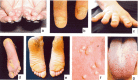
Treatment of Manifestations
Preliminary treatment guidelines have been published [Goldberg et al 2014] and continue to be refined.
The current treatment modalities primarily center on symptomatic relief of pain, hygienic grooming practices including paring of hyperkeratotic areas, treatment of secondary infection when indicated, and use of various walking aids including wheelchairs, crutches, and canes.
Palmoplantar keratoderma. Frequent grooming of the feet is essential and includes paring down the hyperkeratotic areas. However, trimming too aggressively can greatly increase pain. Some find it helpful to soak the feet prior to the paring. The surface of the skin and the instruments used should be clean to avoid infection. Blisters should be punctured with a sterile needle, the fluid drained, and the blister roof left in place until it dries and is shed away.
Topical therapies to remove the hyperkeratosis:
Emollients such as Vaseline® or lanolin-containing products are frequently used. Note: Creams and lotions containing keratolyics such as urea, lactic acid, salicylic acid, or propylene glycol have little effect, with some reporting negative side effects. Occlusive ointments are often poorly tolerated.
Oral retinoids, while reducing the keratoderma, do not affect the underlying blistering and fragility of the skin and sometimes increase the pain. Careful regulation of the dosage is necessary [
Gruber et al 2012].
Special orthotics or insoles, wicking socks, ventilated footwear or cushioned footwear can help to lessen the pain although pain varies from day to day and at times can be intense even at rest.
Maintaining ideal body weight can be a factor in reducing the hyperkeratosis and pain. Limiting walking or standing can help to reduce trauma and slightly diminish the resulting blisters, callus, and pain.
The origin, nature, and underlying mechanism of plantar pain in individuals with PC is poorly understood. Several recent studies suggest that neuropathic pain treatments may be useful in individuals with PC who experience plantar pain [Pan et al 2016; Wallis et al 2016; Authors, unpublished] (see Therapies Under Investigation).
Nail dystrophy. Thickened nails are not typically painful, but become so when infected or traumatized. An effective tool for very thick nails and for children is a guillotine-type pet nail clipper which places no pressure on the nails. Other tools frequently used are razor or surgical blades or sanders such as a Dremel® tool.
If bacterial or fungal infections occur, systemic antibiotics or antifungals are indicated.
Particularly troublesome nails can be successfully removed surgically; however, few affected individuals have had this procedure and in many cases – regardless of the specific pathogenic variant – the nails have regrown [DeKlotz et al 2017].
Oral leukokeratosis. Good oral hygiene and frequent gentle brushing with a toothbrush can significantly improve the appearance of the thick, white patches on the tongue and oral mucosa; however, if done too vigorously, brushing may also traumatize the mucosa resulting in reactive hyperkeratosis.
Some individuals have reported reduction of the leukokeratosis in response to oral antibiotics, suggesting a possible bacterial contribution; more likely, improvement may be a response to the anti-inflammatory properties of the antibiotics.
Follicular hyperkeratosis. Especially bothersome for children and teens, this finding can be treated with alpha-hydroxy acid creams or lotions or keratolytic emollients; however, these treatments may not be especially effective for PC. The use of emollients such as Vaseline® or lanolin-containing products is reported to be as effective.
Leukokeratosis with laryngeal involvement. Reported only in children with PC-K6a, respiratory insufficiency can on occasion become life threatening, requiring emergent surgical intervention to reestablish the airway. The surgical procedures are repeated as necessary to maintain an open airway; however, surgical procedures to the larynx aimed at improving hoarseness should be avoided as they may tend to worsen the condition.
Cysts. Steatocystoma multiplex and other pilosebaceous cysts can be treated by incision with a number 11 blade and subsequent expression of the contents of the cyst ("incision and drainage"). Oral antibiotics may be indicated in the case of secondary infection. A culture should be obtained if infection is a consideration. Intralesional injection of steroid (e.g., triamcinolone) may reduce inflammation of the area if infection is not suspected. If necessary, cysts can be excised.
Failure to thrive. Poor feeding in infancy has been reported to be ameliorated by the use of a soft nipple with an enlarged opening to reduce the sucking required.
Therapies Under Investigation
In 2014, a Phase 1b clinical trial sponsored by PC Project and TransDerm was performed using topical sirolimus. This study included 15 affected individuals and was conducted by Dr Joyce Teng at Stanford University. Background research for this trial was previously published [Hickerson et al 2009].
Short interfering RNA (siRNA) can selectively block expression of a specific K6a-causing pathogenic variant [Hickerson et al 2008, Leachman et al 2008]. The siRNA trial included treatment of a single individual with a specific KRT6A pathogenic variant in a dose-escalation trial of an siRNA directed against the p.Asn171Lys mutated allele [Leachman et al 2010]. The affected individual did not experience any adverse effects from the experimental treatment. The affected person also experienced callus regression on the foot treated with siRNA.
Botulinum toxin has been used to treat pain in several affected individuals, with promising results [Swartling & Vahlquist 2006, Swartling et al 2010, González-Ramos et al 2016]. In addition to these published investigations, a number of other individuals have been treated with botulinum toxin.
Several persons have been treated with statins. The results from studies are mixed. Further research is being conducted and additional animal testing is proposed [Zhao et al 2011].
Other therapies currently under investigation include anti-TNF biologics, duloxetine or duloxetine and a tricyclic combination, gabapentin, topical gabapentin, and capsaicin injections.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.