubMed: 11121511]
- 217.
- Kohm AP, Sanders VM. Norepinephrine: a messenger from the brain to the immune system. Immunol Today. 2000;21:539–542. [PubMed: 11094255]
- 218.
- Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6:318–328. [PMC free article: PMC1783839] [PubMed: 16557263]
- 219.
- Hirano T, Akira S, Taga T, Kishimoto T. Biological and clinical aspects of interleukin 6. Immunol Today. 1990;11:443–449. [PubMed: 2127356]
- 220.
- Rohleder N. Acute and chronic stress induced changes in sensitivity of peripheral inflammatory pathways to the signals of multiple stress systems - 2011 Curt Richter Award Winner. Psychoneuroendocrinology. 2012 Mar;37(3):307–16. [PubMed: 22226321]
- 221.
- Elenkov IJ, Chrousos GP. Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann N Y Acad Sci. 2002;966:290–303. [PubMed: 12114286]
- 222.
- Duperrier K, Velten FW, Bohlender J, Demory A, Metharom P, Goerdt S. Immunosuppressive agents mediate reduced allostimulatory properties of myeloid-derived dendritic cells despite induction of divergent molecular phenotypes. Mol Immunol. 2005;42:1531–1540. [PubMed: 15950746]
- 223.
- Elenkov IJ, Chrousos GP. Stress system--organization, physiology and immunoregulation. Neuroimmunomodulation. 2006;13:257–267. [PubMed: 17709947]
- 224.
- Elenkov IJ, Chrousos GP. Stress Hormones, Th1/Th2 patterns, Pro/Anti-inflammatory Cytokines and Susceptibility to Disease. Trends Endocrinol Metab. 1999;10:359–368. [PubMed: 10511695]
- 225.
- Luther C, Adamopoulou E, Stoeckle C, Brucklacher-Waldert V, Rosenkranz D, Stoltze L, Lauer S, Poeschel S, Melms A, Tolosa E. Prednisolone treatment induces tolerogenic dendritic cells and a regulatory milieu in myasthenia gravis patients. J Immunol. 2009;183:841–848. [PubMed: 19542375]
- 226.
- Harpaz I, Abutbul S, Nemirovsky A, Gal R, Cohen H, Monsonego A. Chronic exposure to stress predisposes to higher autoimmune susceptibility in C57BL/6 mice: glucocorticoids as a double-edged sword. Eur J Immunol. 2013 Mar;43(3):758–69. [PubMed: 23255172]
- 227.
- Liu J, Malkani G, Shi X, Meyer M, Cunningham-Runddles S, Ma X, Sun ZS. The circadian clock Period 2 gene regulates gamma interferon production of NK cells in host response to lipopolysaccharide-induced endotoxic shock. Infect Immun. 2006;74:4750–4756. [PMC free article: PMC1539582] [PubMed: 16861663]
- 228.
- Arjona A, Sarkar DK. Are circadian rhythms the code of hypothalamic-immune communication? Insights from natural killer cells. Neurochem Res. 2008;33:708–718. [PubMed: 17965936]
- 229.
- Reichlin S. Neuroendocrine-immune interactions. N Engl J Med. 1993;329:1246–1253. [PubMed: 8105378]
- 230.
- Akira S, Hirano T, Taga T, Kishimoto T. Biology of multifunctional cytokines: IL 6 and related molecules (IL 1 and TNF). FASEB J. 1990;4:2860–2867. [PubMed: 2199284]
- 231.
- Besedovsky HO, del Rey A. Immune-neuroendocrine circuits: integrative role of cytokines. Front Neuroendocrinol. 1992;13:61–94. [PubMed: 1468599]
- 232.
- Bateman A, Singh A, Kral T, Solomon S. The immune-hypothalamic-pituitary-adrenal axis. Endocr Rev. 1989;10:92–112. [PubMed: 2666113]
- 233.
- Bernardini R, Kamilaris TC, Calogero AE, Johnson EO, Gomez MT, Gold PW, Chrousos GP. Interactions between tumor necrosis factor-alpha, hypothalamic corticotropin-releasing hormone, and adrenocorticotropin secretion in the rat. Endocrinology. 1990;126:2876–2881. [PubMed: 2161738]
- 234.
- Busbridge NJ, Grossman AB. Stress and the single cytokine: interleukin modulation of the pituitary-adrenal axis. Mol Cell Endocrinol. 1991;82:C209–C214. [PubMed: 1794601]
- 235.
- Sapolsky R, Rivier C, Yamamoto G, Plotsky P, Vale W. Interleukin-1 stimulates the secretion of hypothalamic corticotropin-releasing factor. Science. 1987;238:522–524. [PubMed: 2821621]
- 236.
- Hillhouse EW. Interleukin-2 stimulates the secretion of arginine vasopressin but not corticotropin-releasing hormone from rat hypothalamic cells in vitro. Brain Res. 1994;650:323–325. [PubMed: 7953699]
- 237.
- Cambronero JC, Rivas FJ, Borrell J, Guaza C. Interleukin-2 induces corticotropin-releasing hormone release from superfused rat hypothalami: influence of glucocorticoids. Endocrinology. 1992;131:677–683. [PubMed: 1639014]
- 238.
- Arzt E, Stelzer G, Renner U, Lange M, Muller OA, Stalla GK. Interleukin-2 and interleukin-2 receptor expression in human corticotrophic adenoma and murine pituitary cell cultures. J Clin Invest. 1992;90:1944–1951. [PMC free article: PMC443256] [PubMed: 1331177]
- 239.
- Chesnokova V, Auernhammer CJ, Melmed S. Murine leukemia inhibitory factor gene disruption attenuates the hypothalamo-pituitary-adrenal axis stress response. Endocrinology. 1998;139:2209–2216. [PubMed: 9564824]
- 240.
- Auernhammer CJ, Chesnokova V, Bousquet C, Melmed S. Pituitary corticotroph SOCS-3: novel intracellular regulation of leukemia-inhibitory factor-mediated proopiomelanocortin gene expression and adrenocorticotropin secretion. Mol Endocrinol. 1998;12:954–961. [PubMed: 9658400]
- 241.
- Rivier CL, Grigoriadis DE, Rivier JE. Role of corticotropin-releasing factor receptors type 1 and 2 in modulating the rat adrenocorticotropin response to stressors. Endocrinology. 2003;144:2396–2403. [PubMed: 12746300]
- 242.
- Bethin KE, Vogt SK, Muglia LJ. Interleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activation. Proc Natl Acad Sci U S A. 2000;97:9317–9322. [PMC free article: PMC16865] [PubMed: 10922080]
- 243.
- Chesnokova V, Kariagina A, Melmed S. Opposing effects of pituitary leukemia inhibitory factor and SOCS-3 on the ACTH axis response to inflammation. Am J Physiol Endocrinol Metab. 2002;282:E1110–E1118. [PubMed: 11934677]
- 244.
- Chesnokova V, Melmed S. Leukemia inhibitory factor mediates the hypothalamic pituitary adrenal axis response to inflammation. Endocrinology. 2000;141:4032–4040. [PubMed: 11089533]
- 245.
- Kariagina A, Romanenko D, Ren SG, Chesnokova V. Hypothalamic-pituitary cytokine network. Endocrinology. 2004;145:104–112. [PubMed: 14512435]
- 246.
- Mastorakos G, Chrousos GP, Weber JS. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J Clin Endocrinol Metab. 1993 Dec;77(6):1690–4. [PubMed: 8263159]
- 247.
- Mastorakos G, Weber JS, Magiakou MA, Gunn H, Chrousos GP. Hypothalamic-pituitary-adrenal axis activation and stimulation of systemic vasopressin secretion by recombinant interleukin-6 in humans: potential implications for the syndrome of inappropriate vasopressin secretion. J Clin Endocrinol Metab. 1994;79:934–939. [PubMed: 7962300]
- 248.
- Reincke M, Heppner C, Petzke F, Allolio B, Arlt W, Mbulamberi D, Siekmann L, Vollmer D, Winkelmann W, Chrousos GP. Impairment of adrenocortical function associated with increased plasma tumor necrosis factor-alpha and interleukin-6 concentrations in African trypanosomiasis. Neuroimmunomodulation. 1994;1:14–22. [PubMed: 8528879]
- 249.
- Bernardini R, Calogero AE, Ehrlich YH, Brucke T, Chrousos GP, Gold PW. The alkyl-ether phospholipid platelet-activating factor is a stimulator of the hypothalamic-pituitary-adrenal axis in the rat. Endocrinology. 1989;125:1067–1073. [PubMed: 2546734]
- 250.
- Bernardini R, Chiarenza A, Calogero AE, Gold PW, Chrousos GP. Arachidonic acid metabolites modulate rat hypothalamic corticotropin-releasing hormone secretion in vitro. Neuroendocrinology. 1989;50:708–715. [PubMed: 2559344]
- 251.
- Bernton EW, Beach JE, Holaday JW, Smallridge RC, Fein HG. Release of multiple hormones by a direct action of interleukin-1 on pituitary cells. Science. 1987;238:519–521. [PubMed: 2821620]
- 252.
- Fukata J, Usui T, Naitoh Y, Nakai Y, Imura H. Effects of recombinant human interleukin-1 alpha, -1 beta, 2 and 6 on ACTH synthesis and release in the mouse pituitary tumour cell line AtT-20. J Endocrinol. 1989;122:33–39. [PubMed: 2549151]
- 253.
- Salas MA, Evans SW, Levell MJ, Whicher JT. Interleukin-6 and ACTH act synergistically to stimulate the release of corticosterone from adrenal gland cells. Clin Exp Immunol. 1990;79:470–473. [PMC free article: PMC1534960] [PubMed: 2156641]
- 254.
- Gadek-Michalska A, Bugajski J. Role of prostaglandins and nitric oxide in the lipopolysaccharide-induced ACTH and corticosterone response. J Physiol Pharmacol. 2004;55:663–675. [PubMed: 15381835]
- 255.
- Monau TR, Vargas VE, Zhang L, Myers DA, Ducsay CA. Nitric oxide inhibits ACTH-induced cortisol production in near-term, long-term hypoxic ovine fetal adrenocortical cells. Reprod Sci. 2010;17:955–962. [PMC free article: PMC2943550] [PubMed: 20713972]
- 256.
- Elander L, Ruud J, Korotkova M, Jakobsson PJ, Blomqvist A. Cyclooxygenase-1 mediates the immediate corticosterone response to peripheral immune challenge induced by lipopolysaccharide. Neurosci Lett. 2010;470:10–12. [PubMed: 20034541]
- 257.
- Karalis K, Sano H, Redwine J, Listwak S, Wilder RL, Chrousos GP. Autocrine or paracrine inflammatory actions of corticotropin-releasing hormone in vivo. Science. 1991;254:421–423. [PubMed: 1925600]
- 258.
- Crofford LJ, Sano H, Karalis K, Friedman TC, Epps HR, Remmers EF, Mathern P, Chrousos GP, Wilder RL. Corticotropin-releasing hormone in synovial fluids and tissues of patients with rheumatoid arthritis and osteoarthritis. J Immunol. 1993;151:1587–1596. [PubMed: 8335947]
- 259.
- Webster EL, Torpy DJ, Elenkov IJ, Chrousos GP. Corticotropin-releasing hormone and inflammation. Ann N Y Acad Sci. 1998;840:21–32. [PubMed: 9629233]
- 260.
- Elenkov IJ, Webster EL, Torpy DJ, Chrousos GP. Stress, corticotropin-releasing hormone, glucocorticoids, and the immune/inflammatory response: acute and chronic effects. Ann N Y Acad Sci. 1999;876:1–11. [PubMed: 10415589]
- 261.
- Arafah BM. Hypothalamic pituitary adrenal function during critical illness: limitations of current assessment methods. J Clin Endocrinol Metab. 2006;91:3725–3745. [PubMed: 16882746]
- 262.
- Vermes I, Beishuizen A, Hampsink RM, Haanen C. Dissociation of plasma adrenocorticotropin and cortisol levels in critically ill patients: possible role of endothelin and atrial natriuretic hormone. J Clin Endocrinol Metab. 1995;80:1238–1242. [PubMed: 7714094]
- 263.
- Marik PE. Critical illness-related corticosteroid insufficiency. Chest. 2009;135:181–193. [PubMed: 19136406]
- 264.
- Bornstein SR, Engeland WC, Ehrhart-Bornstein M, Herman JP. Dissociation of ACTH and glucocorticoids. Trends Endocrinol Metab. 2008 Jul;19(5):175–80. [PubMed: 18394919]
- 265.
- Bornstein SR. Predisposing factors for adrenal insufficiency. N Engl J Med. 2009;360:2328–2339. [PubMed: 19474430]
- 266.
- Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. 2008;93:1563–1572. [PMC free article: PMC2386273] [PubMed: 18319312]
- 267.
- Labanski A, Langhorst J, Engler H, Elsenbruch S. Stress and the brain-gut axis in functional and chronic-inflammatory gastrointestinal diseases: A transdisciplinary challenge. Psychoneuroendocrinology. 2020;111:104501. [PubMed: 31715444]
- 268.
- Tache Y, Martinez V, Million M, Wang L. Stress and the gastrointestinal tract III. Stress-related alterations of gut motor function: role of brain corticotropin-releasing factor receptors. Am J Physiol Gastrointest Liver Physiol. 2001;280:G173–G177. [PubMed: 11208537]
- 269.
- Collins SM. Stress and the Gastrointestinal Tract IV. Modulation of intestinal inflammation by stress: basic mechanisms and clinical relevance. Am J Physiol Gastrointest Liver Physiol. 2001;280:G315–G318. [PubMed: 11171612]
- 270.
- Scarinci IC, McDonald-Haile J, Bradley LA, Richter JE. Altered pain perception and psychosocial features among women with gastrointestinal disorders and history of abuse: a preliminary model. Am J Med. 1994 Aug;97(2):108–18. [PubMed: 8059776]
- 271.
- De Bellis MD, Chrousos GP, Dorn LD, Burke L, Helmers K, Kling MA, Trickett PK, Putnam FW. Hypothalamic-pituitary-adrenal axis dysregulation in sexually abused girls. J Clin Endocrinol Metab. 1994 Feb;78(2):249–55. [PubMed: 8106608]
- 272.
- Drossman DA. Physical and sexual abuse and gastrointestinal illness: what is the link? Am J Med. 1994 Aug;97(2):105–7. [PubMed: 8059775]
- 273.
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–57. [PubMed: 14744257]
- 274.
- Fukudo S. Role of corticotropin-releasing hormone in irritable bowel syndrome and intestinal inflammation. J Gastroenterol. 2007;42 Suppl 17:48–51. [PubMed: 17238026]
- 275.
- Stratakis CA, Chrousos GP. Neuroendocrinology and pathophysiology of the stress system. Ann N Y Acad Sci. 1995 Dec 29;771:1–18. [PubMed: 8597390]
- 276.
- Valentino RJ, Kosboth M, Colflesh M, Miselis RR. Transneuronal labeling from the rat distal colon: anatomic evidence for regulation of distal colon function by a pontine corticotropin-releasing factor system. J Comp Neurol. 2000;417:399–414. [PubMed: 10701863]
- 277.
- Monnikes H, Schmidt BG, Tebbe J, Bauer C, Tache Y. Microinfusion of corticotropin releasing factor into the locus coeruleus/subcoeruleus nuclei stimulates colonic motor function in rats. Brain Res. 1994;644:101–108. [PubMed: 8032938]
- 278.
- Suto G, Kiraly A, Tache Y. Interleukin 1 beta inhibits gastric emptying in rats: mediation through prostaglandin and corticotropin-releasing factor. Gastroenterology. 1994;106:1568–1575. [PubMed: 8194703]
- 279.
- Kanazawa M, Hongo M, Fukudo S. Visceral hypersensitivity in irritable bowel syndrome. J Gastroenterol Hepatol. 2011;26 Suppl 3:119–121. [PubMed: 21443723]
- 280.
- Nakade Y, Fukuda H, Iwa M, Tsukamoto K, Yanagi H, Yamamura T, Mantyh C, Pappas TN, Takahashi T. Restraint stress stimulates colonic motility via central corticotropin-releasing factor and peripheral 5-HT3 receptors in conscious rats. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1037–G1044. [PubMed: 17158256]
- 281.
- Masere C, Nakade Y, Zheng J, Babygirija R, Ludwig K, Takahashi T. Chronic restraint stress has no more stimulatory effects on colonic motility in rats. Neurosci Lett. 2009;453:147–150. [PubMed: 19429023]
- 282.
- Zheng J, Babygirija R, Bulbul M, Cerjak D, Ludwig K, Takahashi T. Hypothalamic oxytocin mediates adaptation mechanism against chronic stress in rats. Am J Physiol Gastrointest Liver Physiol. 2010;299:G946–G953. [PMC free article: PMC2957337] [PubMed: 20689056]
- 283.
- Stengel A, Wang L, Tache Y. Stress-related alterations of acyl and desacyl ghrelin circulating levels: mechanisms and functional implications. Peptides. 2011;32:2208–2217. [PMC free article: PMC3220774] [PubMed: 21782868]
- 284.
- Petrella C, Agostini S, Guerrini R, Calo G, Giaquinto A, De NC, Improta G, Broccardo M. Neuropeptide S inhibits stress-stimulated faecal output in the rat. Pharmacol Res. 2011;64:471–477. [PubMed: 21708257]
- 285.
- Selye H. A syndrome produced by diverse nocuous agents. J. Neuropsychiatry Clin. Neurosci. 1936;138:230–231. [PubMed: 9722327]
- 286.
- Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (2). N Engl J Med. 1988;319:413–420. [PubMed: 3041279]
- 287.
- Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (1). N Engl J Med. 1988;319:348–353. [PubMed: 3292920]
- 288.
- Gold PW, Chrousos GP. Organization of the stress system and its dysregulation in melancholic and atypical depression: high vs low CRH/NE states. Mol Psychiatry. 2002;7:254–275. [PubMed: 11920153]
- 289.
- Tsigos C, Chrousos GP. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res. 2002;53:865–871. [PubMed: 12377295]
- 290.
- Raadsheer FC, Hoogendijk WJ, Stam FC, Tilders FJ, Swaab DF. Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology. 1994;60:436–444. [PubMed: 7824085]
- 291.
- Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A. 1996;93:3908–3913. [PMC free article: PMC39458] [PubMed: 8632988]
- 292.
- Drevets WC, Price JL, Simpson JR Jr, Todd RD, Reich T, Vannier M, Raichle ME. Subgenual prefrontal cortex abnormalities in mood disorders. Nature. 1997;386:824–827. [PubMed: 9126739]
- 293.
- Gold PW, Pigott TA, Kling MA, Kalogeras K, Chrousos GP. Basic and clinical studies with corticotropin-releasing hormone. Implications for a possible role in panic disorder. Psychiatr Clin North Am. 1988;11:327–334. [PubMed: 3047702]
- 294.
- Insel TR, Kalin NH, Guttmacher LB, Cohen RM, Murphy DL. The dexamethasone suppression test in patients with primary obsessive-compulsive disorder. Psychiatry Res. 1982;6:153–160. [PubMed: 6953457]
- 295.
- De Bellis MD, Chrousos GP, Dorn LD, Burke L, Helmers K, Kling MA, Trickett PK, Putnam FW. Hypothalamic-pituitary-adrenal axis dysregulation in sexually abused girls. J Clin Endocrinol Metab. 1994;78:249–255. [PubMed: 8106608]
- 296.
- Wand GS, Dobs AS. Alterations in the hypothalamic-pituitary-adrenal axis in actively drinking alcoholics. J Clin Endocrinol Metab. 1991;72:1290–1295. [PubMed: 2026749]
- 297.
- von BU. Heuser I, Holsboer F. Human CRH stimulation response during acute withdrawal and after medium-term abstention from alcohol abuse. Psychoneuroendocrinology. 1989;14:441–449. [PubMed: 2560222]
- 298.
- Risher-Flowers D, Adinoff B, Ravitz B, Bone GH, Martin PR, Nutt D, Linnoila M. Circadian rhythms of cortisol during alcohol withdrawal. Adv Alcohol Subst Abuse. 1988;7:37–41. [PubMed: 3223434]
- 299.
- Gold PW, Gwirtsman H, Avgerinos PC, Nieman LK, Gallucci WT, Kaye W, Jimerson D, Ebert M, Rittmaster R, Loriaux DL, et al. Abnormal hypothalamic-pituitary-adrenal function in anorexia nervosa. Pathophysiologic mechanisms in underweight and weight-corrected patients. N Engl J Med. 1986;314:1335–1342. [PubMed: 3010109]
- 300.
- Kaye WH, Gwirtsman HE, George DT, Ebert MH, Jimerson DC, Tomai TP, Chrousos GP, Gold PW. Elevated cerebrospinal fluid levels of immunoreactive corticotropin-releasing hormone in anorexia nervosa: relation to state of nutrition, adrenal function, and intensity of depression. J Clin Endocrinol Metab. 1987;64:203–208. [PubMed: 3491830]
- 301.
- Streeten DH. Is hypothalamic-pituitary-adrenal hyperactivity important in the pathogenesis of excessive abdominal fat distribution? J Clin Endocrinol Metab. 1993;77:339–340. [PubMed: 8345038]
- 302.
- Roy MS, Roy A, Gallucci WT, Collier B, Young K, Kamilaris TC, Chrousos GP. The ovine corticotropin-releasing hormone-stimulation test in type I diabetic patients and controls: suggestion of mild chronic hypercortisolism. Metabolism. 1993;42:696–700. [PubMed: 8389960]
- 303.
- Tsigos C, Young RJ, White A. Diabetic neuropathy is associated with increased activity of the hypothalamic-pituitary-adrenal axis. J Clin Endocrinol Metab. 1993;76:554–558. [PubMed: 8383141]
- 304.
- Demitrack MA, Dale JK, Straus SE, Laue L, Listwak SJ, Kruesi MJ, Chrousos GP, Gold PW. Evidence for impaired activation of the hypothalamic-pituitary-adrenal axis in patients with chronic fatigue syndrome. J Clin Endocrinol Metab. 1991;73:1224–1234. [PubMed: 1659582]
- 305.
- Joseph-Vanderpool JR, Rosenthal NE, Chrousos GP, Wehr TA, Skwerer R, Kasper S, Gold PW. Abnormal pituitary-adrenal responses to corticotropin-releasing hormone in patients with seasonal affective disorder: clinical and pathophysiological implications. J Clin Endocrinol Metab. 1991;72:1382–1387. [PubMed: 1851185]
- 306.
- Griep EN, Boersma JW, de Kloet ER. Altered reactivity of the hypothalamic-pituitary-adrenal axis in the primary fibromyalgia syndrome. J Rheumatol. 1993;20:469–474. [PubMed: 8386766]
- 307.
- Kamilaris TC, DeBold CR, Pavlou SN, Island DP, Hoursanidis A, Orth DN. Effect of altered thyroid hormone levels on hypothalamic-pituitary-adrenal function. J Clin Endocrinol Metab. 1987;65:994–999. [PubMed: 3499449]
- 308.
- Puddey IB, Vandongen R, Beilin LJ, English D. Haemodynamic and neuroendocrine consequences of stopping smoking--a controlled study. Clin Exp Pharmacol Physiol. 1984;11:423–426. [PubMed: 6394183]
- 309.
- Elgerot A. Psychological and physiological changes during tobacco-abstinence in habitual smokers. J Clin Psychol. 1978;34:759–764. [PubMed: 690223]
- 310.
- Doherty GM, Nieman LK, Cutler GB Jr, Chrousos GP, Norton JA. Time to recovery of the hypothalamic-pituitary-adrenal axis after curative resection of adrenal tumors in patients with Cushing's syndrome. Surgery. 1990;108:1085–1090. [PubMed: 2247834]
- 311.
- Gomez MT, Magiakou MA, Mastorakos G, Chrousos GP. The pituitary corticotroph is not the rate limiting step in the postoperative recovery of the hypothalamic-pituitary-adrenal axis in patients with Cushing syndrome. J Clin Endocrinol Metab. 1993;77:173–177. [PubMed: 8392083]
- 312.
- Sternberg EM, Glowa JR, Smith MA, Calogero AE, Listwak SJ, Aksentijevich S, Chrousos GP, Wilder RL, Gold PW. Corticotropin releasing hormone related behavioral and neuroendocrine responses to stress in Lewis and Fischer rats. Brain Res. 1992;570:54–60. [PubMed: 1319794]
- 313.
- Sternberg EM, Chrousos GP, Wilder RL, Gold PW. The stress response and the regulation of inflammatory disease. Ann Intern Med. 1992;117:854–866. [PubMed: 1416562]
- 314.
- Sternberg EM, Hill JM, Chrousos GP, Kamilaris T, Listwak SJ, Gold PW, Wilder RL. Inflammatory mediator-induced hypothalamic-pituitary-adrenal axis activation is defective in streptococcal cell wall arthritis-susceptible Lewis rats. Proc Natl Acad Sci U S A. 1989;86:2374–2378. [PMC free article: PMC286915] [PubMed: 2538840]
- 315.
- Sternberg EM, Young WS III, Bernardini R, Calogero AE, Chrousos GP, Gold PW, Wilder RL. A central nervous system defect in biosynthesis of corticotropin-releasing hormone is associated with susceptibility to streptococcal cell wall-induced arthritis in Lewis rats. Proc Natl Acad Sci U S A. 1989;86:4771–4775. [PMC free article: PMC287355] [PubMed: 2786636]
- 316.
- Chikanza IC, Petrou P, Kingsley G, Chrousos G, Panayi GS. Defective hypothalamic response to immune and inflammatory stimuli in patients with rheumatoid arthritis. Arthritis Rheum. 1992;35:1281–1288. [PubMed: 1445443]
- 317.
- Chikanza IC, Chrousos G, Panayi GS. Abnormal neuroendocrine immune communications in patients with rheumatoid arthritis. Eur J Clin Invest. 1992;22:635–637. [PubMed: 1459167]
- 318.
- Zoumakis E, Chrousos GP. Corticotropin-releasing hormone receptor antagonists: an update. Endocr Dev. 2010;17:36–43. [PubMed: 19955754]
- 319.
- Habib KE, Weld KP, Rice KC, Pushkas J, Champoux M, Listwak S, Webster EL, Atkinson AJ, Schulkin J, Contoreggi C, Chrousos GP, McCann SM, Suomi SJ, Higley JD, Gold PW. Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc Natl Acad Sci U S A. 2000;97:6079–6084. [PMC free article: PMC18561] [PubMed: 10823952]
- 320.
- Webster EL, Lewis DB, Torpy DJ, Zachman EK, Rice KC, Chrousos GP. In vivo and in vitro characterization of antalarmin, a nonpeptide corticotropin-releasing hormone (CRH) receptor antagonist: suppression of pituitary ACTH release and peripheral inflammation. Endocrinology. 1996;137:5747–5750. [PubMed: 8940412]
- 321.
- Bornstein SR, Webster EL, Torpy DJ, Richman SJ, Mitsiades N, Igel M, Lewis DB, Rice KC, Joost HG, Tsokos M, Chrousos GP. Chronic effects of a nonpeptide corticotropin-releasing hormone type I receptor antagonist on pituitary-adrenal function, body weight, and metabolic regulation. Endocrinology. 1998;139:1546–1555. [PubMed: 9528933]
- 322.
- Deak T, Nguyen KT, Ehrlich AL, Watkins LR, Spencer RL, Maier SF, Licinio J, Wong ML, Chrousos GP, Webster E, Gold PW. The impact of the nonpeptide corticotropin-releasing hormone antagonist antalarmin on behavioral and endocrine responses to stress. Endocrinology. 1999;140:79–86. [PubMed: 9886810]
- 323.
- Webster EL, Barrientos RM, Contoreggi C, Isaac MG, Ligier S, Gabry KE, Chrousos GP, McCarthy EF, Rice KC, Gold PW, Sternberg EM. Corticotropin releasing hormone (CRH) antagonist attenuates adjuvant induced arthritis: role of CRH in peripheral inflammation. J Rheumatol. 2002;29:1252–1261. [PubMed: 12064844]
- 324.
- Gabry KE, Chrousos GP, Rice KC, Mostafa RM, Sternberg E, Negrao AB, Webster EL, McCann SM, Gold PW. Marked suppression of gastric ulcerogenesis and intestinal responses to stress by a novel class of drugs. Mol Psychiatry 2002;7:474-83, 433. [PubMed: 12082565]
- 325.
- Wong ML, Webster EL, Spokes H, Phu P, Ehrhart-Bornstein M, Bornstein S, Park CS, Rice KC, Chrousos GP, Licinio J, Gold PW. Chronic administration of the non-peptide CRH type 1 receptor antagonist antalarmin does not blunt hypothalamic-pituitary-adrenal axis responses to acute immobilization stress. Life Sci. 1999;65:L53–L58. [PubMed: 10421433]
- 326.
- Ising M, Holsboer F. CRH-sub-1 receptor antagonists for the treatment of depression and anxiety. Exp Clin Psychopharmacol. 2007;15:519–528. [PubMed: 18179304]
- 327.
- Lawrence AJ, Krstew EV, Dautzenberg FM, Ruhmann A. The highly selective CRF(2) receptor antagonist K41498 binds to presynaptic CRF(2) receptors in rat brain. Br J Pharmacol. 2002;136:896–904. [PMC free article: PMC1573413] [PubMed: 12110614]
- 328.
- Sajdyk TJ, Gehlert DR. Astressin, a corticotropin releasing factor antagonist, reverses the anxiogenic effects of urocortin when administered into the basolateral amygdala. Brain Res. 2000;877:226–234. [PubMed: 10986336]
- 329.
- Henckens MJ, Deussing JM, Chen A. Region-specific roles of the corticotropin-releasing factor-urocortin system in stress. Nat Rev Neurosci. 2016 Oct;17(10):636–51. [PubMed: 27586075]
- 330.
- Pan W, Kastin AJ. Urocortin and the brain. Prog Neurobiol. 2008;84:148–156. [PMC free article: PMC2267723] [PubMed: 18078706]
- 331.
- Huang Y, Yao XQ, Lau CW, Chan YC, Tsang SY, Chan FL. Urocortin and cardiovascular protection. Acta Pharmacol Sin. 2004 Mar;25(3):257–65. [PubMed: 15000874]
- 332.
- Abuirmeileh A, Harkavyi A, Lever R, Biggs CS, Whitton PS. Urocortin, a CRF-like peptide, restores key indicators of damage in the substantia nigra in a neuroinflammatory model of Parkinson's disease. J Neuroinflammation. 2007;4:19. [PMC free article: PMC1976313] [PubMed: 17659087]
- 333.
- Liew HK, Pang CY, Hsu CW, Wang MJ, Li TY, Peng HF, Kuo JS, Wang JY. Systemic administration of urocortin after intracerebral hemorrhage reduces neurological deficits and neuroinflammation in rats. J Neuroinflammation. 2012;9:13. [PMC free article: PMC3271957] [PubMed: 22257737]
- 334.
- Monteiro-Pinto C, Adão R, Leite-Moreira AF, Brás-Silva C. Cardiovascular Effects of Urocortin-2. Pathophysiological Mechanisms and Therapeutic Potential. Cardiovasc Drugs Ther. 2019;33(5):599–613. [PubMed: 31512017]
- 335.
- Mackay KB, Stiefel TH, Ling N, Foster AC. Effects of a selective agonist and antagonist of CRF2 receptors on cardiovascular function in the rat. Eur J Pharmacol. 2003;469:111–115. [PubMed: 12782192]
- 336.
- Basman C, Agrawal P, Knight R, et al. Cardioprotective Utility of Urocortin in Myocardial Ischemia- Reperfusion Injury: Where do We Stand?. Curr Mol Pharmacol. 2018;11(1):32–38. [PubMed: 28228090]
- 337.
- Kim BJ, Kayembe K, Simecka JW, Pulse M, Jones HP. Corticotropin-releasing hormone receptor-1 and 2 activity produces divergent resistance against stress-induced pulmonary Streptococcus pneumoniae infection. J Neuroimmunol. 2011;237:57–65. [PMC free article: PMC5715473] [PubMed: 21774994]
- Review Stress, visceral obesity, and metabolic complications.[Ann N Y Acad Sci. 2006]Review Stress, visceral obesity, and metabolic complications.Kyrou I, Chrousos GP, Tsigos C. Ann N Y Acad Sci. 2006 Nov; 1083:77-110.
- Review Immune System Effects on the Endocrine System.[Endotext. 2000]Review Immune System Effects on the Endocrine System.Yavropoulou MP, Sfikakis PP, Chrousos GP. Endotext. 2000
- Review Stress mechanisms and metabolic complications.[Horm Metab Res. 2007]Review Stress mechanisms and metabolic complications.Kyrou I, Tsigos C. Horm Metab Res. 2007 Jun; 39(6):430-8.
- Review Stress and disorders of the stress system.[Nat Rev Endocrinol. 2009]Review Stress and disorders of the stress system.Chrousos GP. Nat Rev Endocrinol. 2009 Jul; 5(7):374-81. Epub 2009 Jun 2.
- Review AIDS and HPA Axis.[Endotext. 2000]Review AIDS and HPA Axis.Nicolaides NC, Kino T, Chrousos G. Endotext. 2000
- Stress: Endocrine Physiology and Pathophysiology - EndotextStress: Endocrine Physiology and Pathophysiology - Endotext
- Mir7677 microRNA 7677 [Mus musculus]Mir7677 microRNA 7677 [Mus musculus]Gene ID:102465779Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...