

dbVar Help & FAQ
- Introduction
- dbVar Study Browser
- dbVar Study Page
- dbVar Variant Page
- Variant Call and Region Types
- dbVar Variant Rendering
- dbVar Placements
- FTP Site
- dbVar Entrez Search
- FAQ
Last updated June 2020
Introduction
dbVar is a database of human genomic structural variation where users can search, view, and download data from submitted studies. dbVar stopped supporting data from non-human organisms on November 1, 2017; however existing non-human data remains available via FTP download. In keeping with the common definition of structural variation, most variants are larger than 50 basepairs in length - however a handful of smaller variants may also be found. dbVar provides access to the raw data whenever available, as well as links to additional resources, from both NCBI and elsewhere. For more information on structural variation see the Overview of Structural Variation page. For frequently asked questions see the dbVar Help & FAQ page.
dbVar is a free resource that is developed and maintained by the National Center for Biotechnology Information (NCBI) at the U.S. National Library of Medicine (NLM), located at the National Institutes of Health (NIH) in Bethesda, Maryland.
dbVar Study Browser
Data in dbVar is organized by Study. "Study" typically refers to a publication, however some Studies are community resources which are updated with new data on a regular basis (e.g., Clinical Structural Variants - ClinVar, nstd102). Each Study is given an accession that begins with -std: nstd if the study was submitted to NCBI, estd if submitted to EBI, and dstd if submitted to DDBJ. The dbVar Study Browser (Figure 1) provides a summary of the studies in dbVar. The browser can be sorted by Study accession, Organism, Study Type, Method, Number of Variant Regions, Number of Variant Calls, or Publication. Links to the Study Page and PubMed summary for each study are provided. Filters to the right of the browser allow you to narrow content by Study Type, Method, or Number of Variant Regions.
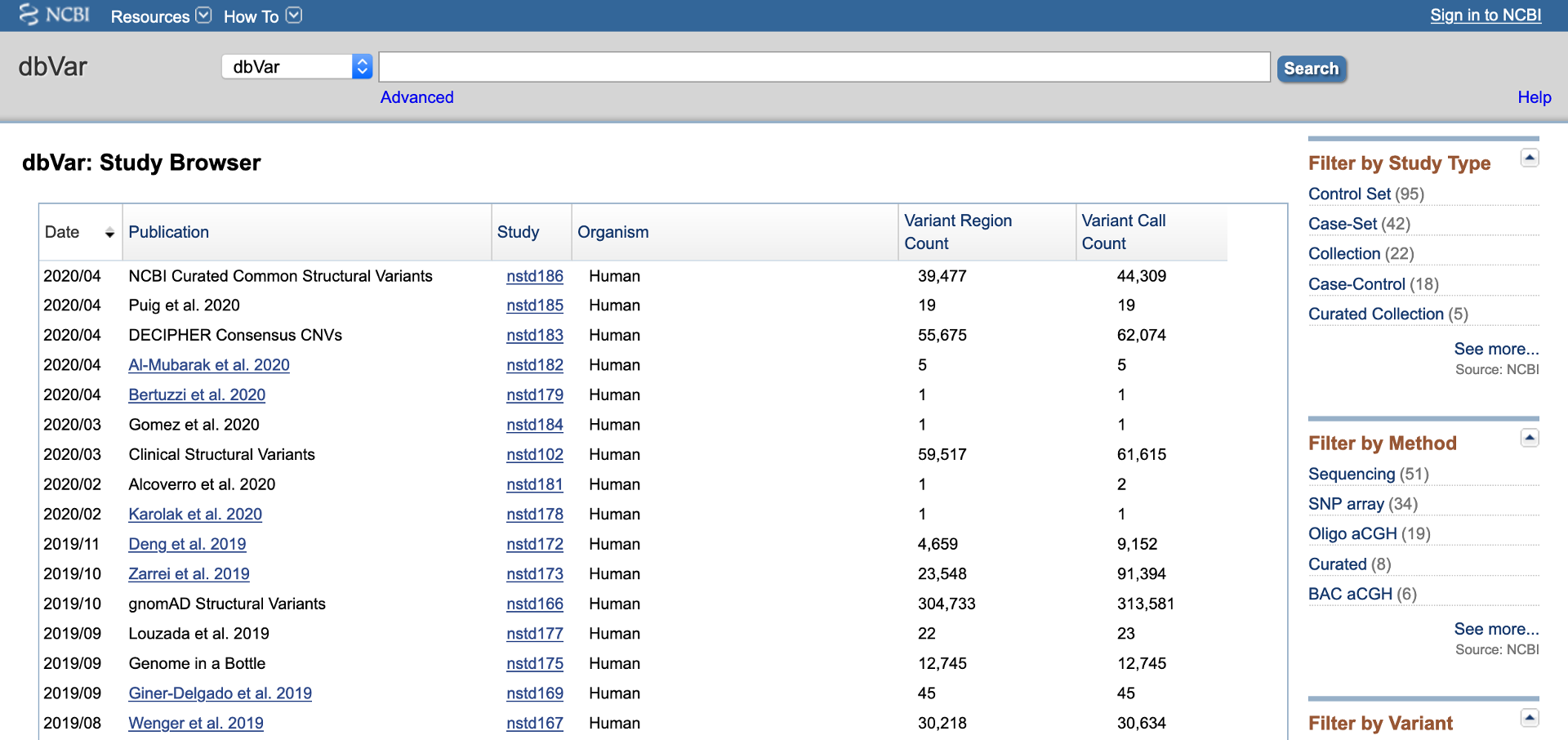
Figure 1: dbVar Study Browser
dbVar Study Page
Each Study in dbVar has a Study Page that displays basic information about the study. At the top is a general information section describing basic information about the study, including links to BioProjects, PubMed, dbGaP, and the submitter's lab page (if available). This is followed by a Detailed Information section where you can download variant data for the current study (Variant Regions, Variant Calls, Both, or all via FTP) or browse details about Variant Summary, Samplesets, Experimental Details, or Validations in a tabbed format.
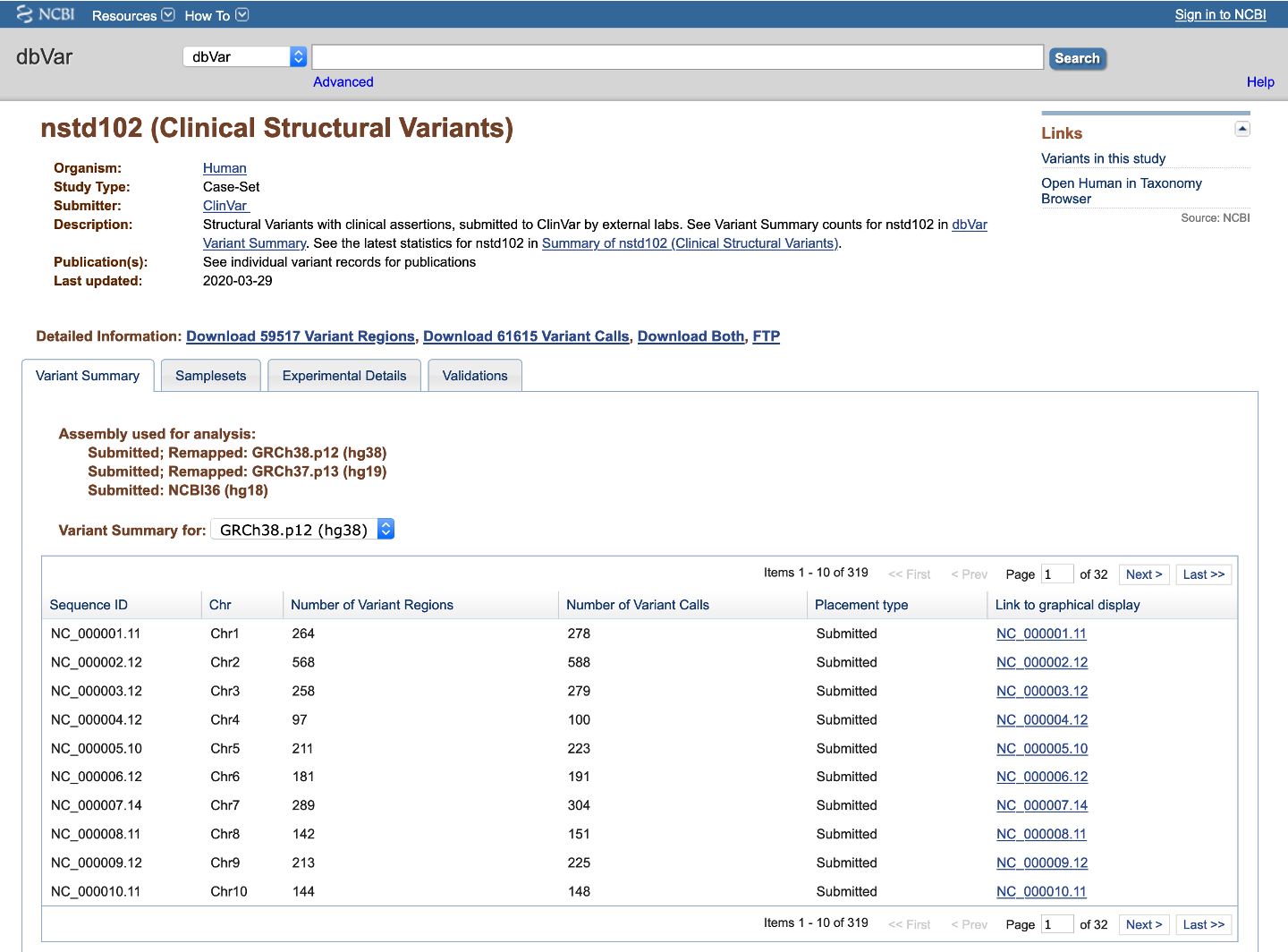
Figure 2a: General Information and Variant Summary Tab (Study Page) - The Variant Summary tab displays the number of Variant Calls and Variant Regions for the current study on each chromosome, Placement type (i.e., whether data is available on the Submitted assembly and/or Remapped assemblies), and links to a graphical display of the variants on NCBI's Sequence Viewer. A pull-down menu above the table allows you to display the current study's data on the submitted assembly, or on any assemblies to which the data has been remapped.
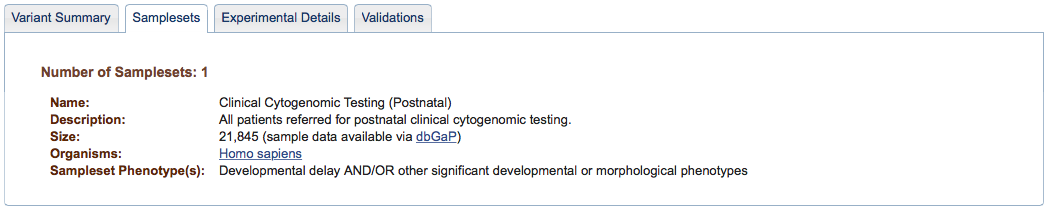
Figure 2b: Samplesets Tab (Study Page) - The Samplesets tab describes any logical division of samples in the study. Details include Name, Description, Size, and relevant Phenotypes, if any. Note that additional details about individual subjects in a sampleset can be found by downloading the Samples data. If subjects have not been consented to have their data displayed publicly (as in the example above), clinical information and links between genetic variants are stored behind controlled access at NCBI's dbGaP. Samples and variants are then anonymized before being forwarded to dbVar.
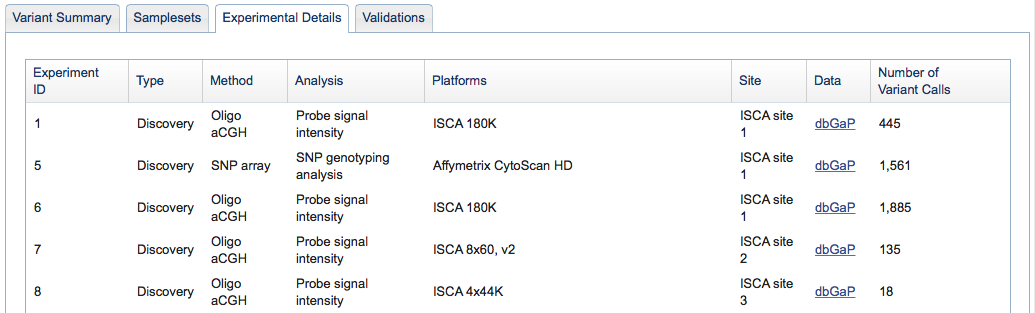
Figure 2c: Experimental Details Tab (Study Page) - The Experimental Details tab provides information on the methods and analyses that were used in the study. Each unique combination of method and analysis is named an "Experiment." A complete list of allowable Method Types and Analysis Types can be found in the Excel submission template, or by clicking here. Experiments can be one of three types: Discovery, Validation, or Genotyping. The Experimental Details tab is also where you can access links to the study's raw data (e.g., sequence traces or array data stored in an external database such as Trace or Array Express).
Figure 2d: Validations Tab (Study Page) - If validation experiments were performed to confirm variant calls generated by discovery experiments, the validation experiments are assigned unique IDs and are listed along with the relevant method in the Validations tab. Individual validation results can be accessed by downloading the variant data itself from the Study Page. If no validation experiments were performed for a given study, the Validations tab will indicate that no validation data were submitted to dbVar.
dbVar Variant Page
Each dbVar Variant page displays detailed information about an accessioned Variant Region. At the top of the page is a section with general information about the Variant Region (Study, number of supporting Variant Calls, Region size, etc. - see Figure 3a), and a chromosome ideogram displaying the variant's location on the genome. This is followed by a tabbed section with complete variant details: Genome View, Variant Region Details and Evidence, Validation Information, Clinical Assertions, and Genotype Information (see figure legends below for information on each of these tabs).
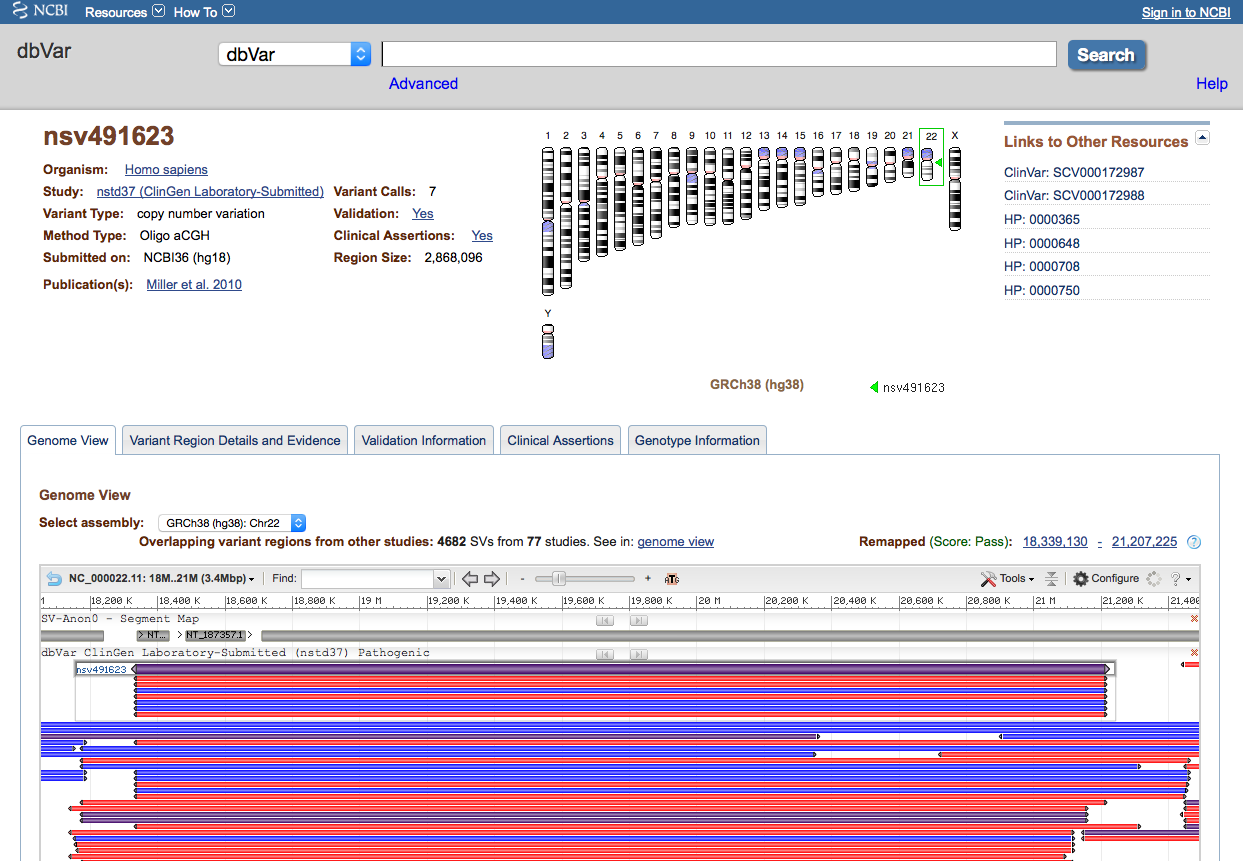
Figure 3a: General Information (top) and Genome View Tab (Variant Page) - The Genome View Tab shows the current variant in NCBI’s Sequence Viewer in the context of known genes and other variant data from the current study. Use the pull-down menu to select the assembly you want to view. Each black bar represents a Variant Region (the current region is centered and highlighted), while the colored bars represent Variant Calls that support each region. The display default is to collapse all supporting variant calls, but this can be adjusted (for example, to show all individual calls) by clicking Configure… Variations… Show All. Clicking on any Variant Region or Variant Call will reveal a pop-up tooltip with information about the variant and a link to its dbVar page.
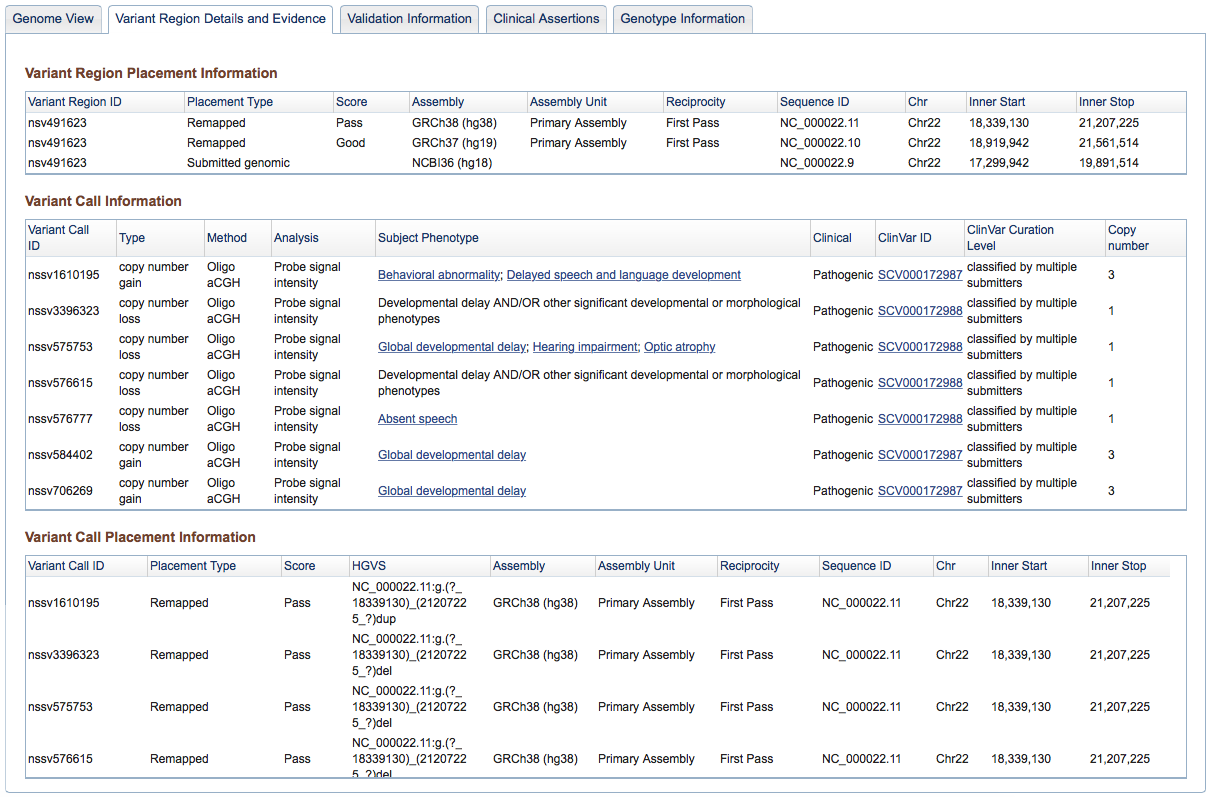
Figure 3b: Variant Region Details and Evidence Tab (Variant Page) - This tab contains placement coordinates for the variant region on the assembly on which the region was originally submitted as well as assemblies to which it was subsequently mapped. In the case of remapped placements, the Score column indicates the quality of the remap – Perfect, Good, or Pass (for details see dbVar Placements section below). Below the Variant Region information are details on the supporting Variant Calls that were merged to define the Region, including their placements (on Submitted and/or Remapped assemblies). Complete details can always be downloaded using the links above the tabbed section of the Variant Page.
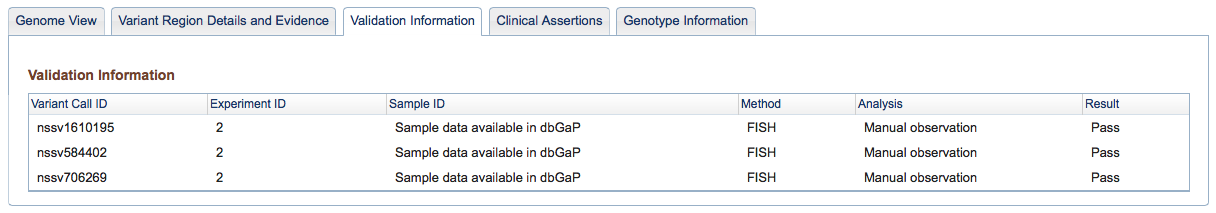
Figure 3c: Validation Information Tab (Variant Page) - If any Variant Call results were validated with additional methods, this tab provides details on the methods and analyses used in the validation, and the results (Pass or Fail for each call tested).
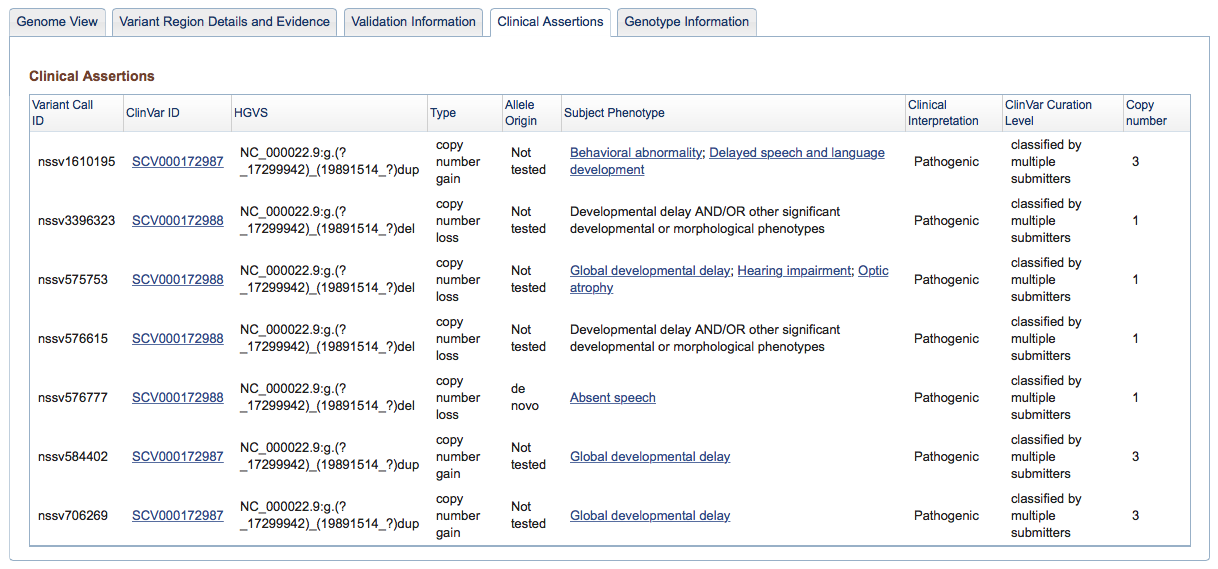
Figure 3d: Clinical Assertions Tab (Variant Page) - If in the course of a study an association was established between a Variant Call and a phenotype observed in a Subject, details are provided here. The Variant Call ID is followed by the sample in which it was observed (with a link if the sample is publicly available), the type of event (insertion, deletion, copy number gain, etc.), the parental origin of the variant if it was determined, the phenotype associated with the variant, and the authors’ assessment of its likely pathogenicity. For a reminder of how studies involving sensitive clinical and genetic information are processed, please refer to the Figure 2b legend above.
Variant Call and Region Types
dbVar supports the types of structural variation listed in Table 1 below. Each variant call is submitted using one of the Variant Call Types in the first column; corresponding Sequence Ontology IDs and definitions are listed in the middle column; and Variant Region Types (for regions formed by merging similar calls) are indicated in the last column. (For an explanation of the differences between calls and regions, see our Overview of Structural Variation page.)
| Variant Call Type | Sequence Ontology ID | Variant Region Type |
|---|---|---|
|
complex substitution |
|
complex substitution |
|
copy number gain |
|
copy number variation |
|
copy number loss |
|
copy number variation |
|
copy number variation |
|
copy number variation |
|
deletion |
|
copy number variation |
|
duplication |
|
copy number variation |
|
delins |
|
delins |
|
insertion |
|
insertion |
|
interchromosomal translocation |
|
translocation, complex chromosomal rearrangement |
|
intrachromosomal translocation |
|
translocation, complex chromosomal rearrangement |
|
inversion |
|
inversion |
|
mobile element insertion |
|
mobile element insertion |
|
Alu insertion |
|
mobile element insertion |
|
LINE1 insertion |
|
mobile element insertion |
|
SVA insertion |
|
mobile element insertion |
|
HERV insertion |
|
mobile element insertion |
|
novel sequence insertion |
|
novel sequence insertion |
|
mobile element deletion |
|
mobile element deletion |
|
Alu deletion |
|
mobile element deletion |
|
LINE1 deletion |
|
mobile element deletion |
|
SVA deletion |
|
mobile element deletion |
|
HERV deletion |
|
mobile element deletion |
|
sequence alteration |
|
sequence alteration |
|
short tandem repeat variation |
|
short tandem repeat variation |
|
tandem duplication |
|
tandem duplication |
dbVar Variant Rendering

Figure 4a: Rendering breakpoint ambiguity. There are four common scenarios for most variants (either SVs or SSVs) as shown in the table above. However, mixed cases with a defined breakpoint at one end and an undefined breakpoint range at the other end are possible as well. Here, we use (CNV SV) as examples.

Figure 4b: Rendering variant types. Variants are visually represented in several places: the Genome View tab of variant pages; the dbVar Genome Browser; and NCBI's Sequence Viewer. Colors distinguish types of structural variant (copy number gain/loss, insertion, inversion, etc.). Breakpoint ambiguity is represented by translucency and/or arrows at variant ends.
dbVar Placements
Most variants are submitted to dbVar as asserted locations on a particular assembly; this assembly is not always the current assembly. Additionally, all studies have not used the same assembly to do their analysis. In order to compare data from different studies, it is necessary to obtain placements for all variants on the same set of assemblies. For most variants, we do this using in-house remapping software with the following parameters:
- Minimum ratio of bases that must be remapped: 0.5
- Maximum ratio for difference between source length and target length: 4.0
- The 'Merge' function is turned on.
- Multiple locations can be returned.
We use relatively permissive parameters as many structural variants fall within regions of the genome that are likely to change from assembly to assembly. A coverage score is calculated by taking the length of the feature in the target assembly and dividing it by the length of the feature in the source assembly. A score of 1 suggests the region is relatively unchanged between assemblies (note: single bases aren't assessed); a score of >1 suggests an insertion in the target assembly and a score of <1 suggests a deletion in the target assembly. We provide a qualitative assessment of the remapping status on our web pages:
- Perfect: coverage score equal to 1
- Good: coverage score within 5% of 1
- Pass: coverage deviates from 1 by greater than 5%
A relatively small number of variants submitted to dbVar are defined by sequence. These are typically insertion sequences that could not be placed on the assembly used in the analysis of the study. Such sequences are submitted to GenBank/EMBL/DDBJ and tracked in dbVar using the assigned accession.version. These sequences are aligned to newer assemblies using a process developed in-house called the NG Aligner (unpublished). Sequences that are aligned with >95% coverage and 98% identity are considered placed.
It is in the nature of evolving assemblies that variants called on an earlier assembly sometimes will not map cleanly to a newer (presumably more accurate) assembly. In these situations the Remapper may output multiple alternative placements and, even though they are scored, one cannot know with certainty exactly where, and with what degree of certainty, a variant can be placed on the newer assembly. Therefore dbVar implements a set of rules for filtering remap results, with the goal of providing users our best estimates based on imperfect remapping data. In some cases variants may fail to remap altogether – these are indicated in dbVar FTP download files as having failed remap – and should be considered suspect.
Assemblies in scope
dbVar displays placement data for the assembly used in the analysis of a study (the 'Submitted' placement). For most organisms, we will also attempt to find a placement for a variant on the 'current' assembly, if available. When an assembly is updated we will support the 'current' and 'previous' assembly for up to a year. For example, we currently support placements for human data on GRCh37 and GRCh38.
FTP Site
All data in dbVar can be downloaded from our FTP site: https://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/. Data is organized by assembly and by study. A complete listing of available downloads is available online. If you download by assembly, please be aware that some Variant Calls or Variant Regions may have been submitted on an assembly other than the one being downloaded; these variants will have been remapped to reflect their corresponding placements on the downloaded assembly, placements for which can be found in a separate file in the FTP directory. For example, the GRCh37 assembly download includes all variants that were submitted on GRCh37, plus variants submitted on other assemblies and that were remapped to GRCh37. Remapped status is indicated in the download files.
More information about FTP site organization can be found here: https://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/README.txt.
dbVar Entrez Search
A search bar is provided on the top of each dbVar page with a drop-down menu to search dbVar and other NCBI resources using Entrez (See Entrez Help for further details).
A dbVar search returns Studies, with links to the Study page, and Variants, with links to the Variant page. Filters on the left side of the page allow the user to narrow their results based on a wide variety of data fields.
Advanced Search
The dbVar Advanced search page allows the user to build Entrez search queries by selecting search fields and values from drop-down menus.
dbVar Search Fields
The following fields are available to search dbVar:
| Field name | Field aliases | Description |
|---|---|---|
| Accession | ACC, ACCN | Accession of any internal or external identifier. Versions removed from GENBANK accessions. |
| Age of Subject | AGE | Age of subject in years |
| All Fields | ALL, * | All terms from all searchable fields |
| Assembly | ASSM | Accession or name of placement assembly |
| Author | AUTH, AU | All authors included in journal |
| BioProject | PROJ | Accession or Name of associated BioProject |
| Chr | CH, CHROM, CHROMOSOME | Chromosome of placement |
| ChrEnd | CHR_END, CHR_STOP, END | End of placement on chromosome |
| ChrPos | BASE, BASE_POSITION, CHR_POS, CHR_START, START | Start of placement on chromosome |
| Clinical Interpretation | CLIN, CLINICAL_ASSERTION | Clinical interpretation of a variant (controlled vocabulary) |
| Clinical Phenotype | DISEASE, PHENO, PHENOTYPE, PTYPE | Phenotype of sample/subject study or reference specimen |
| Filter | FILT, FLTR, SUBSET, SB, FIL | Limits the records |
| Frequency | FREQ, GLOB_FREQ | Global Allele Frequency |
| Gene | GENE_NAME, SYM | Name or alias of gene at same location as variant |
| Has Clinical | HASCLIN | Flag indicating subject has Clinical Interpretation: 0=no; 1 = yes |
| MeSH | MH | Medical Subject Headings assigned to publication |
| MeSH ID | MHID | NLM MeSH Browser Unique ID |
| Method Type | METH, METHOD | Method type (controlled vocabulary) |
| OMIM | MIM | Online Mendelian Inheritance in Man |
| Object Type | OT | Object type in dbVar (STUDY, VARIANT) |
| Organism | ORG, ORGN | Organism name (exploded) |
| Origin | ALLELE_ORIGIN | Allele origin (controlled vocabulary), including Both=Germline+Somatic |
| Pathogenic Overlap Range | PATHO_RNG | Range of Reciprocal Overlap with a Pathogenic Variant |
| Population | POP | Subject population (major continental group) of calls with frequency data |
| Publication | PMID, PUBMED_ID | Unique identifier from PubMed |
| Publication Date | PDAT, PUBDATE | Journal Publication date |
| Sample | SMPL | Sample/subject ID of study or reference specimen |
| Sample Count | SC, SCOUNT | Number of samples in study |
| Sex of Subject | GENDER, SEX | Sex of Subject (controlled vocabulary) |
| Study | STDY | Study ID, alias, display name, or publication name |
| Study Type | STYPE | Study type assigned by NCBI |
| Subject Phenotype Status | SUBPSTAT | Flag indicating subject has a phenotype: 0=no; 1 = yes |
| Taxonomy ID | TAX, TAX_ID, TAXID, TAXONOMY | Taxonomy ID |
| UID | UID | Unique number assigned to study or variant in Entrez |
| Variant Size | VLEN, VSIZE | Size of variant |
| Variant Type | VT, VTYPE, VARTYPE | Type of variant region or call (controlled vocabulary) |
| Zygosity | ZYG | Zygosity of a variant (controlled vocabulary) |
dbVar FAQs
Frequently asked questions (FAQs):
- What is 'dbVar'?
- How does dbVar differ from the Database of Genomic Variants (DGV)?
- What is ‘structural variation’?
- What types of structural variation data does dbVar accept?
- Does it matter how I detected the structural variation?
- Can I submit structural variation data from any organism?
- Can I submit structural variation data from human clinical or cancer studies?
- Does dbVar distinguish between pathogenic and benign variants?
- Does dbVar accept genotype data?
- Will I get a unique dbVar ID to use in publications?
- What is the difference between the dbVar accessions?
- Why is the data in dbVar different from that provided in the publication?
- Why do some dbVar variants have >1 location?
- What's the difference between an nsv and an nssv?
- Can I download the whole database?
- How do I know if a given variant(s) in dbVar is real (or of high quality)?
- How do I download all the data from a given region or gene?
- Is there a way to integrate dbVar data with dbSNP data?
- Can I include any clinical information in my data submission?
- My submission contains sensitive private clinical information. How does dbVar guarantee the information will remain private?
- What is the smallest size structural variant dbVar accepts?
- My study has identified novel insertion sequences and I don't have a genomic coordinate for these. How can I submit these to dbVar?
- The number of "Other Calls in this Sample" in the Variant Call Information table of the Variant Page is different from the number of variants that are returned when I do a dbVar search for the same study and sample ID. What's going on?
- How does dbVar place data submitted on one assembly (e.g. NCBI36) on other assemblies (e.g. GRCh37)?
- How do I submit to dbVar?
- How should I cite dbVar?
-
What is 'dbVar'?
dbVar is the NCBI database of human genomic structural variation. For information on how to navigate dbVar see the dbVar Help page.
-
How does dbVar differ from the Database of Genomic Variants (DGV)?
DGV has been a useful resource for the human genetics community with respect to collecting and curating structural variation data for human. DGV, dbVar and its European counter-part DGVa, contain data for healthy control human samples, while the latter two also include clinically relevant structural variation data from ClinVar.
-
What is ‘structural variation’?
Structural variation (SV) is generally defined as any region of DNA involved in inversions and balanced translocations or genomic imbalances (insertions and deletions), commonly referred to as copy number variants (CNVs). For more information see the Overview of Structural Variation page.
-
What types of structural variation data does dbVar accept?
dbVar is a structural variation database designed to store data on variant DNA ≥ 1 bp in size. Practically speaking, we recommend submitting variation data that is > 50bp to dbVar and variation data that is ≤ 50bp to dbSNP. We can accept diverse types of events, including inversions, insertions and translocations. We discourage the submission of somatic and cancer-related variants, as they tend to be complex and sample-specific, and are more appropriately stored in custom databases.
-
Does it matter how I detected the structural variation?
dbVar accepts submissions based on analysis of whole-genome or whole-exome next-generation sequencing (NGS) (including paired-end mapping, split-read pair, and read depth) as well as microarray-based experiments including array-CGH and SNP genotyping. A full list of supported methods and analyses can be found on the dbVar Help page.
-
Can I submit structural variation data from any organism?
Beginning on September 1, 2017 dbVar stopped accepting submissions for any non-human organisms. Non-human SV data can be submitted to DGVa.
-
Can I submit structural variation data from clinical or cancer studies?
No. All clinically relevant structural variation should be submitted to ClinVar or dbGaP. We discourage the submission of somatic and cancer-related SV to dbVar.
-
Does dbVar distinguish between pathogenic and benign variants?
Yes, dbVar will clearly mark and allow searching for variants that are known to be pathogenic, providing links to OMIM when available.
-
Does dbVar accept genotype data?
Yes, dbVar can accept genotype data.
-
Will I get a unique dbVar ID to use in publications?
Yes, dbVar will provide a unique accession number for each study, each submitted variant region and each supporting level variant.
-
What is the difference between the dbVar accessions?
dbVar collaborates with DGVa at EBI and with TogoVar-repository at DDBJ to accession genomic structural variants. Accessions prefixed with 'n' have been processed by NCBI (dbVar, with 'e' by EBI (DGVa), and with 'd' by (DDBJ. NCBI, EBI, and DDBJ provide three levels of accessions:
- (n|e|d)std: the study id - this identifies a submitted study
- (n|e|d)sv: the structural variant id - this identifies the submitted region of variation
- (n|e|d)ssv: the supporting structural variant id - this identifies the supporting regions of variation (often sample-specific) that were used to call the submitted region of variation
-
Why is the data in dbVar sometimes different from that provided in the publication?
The loading of studies that are submitted to dbVar after publication may highlight errors during our quality control checks. In these cases submitters will be contacted and the errors corrected. In other cases a submitter may detect errors before submitting or may decide to edit their data. Often this will be documented in the study record.
-
Why do some dbVar variants have more than one location?
There are two reasons:
When a submitter gives us data on an assembly obtained from UCSC, we translate this into native assembly coordinates and map sequences (chromosomes and unplaced/unlocalized sequences) to their accession.versions. UCSC concatenates unplaced/unlocalized sequences into pseudo-scaffold objects they call chr*_random. In some cases, submitters have provided data that cross gaps on the 'chr*_random' sequences, meaning that the feature actually maps to two different, unrelated sequences.
More than one location may also be provided if the variant is the result of a transposition event. In this case, coordinates from both the donor and recipient sites are provided, to retain as much information about the variant event as possible.
-
What's the difference between an nsv and an nssv?
nsv and nssv are accession prefixes for variant regions and variant calls (or instances), respectively. Typically, one or more variant instances (nssv – variant calls based directly on experimental evidence) are merged into a variant region (nsv – a pair of start-stop coordinates reflecting the submitters’ assertion of the region of the genome that is affected by the variant instances). The ‘n’ preceding sv or ssv indicates that the variants were submitted to NCBI (dbVar). esv and essv represent the same variant entities, but those that were submitted to EBI (DGVa); similarly, dsv and dssv were submitted to DDBJ (TogoVar-repository).
Please see Overview of Structural Variation for more information.
-
Can I download the whole database?
All data is available on our FTP site. Data are available on a per study and per assembly basis.
-
How do I know if a given variant(s) in dbVar is real / of high quality?
dbVar is an archive. We report variants as they are submitted to us, usually in association with a peer-reviewed publication. Responsibility for data reproducibility lay with the submitting investigator. dbVar encourages, but does not require, the validation of all variants by at least one alternative method, and we provide a mechanism to include validation results in the submission template. Any submitted validation data are presented as an integral part of the study data. To be listed as “validated” a variant must have been confirmed with at least one, or possibly more, additional independent methods. If, as a consumer of dbVar data, have concerns regarding a particular variant or data set, we recommend you contact the relevant submitter for more supporting information.
-
How do I find data from a given region or gene?
You can perform searches using gene names. The results that are returned will include all variants that overlap the gene, and the studies with which the variants are associated.
-
Is there a way to integrate dbVar data with dbSNP data?
Yes, Variation Viewer provides some functionality in this regard. Alternatively, the user can integrate the data manually with avilable bioinformatics tools.
-
Can I include any clinical information in my data submission?
No, all submissions with clinically relevant structural variation should be submitted to ClinVar.
-
My submission contains sensitive private clinical information. How does dbVar guarantee the information will remain private?
Submisions with sensitive patient information should be submitted to dbGaP.
-
What is the smallest size structural variant dbVar accepts?
There are no size restrictions on structural variation data. We recommend that variants smaller than 50 bp be submitted to dbSNP but we will accept variants as small as a single basepair as long as the variant is an insertion or deletion, not a single nucleotide change.
-
My study has identified novel insertion sequences and I don't have a genomic coordinate for these. How can I submit these to dbVar?
If you have novel insertion sequence data, please submit it first as a WGS Project. This will give all of your novel insertion sequences unique identifiers that can then be tracked. You can then submit your data to dbVar and these novel insertion sequences can reference the sequence identifiers obtained from the WGS submission. With stable sequence identifiers, we may be able to map the sequence to updated assemblies and obtain a chromosome context for this sequence.
-
The number of "Other Calls in this Sample" in the Variant Call Information table of the Variant Page is different from the number of variants that are returned when I do a dbVar search for the same study and sample ID. What's going on?
The number in the table on the Variant Page indicates the number of Variant Calls (SSVs) in the sample. A search for the study and sample ID returns the number of Variant Regions (SVs) after similar calls have been merged into regions.
-
How does dbVar place data submitted on one assembly (e.g., GRCh37) on other assemblies (e.g., GRCh38)?
dbVar uses in-house software to map variants between assemblies. All variants reported in human-based studies are automatically remapped to both GRCh37 and GRCh38.
-
How do I submit to dbVar?
We encourage you to submit data to dbVar by completing one of the templates we provide (Excel, Tab-delimited, XML) or by VCF, and emailing it to dbvar@ncbi.nlm.nih.gov. Please see dbVar Submission Information for more information.
-
How should I cite dbVar?
Please reference the following publication when citing dbVar:
Lappalainen I, Lopez J, Skipper L, Hefferon T, Spalding JD, Garner J, Chen C, Maguire M, Corbett M, Zhou G, Paschall J, Ananiev V, Flicek P, Church DM. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2013 Jan;41(Database issue):D936-41. doi: 10.1093/nar/gks1213. Epub 2012 Nov 27. PMID: 23193291; PMCID: PMC3531204.
If you wish to reference a specific submission, cite the study accession, e.g., nstd166; if you wish to reference a specific variant, cite the variant region or variant call accession, e.g., nsv4136077 or nssv15910013.