

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Fitridge R, Thompson M, editors. Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet]. Adelaide (AU): University of Adelaide Press; 2011.

Mechanisms of Vascular Disease: A Reference Book for Vascular Specialists [Internet].
Show detailsDefinition
Compartment syndrome is a clinical and pathological syndrome where the pressure within an anatomical tissue compartment rises above the normal physiological value for that compartment and detrimentally alters the function of the tissues either temporarily or permanently. Acute compartment syndromes affecting the abdominal cavity and the fascial compartments of the limbs are those encountered in vascular surgery.
Acute Limb Compartment Syndrome
The importance of acute limb compartment syndrome (LCS) is that, if left untreated, it results in rhabdomyolysis with resultant release of potassium, myoglobin and other toxins into the systemic circulation, which can lead to renal and/or multi-organ failure. The mortality of acute renal failure and multi-organ failure is high. These patients require critical care which may involve dialysis and other organ support. Untreated LCS often necessitates amputation to prevent further acute systemic deterioration. If the immediate insult is survived without the need for amputation a permanently disabling ischaemic contracture may result.1
LCS arises due to the anatomical arrangement of muscles surrounded by restrictive, inelastic osteofascial envelopes. Increased pressure within these fascial compartments can occur as a result of extrinsic compression such as that from plaster casts or bandages, or increased volume of the contents of the compartment. The volume within the compartment can be increased as a result of enlargement of those tissues contained within the compartment (e.g.muscle oedema) or due to the presence of a pathological spaceoccupying mass (such as a haematoma or abscess). In the field of vascular surgery LCS is most commonly encountered following delayed revascularisation of an ischaemic limb, fractures with/without vascular injury or following radiological complications such as perforation during angioplasty. It is occasionally seen in conditions such as phlegmasia caerulea dolens where venous hypertension exists.
Incidence
The incidence of LCS after revascularisation depends primarily on the type of insult. After elective vascularisation of chronically ischaemic limbs the incidence is very low, from 0% to 0.5%.2,3 the incidence increases in revascularised acutely ischaemic limbs to approximately 10% to 20%.4-6 Revascularisation following vascular trauma is the most significant risk factor for LCS, with a reported incidence of up to 62%,7 particularly if this is associated with either a vascular injury at or below the popliteal artery, or a fracture (Figure 19.1).
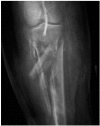
FIGURE 19.1
Severe comminuted fracture of the tibia and fibula with distal popliteal artery injury demonstrated angiographically. An injury with a high risk of acute limb compartment syndrome.
Anatomy/Physiology
LCS develops in the limbs because of their particular anatomical and physiological characteristics. Anatomically this is principally the arrangement of muscles within envelopes of dense, inelastic fibrous fascia. In addition to muscles, each compartment also contains peripheral nerves and blood vessels that traverse these compartments to supply distal parts of the limb and/or supply the structures within that compartment.
The anatomical arrangement of the capillary beds and physiological processes occurring within these also contributes to the development of LCS. Figure 19.2a shows the normal anatomical arrangement and physiological passage of fluid across the capillary wall. Fluid exchange across a capillary wall is affected by hydrostatic pressure and oncotic pressure. The extracellular compartment has low hydrostatic and oncotic pressure due to the draining action of lymphatics. In capillaries hydrostatic and oncotic pressure varies along their length. At the arterial end of the capillary, hydrostatic pressure (35mmh g) is greater than tissue hydrostatic pressure (0mmhg). Oncotic pressure within the capillary (28mmHg) is higher than tissue oncotic pressure (3mmh g) (therefore acting to draw fluid back into the capillary) but since the net hydrostatic pressure acting to filter fluid out of the capillary is greater than the net oncotic pressure, fluid is filtered into the extracellular space. As blood passes along the capillary to the venous end fluid is lost. At the venous end of the capillary, hydrostatic pressure is much lower (15mmhg) and the resultant net force now acts to draw fluid back into the capillary (net oncotic pressure is greater than the net hydrostatic pressure).
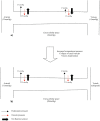
FIGURE 19.2
Capillary fluid exchange, a) in normal physiological circumstances, b) in the case of raised extracellular tissue pressure, as in acute limb compartment syndrome.
Aetiology/Pathophysiology
LCS causes tissue damage due to ischaemia within the affected compartment. This ischaemia is not due to interruption of the regional blood supply but due to the failure of the micro-circulation caused by the elevated compartment pressure (the initial insult may, of course, be due to regional ischaemia). The initial injury causes swelling of the tissues within the compartment that, in turn, results in increased intracompartmental pressure (ICP).
AS ICP increases above physiological levels the first part of the circulation to be affected are the small venules, since these vessels have the lowest pressure. These venules collapse and since the arterial side of the capillary beds remains open, the hydrostatic pressure within the capillaries continues to filter plasma out of the blood vessels into the tissues. However, since the venous side of the capillary bed is closed, the normal return of extracellular fluid back into the circulation by combined oncotic and hydrostatic forces cannot occur and actually reverses due to the venous hypertension in the capillary (Figure 19.2b). Lymphatic drainage is also impaired by the high tissue pressures. The net result is further tissue swelling, with a subsequent further increase in ICP and thus the initiation of a vicious cycle. AS ICP increases further the arterial side of the capillary bed and eventually the arterioles become affected causing frank ischaemia and permanent tissue damage shortly follows.
In the initial stages of LCS the principal pathological changes in muscles affected is oedema within and around muscle fascicles. As LCS progresses frank ischaemic changes occur in the muscle fibres.8
The most common cause of compartment syndrome in vascular surgery is tissue oedema due to the ischaemia-reperfusion injury caused by limb revascularisation. The re-establishment of a blood supply to ischaemic tissues has been observed to cause more damage than ischaemia alone. During ischaemia, anti-oxidant mechanisms are capable of dealing with any oxygen free radicals produced. However, the return of oxygenated blood to ischaemic tissues results in a burst in production of oxygen free radicals due to the action of xanthine oxidase (Xo) on tissue hypoxanthine, both of which are produced in ischaemic tissues. The return of oxygen upon reperfusion supplies the final substrate for this reaction to proceed and this produces a burst of superoxide production (Figure 19.3). Free radicals are produced from this superoxide and mediate cell damage largely via lipid peroxidation of cell membranes. Oxygen free radicals also cause activation of microvascular neutrophil polymorphs and endothelial cells. Activated endothelium produces arachidonic acid metabolites, nitric oxide (no), endothelins, complement and cytokines. These various mediators contribute to the continuation and extension of a local inflammatory response and the production of a cellular and acellular inflammatory infiltrate with corresponding tissue swelling (Figure 19.4).9
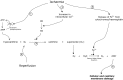
FIGURE 19.3
Xanthine Oxidase (XO) pathway activation by ischaemia-reperfusion and the production of reactive oxygen species. Ischaemia prevents oxidative phosphorylation (1) and cellular ATP cannot be regenerated. This leads to the accumulation of AMP and, in turn, (more...)
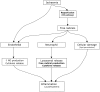
FIGURE 19.4
Pathways and effects of ischaemia-reperfusion injury. XO: xanthine oxidase, NO: nitric oxide.
Clinical presentation
Different tissues within the osteo- fascial compartments of the limbs are able to tolerate ischaemia to different degrees. The most sensitive to ischaemia are unmyelinated nerve fibres followed by myelinated nerve fibres, skeletal muscle, skin and then bone. Large arteries are relatively resistant to acute hypoxia and blood flow within them is maintained until late after the onset of LCS since ICP only exceeds systemic blood pressure at the latter stages of the disease process. It is these different hypoxic tolerances of each tissue that lead to the symptoms and clinical signs of LCS.
The most common symptom of LCS is severe pain that is unresolved by analgesics and the degree of which is out of proportion to the injury sustained. Symptoms due to neurological dysfunction include weakness and paraesthesia in the myotomes and dermatomes associated with the peripheral nerves that pass through the affected compartment. These symptoms will progress steadily over a short period of time.
Clinical signs associated with LCS are pain on passive movement of the muscles in the affected compartment, tenderness of the muscle bellies lying within the affected compartment and tenseness of the compartment. There may be muscle weakness and sensory loss (particularly two-point discrimination due to the early loss of the unmyelinated nerve fibres). Commonly foot drop occurs in LCS affecting the lower leg. There may be signs of the injury that has caused LCS to develop such as a fracture, surgical dressings or bruising. Peripheral pulses will be maintained until long after LCS has become established. Signs suggestive of irreversible ischaemic change include fixed, non-blanching skin staining or frank gangrene. In these cases therapy should be directed towards preventing systemic complications and death.
Clinical assessment of suspected LCS is difficult. The majority of symptoms and signs are only reliably assessed in a fully conscious patient; those at highest risk of LCS often have an altered level of consciousness due to the injury or operation that has placed them at risk. Also, many patients may have some of these clinical signs present due to the injury that has caused the LCS such as the pulseless, painful, paraesthetic limb of acute ischaemia. The most useful guide to LCS is to maintain a high index of clinical suspicion.
Investigation
Since clinical assessment of a limb at risk of LCS is difficult, a test to give an objective measurement of ICP is desirable. Many methods for measuring ICP exist. Described techniques are wick catheters,10 slit catheters,11 needle manometry12 and infra-red spectroscopy (Figure 19.5).13 These various methods all have relative advantages and disadvantages. Catheter techniques allow continuous monitoring of limbs at risk and are more accurate than simple needle manometry10 but they are more complex and require prior training in their use. Needle manometry can be simply performed using an 18g needle connected to a mercury manometer, or small handheld electronic devices are available. Near-infrared spectroscopy is still under evaluation. Whilst this technique is non-invasive it requires relatively expensive equipment and the interpretation of the readings from this method are less intuitive than a figure for absolute compartment pressure expressed in mmHg.
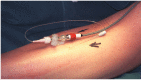
FIGURE 19.5
Slit catheter inserted into anterior tibial compartment and connected to a pressure transducer. (Photograph courtesy of Mr MJ Allen.).
Normal resting ICP is 010mmHg.2,14 Capillary blood pressure varies from 30– 40mmHg at the arterial side of the capillary bed to 10–15mmhg at the venous side.15 Given the suggested pathophysiological processes underlying the development of LCS, it would be expected that symptoms would first occur at compartment pressures somewhere between these two values. Many authors have suggested absolute cut-off levels of ICP to diagnose LCS. Mubarak suggested a value of 30mmhg16 whilst a llen suggested a value of 50mmHg g or a value above 40mmHg for longer than 6 hours.17 However, in several clinical studies clinical symptoms and signs of LCS have not correlated well with measured compartment pressures. Tissue perfusion is dependent, not only on the interstitial pressure but also the arterial perfusion pressure. Because of this, Whitesides et al suggested that the difference between diastolic blood pressure and ICP should be used to diagnose LCS, with a difference of less than 30mmHg being diagnostic.18 It has been shown that this definition for diagnosing LCS results in less unnecessary fasciotomies that using absolute ICP levels of either 30mmHg or 40mmHg.19 an alternative method is to calculate the difference between mean blood pressure and ICP, with a value of less than 40mmHg as a diagnostic cutoff.20
The measurement of ICP may allow more accurate diagnosis in those patients in whom clinical assessment is difficult due to co-existent injury or physical attributes but time should not be wasted on ICP measurement in patients who have clinically obvious LCS. In addition, many hospitals will not have the equipment or expertise available to accurately measure ICP. It is also thought that measured ICP may only reflect the ICP at the tip of the needle/catheter and not reflect the pressure change in the whole compartment. Since any delay in initiating treatment for LCS may result in a worse outcome once diagnosed, LCS should be treated expediently.
Apart from the measurement of ICP there are no specific tests to diagnose LCS. In clinically advanced cases where there is tissue damage, the resulting inflammation may be manifest as a leucocytosis or, if there is significant tissue necrosis, serum creatine phosphokinase will be elevated and a metabolic acidosis occurs.
Treatment
The treatment of acute LCS is urgent fasciotomy of all affected compartments (Figure 19.6). In patients who present acutely (within 12 hours of onset of LCS) this should be performed immediately. Delayed fasciotomy (longer than 12 hours after the onset of LCS) results in a significantly poorer outcome in terms of functional loss.21 In those patients whose presentation is delayed, consideration has to be given as to whether the limb is unsalvageable and the possibility that a fasciotomy in this group of patients may lead to significant morbidity without ultimately improving functional outcome. If fasciotomy is delayed longer than 12 hours but less than 36 hours infection rates increase but limb salvage rates are similar.22 Beyond 36 hours rates of amputation, infection, neurological injury and death increase such that early amputation rather than futile attempts at limb salvage should be considered in this group.22,23
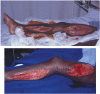
FIGURE 19.6
a) Medial thigh and calf fasciotomies following lower limb ischaemia after traumatic vascular injury. b) The same wounds 5 days showing healthy granulation tissue.
Fasciotomy should be performed in such a manner so that all constrictive elements surrounding a compartment are released. In the limbs this is the skin and the deep fascia, which encloses four separate compartments, the anterior, peroneal (lateral), superficial posterior and deep posterior. These can be decompressed either via a single lateral incision24 or via two incisions, one lateral and one medial (Figure 19.7).25The lateral incision is made over the peroneal compartment one finger-breadth anterior to the fibula from the fibular head to the ankle. The anterior and peroneal compartments are then opened along the length of the incision. The posterior compartments can then be opened either through this incision by dissecting behind the fibula or by removing a piece of the fibula. Alternatively the deep posterior compartment can be opened by dissecting anteriorly to the fibula through the interosseous membrane. If two incisions are used the second is made on the medial aspect of the lower leg and the two posterior compartments decompressed. The thigh is usually decompressed medially and/or laterally.
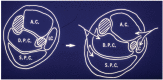
FIGURE 19.7
Diagrammatic cross-section through mid-calf showing four osteo-fascial compartments (left), arrows indicating medial and lateral incisions required for four-compartment fasciotomy. Shaded areas represent the tibia and fibula. AC: anterior compartment, (more...)
In the upper limb the forearm is the most commonly affected by LCS. Both the volar and dorsal compartments can usually be decompressed via a single volar incision over the whole length of the forearm made in a curved fashion to avoid contractures. In other areas of the limbs the incisions should be made based on the anatomy of that region and positioned so as to open the whole length of the affected compartment. After the fasciotomy has been performed any devitalised or necrotic muscle should be debrided.
Several alternatives exist for the management of fasciotomy wounds after the compartment syndrome has resolved: skin grafting, delayed primary closure, secondary closure, healing by secondary intention and the use of skin flaps. Skin grafting is usually performed between 7 and 21 days and has been suggested to reduce wound complications when compared to other methods (Figure 19.8).26

FIGURE 19.8
Split skin grafting to calf fasciotomy 2 weeks post-injury (same patient as figure 19.6). The thigh fasciotomy has been treated by delayed closure leaving a small defect to heal by secondary intention.
In addition to fasciotomy, methods directed towards reducing the degree of initial tissue injury causing LCS have been suggested. Free radical scavengers such as mannitol and superoxide dismutase have been shown to be of benefit in LCS caused by ischaemia-reperfusion injury in animals.27,28 Some benefit has been shown using these agents in humans29 although no comparative studies between these treatments and fasciotomy have been performed. Lysineacetyl-salicylate, a thromboxane A2 inhibitor has shown some benefit in animals but has not been studied in humans.30 At present there is not enough evidence to justify the routine use of these compounds in clinical practice. Hyperbaric oxygen therapy has been suggested to be of use in improving the outcome of fasciotomy.31
Complications of LCS
Locally, LCS, if left untreated, will cause ischaemia and subsequent limb loss. If infection occurs in the devitalised tissues systemic sepsis can occur and may result in multi-organ failure. In addition to infective complications the metabolic consequences of a devitalised limb have to be considered. As skeletal muscle becomes ischaemic and necroses, myoglobin and potassium are released into the circulation. These have nephrotoxic and cardiotoxic effects if released in large enough quantities, and may cause remote organ dysfunction or failure.
Following decompression of LCS, devitalised muscle released myoglobin can cause renal failure due to tubular blockage. Treatment involves optimal fluid therapy and consideration of alkalisation of the urine using intravenous sodium bicarbonate, aiming for a urinary pH greater than 6.5 and a plasma pH of greater than 7.4.
In some situations, amputation above the level of ischaemia may be required, a procedure associated with a high risk of mortality and morbidity in an already severely compromised patient.
Outcome
Whilst fasciotomy wounds are associated with a moderate degree of morbidity,26 fasciotomy does not appear to have any effect on long-term calf-pump function.32 If fasciotomy is performed without delay in LCS the outcome in terms of preventing limb loss, systemic complications and long term functional disability is good.33,34 Failure to promptly treat LCS risks the development of systemic complications such as multiorgan failure with a corresponding high risk of death.
Acute Abdominal Compartment Syndrome
The abdominal compartment syndrome (ACS) was first described by Kron in 1984.35 ACS is a clinical syndrome characterised by progressive intra-abdominal organ dysfunction resulting from increased intra-abdominal pressure (IAP. In 2004, the World Society of Abdominal Compartment Syndrome gathered at the International ACS Consensus Definitions Conference in 2004 to produce internationally accepted definitions.36,37 The consensus statement defined intra-abdominal hypertension (IAH) as IAP more than or equal to 12mmHg and ACS as a sustained IAP more than or equal to 20mmHg that is associated with new organ dysfunction or failure. The severity of IAH was characterised into 4 grades (summarised in Table 19.1). The concept of intra-abdominal perfusion pressure (IAPP) was also defined by the society as mean arterial pressure minus the IAP, as a measure of net pressure available for perfusion of intra-abdominal organs. The normal value of IAPP is greater than 60mmHg.
TABLE 19.1
Intra-abdominal hypertension grades as defined by the World Society of Abdominal Compartment Syndrome.
Acute elevation of intra-abdominal pressure causes not only dysfunction of those organs within the abdominal cavity (hepatic, gastro-intestinal and renal dysfunction) but has effects on more distant organ systems such as the cardiovascular, respiratory and central nervous systems. Whilst it has most commonly been described following abdominal trauma,38 vascular surgical procedures are the next most common cause.39 ACS is important since it results in dysfunction of multiple organ systems in patients who are already significantly compromised and is a contributory factor in the development of multi-organ failure.
In addition to acute ACS, intra-abdominal pressure may become chronically elevated in patients with obesity or ascites. In this situation, the rise in intra-abdominal pressure occurs over a prolonged period of time and abdominal wall compliance increases concurrently, thus preventing the detrimental physiological effects of acute ACS.40 This condition is largely irrelevant, except that it may result in falsely high readings when assessing acute changes in intra-abdominal pressure in these patients.
Incidence
Papavasiliou et al41 reported that IAP was significantly higher after ruptured AAA (ruptured AAA) repair than either open or endovascular elective repair of nonruptured AAA. In the ruptured AAA group, 55% developed IAP values of greater than 15mmHg. Djavani et al42 reported that in their series of 17 ruptured AAA patients who underwent IAP monitoring, 9 (53%) patients developed IAP pressures greater than 20mmHg (IAH grade III and above) and 7 (41%) developed clinical ACS. However a limitation of this study was that IAP monitoring was restricted to complicated cases. As not all patients had IAP monitoring, the true incidence of ACS may be overestimated in their study. Mehta et al43 reported a 20% incidence of ACS in their 30 patients who underwent endovascular repair for ruptured AAA, and management of this syndrome is assuming increasing importance after endovascular surgery for ruptured AAA. ACS is rare following elective aortic surgery.44
Aetiology
In a similar fashion to LCS, raised intra-abdominal pressure can occur as a result of increased intra-abdominal volume (either retroperitoneal or intra-peritoneal) or extrinsic compression, which is usually due to changes in the abdominal wall, either iatrogenic or pathological.
Expansion of retroperitoneal volume can be caused by traumatic bleeding, pancreatitis or sepsis.45,46 More commonly intra-abdominal volume expansion is caused by intra-peritoneal expansion either by traumatic or iatrogenic bleeding, peritonitis, visceral oedema, or intra-abdominal packing for uncontrollable haemorrhage.47-51 Rarely visceral oedema can occur following nonabdominal trauma that is thought to be due to large volume fluid resuscitation.52
Extrinsic compression of the abdominal cavity can be caused by tight abdominal closure following laparotomy incisions, burns eschars, pneumatic anti-shock trousers and the repair of large hernias which results in an effective reduction of abdominal cavity volume.51,53-55 In addition high intra-thoracic pressure may lead to high intra-abdominal pressure.56
The pathophysiology of a ACS after ruptured AAA is multifactorial.41,57-59 the space occupying effect of large retroperitoneal haematoma (primary ACS) is a significant factor contributing to IAH, as dictated by the inverse relation between pressure and volume. In endovascular treatment of ruptured AAA, any ongoing type II endoleak bleeding from the lumbar and inferior mesenteric arteries into the disrupted aneurysm sac may contribute to the size of the haematoma and exacerbate the existing IAH. This is aggravated in the setting of severe coagulopathy.43 Furthermore, modifications in microvascular permeability associated with the shock state in ruptured AAA can lead to visceral and soft tissue oedema, worsening the IAH. 41,57-59 Secondary ACS (where pathology lies outside of the abdomen) is caused by massive fluid resuscitation-induced bowel oedema, ascites or through reperfusion injury associated with ruptured AAA.60
Pathological effects of raised intra-abdominal pressure
Elevated intra-abdominal pressure causes a reduction in mesenteric and hepatic arterial, intestinal and hepatic micro-circulatory, and portal venous blood flow.61,62 This reduction in the visceral blood flow occurs at pressures as low as 10mmHg and further impairment occurs with further increases of intra-abdominal pressure. As visceral blood flow reduces, ischaemia occurs, resulting in impaired cellular respiratory function and subsequent cellular damage. The acidosis which follows can be assessed by gastric tonometry. Reduction in gastric pH has been shown to occur early in ACS and this can be reversed by abdominal decompression.63 Whilst severe tissue damage has been shown to only occur at high intra-abdominal pressures (>40mmhg),64 gastrointestinal bacterial translocation occurs at much lower pressures (25mmhg).65 Since bacterial translocation has been implicated as a significant contributory factor to the development of multi-organ failure, this is an important effect of relatively low pressure ACS.
Renal impairment was one of the earliest noted effects of ACS.35 Progressive deterioration in renal function occurs as intra-abdominal pressure increases. Oliguria occurs at pressures above 15mmHg whereas pressures of greater than 30mmHg cause absolute anuria.44,66,67 Compression of the renal veins and direct renal parenchymal compression causes increased vascular resistance with a secondary reduction in renal perfusion.46,66 These changes cause a reduction in glomerular filtration rate and a subsequent increase in renin, aldosterone and anti-diuretic hormone occurs. Further increases in renal vascular resistance occur as a result and lead to retention of sodium and water. Ureteral compression does not appear to cause renal dysfunction in ACS since the placement of ureteral stents has been shown not to improve renal function in a ACS.68
Elevated intra-abdominal pressure affects not only those organs within the abdominal cavity but also has detrimental effects on distant organ systems. Cardiac output decreases as intra-abdominal pressure increases as a result of decreasing preload and increasing afterload.57 Preload is reduced due to direct compression of the abdominal inferior vena cava and compression of the superior vena cava due to increased intra-thoracic pressure as a result of direct transmission of elevated intra-abdominal pressure across the diaphragm. Elevated intrathoracic pressure also directly compresses the heart, reducing end diastolic volume. All of the above results in reduced stroke volume. A compensatory increase in heart rate occurs which only partially restores cardiac output.69 The reduced cardiac output caused by ACS also causes further impairment of renal function beyond that caused by ACS alone.
Respiratory dysfunction also occurs as intra-abdominal pressure increases.69 Direct transmission of elevated intra-abdominal pressure across the diaphragm causes elevations in intra-thoracic pressure, which in turn increases pulmonary vascular resistance. The volume of the thoracic cavity is also decreased due to elevation of the diaphragm, compressing the lungs. This compression results in reduced lung volume and compliance.70 These changes in the vasculature and physical properties of the lungs reduce respiratory function.
Raised intra-abdominal pressure causes increased intra-cerebral pressure and reduced cerebral perfusion that is thought to be due to reduced cerebral venous drainage.71,72 Also, abdominal wall blood flow is reduced in ACS due to direct compression and leads to ischaemia and muscle swelling.73 This, in turn, reduces abdominal wall compliance, exacerbating ACS.74
Clinical presentation
Clinical evaluation of patients with ACS is not reliable75 and the only physical sign due to ACS per se may be a tense, distended abdomen. The majority of clinical signs of ACS are due to the compromise of those organs systems affected by ACS – respiratory, renal, gastrointestinal and cardiovascular dysfunction. The classic collection of clinical presentations associated with ACS includes a tense abdomen on physical exam with oliguria and increased airway pressure.35 However these signs and symptoms are extremely nonspecific in critically ill patients, in whom ACS occurs most frequently. Critically ill patients frequently undergo large-volume fluid resuscitation and therefore commonly have impaired tissue perfusion, hypotension, and oliguria. Acute lung injury or pulmonary oedema are often seen in these same patients, either of which may result in increased airway pressure.76 Clinical examination therefore has a limited role in diagnosing ACS. In these patients the most important factor to consider is a history of an abdominal injury or intervention that places them at risk of developing ACS leading to active monitoring of IAP to detect ACS.
Investigation
The investigation of choice in a patient with suspected ACS is the measurement of intra-abdominal pressure. The most commonly applied technique is that described by Kron.35 This utilises an indwelling urinary catheter to obtain a direct measurement of intra-vesical pressure and has been shown to correlate well with intra-abdominal pressure.77 50ml of saline is introduced into the bladder via the aspiration port of the catheter which is clamped distal to this point. After allowing the pressure within the bladder to equilibrate with that in the abdominal cavity a pressure transducer (such as that used for measuring central venous pressure) is attached to an 18g needle inserted into the aspiration port and the pressure measured using the symphysis pubis as a reference point (‘zero’). The intra-abdominal pressure can then be measured. Modifications to this technique have been proposed to avoid the repeated disturbance of a closed system with the potential to introduce infection.78 This method has been shown to be inaccurate at low pressures (<15mmHg).79 Alternative techniques include intra-gastric pressure measurement80 and inferior vena caval pressure measurement via femoral vein catheterisation.81 Whilst gastric pressure has shown good correlation with bladder pressure measurements at low intra-abdominal pressure neither of these methods have been validated against the high bladder pressure measurement in humans with established ACS.82
Treatment
The principles of management for ACS are prevention, early recognition and definitive treatment of fully manifested ACS by surgical decompression. Prevention of ACS involves identification of at-risk patients by appreciating the risk factors, setting, and pathogenesis of ACS. Risk factors for ACS in vascular cases are massive fluid resuscitation (>5L/24hr), sepsis or bacteraemia, mechanical ventilation including use of positive end expiratory pressure, polytransfusion (>10U packed red blood cells/24hr) and acidosis.37 Intra operative risk factors for grade III or IV IAH for open ruptured AAA repair were noted to be longer cross clamping time, increased operative bleeding and increased operative time,42 and the following risk factors identified for endovascular repair of ruptured AAA: use of aortic occlusion balloon, presence of severe coagulopathy, massive transfusion requirements, and the emergent conversion of modular bifurcated stent grafts to aorto-uniiliac devices.43 Presence of these risk factors should alert the clinician to more aggressive monitoring of IAP and the introduction of preventive measures before fullscale ACS develops. In general the at-risk patients in whom IAP should be monitored are detailed in Table 19.2 as set out by the World Society of Abdominal Compartment Syndrome.37
TABLE 19.2
Indications for intra-abdominal pressure monitoring as defined by the world consensus definitions.
There are various nonsurgical treatment options available for dealing with elevated IAP83 to prevent manifestation of ACS. Some of these methods (nasogastric decompression, effective pain management and sedation) are used routinely in critically ill patients. Body positioning (avoidance of acute flexion at the hips and reverse Trendelenburg) can relieve pressure on the abdomen. Neuromuscular blockade can alleviate abdominal wall tensions leading to dramatic decrease in IAP.84,85 this can be administered quickly and safely to intubated ICU patients84,85 and is a useful adjunct to other nonoperative measures, allowing measures such as removal of excess fluid to become effective. Rectal decompression with enemas or rectal tubes, and prokinetic agents may potentially be used in selective cases such as suspected toxic megacolon or inflammatory bowel syndrome.
Other preventive measures include avoidance of excess fluid resuscitation therapy. The end points of resuscitation need to be accurately determined and unnecessary over-vigorous fluid administration should be averted by meticulous and precise intensive care monitoring.86 Fluid resuscitation should be goal-directed and titrated aggressively against achieving end points such as decrease in lactate, adequate mixed venous oxygen saturation and reductions in base deficit. Another important preventive measure against ACS is the use of prophylactic delayed abdominal closure and maintenance of an open abdomen. Delayed wound closure in ruptured AAA repair patients in whom primary closure was not possible demonstrated a trend towards decreased mortality from 73% in primary closure to 50% with delayed closure.87 An in-vitro abdominal simulation model demonstrated that the ‘Bogota bag’ technique which involves utilising a sterile fluid administration bag, cut flat and sewn to close the defect was most effective at preventing increases in abdominal pressures and proposed that vacuum dressing is contraindicated in the initial phase following decompression.88 However vacuum-pack temporary closure has advantages of ease of mastery, effectiveness in patient care and comfort, consistently low associated complication rate, and low cost in both general and vascular surgery and trauma patients. A series involving 258 surgical patients undergoing open abdomen management with the sutureless negative pressure dressing reported an overall primary fascial closure rate of 68.4%.89 It is still imperative that the ICU team avoid overresuscitation in patients who are admitted to the ICU with a temporary abdominal closure. Monitoring the IAPP during this period may provide guidance and if it is not possible to maintain an IAPP >60, then additional surgical intervention may be necessary.
The definitive treatment of ACS is expedient decompressive laparostomy. This has been shown to rapidly and effectively reverse the detrimental effects of ACS in the gastrointestinal, renal, cardiovascular, respiratory and central nervous systems.45,46,57,69,90-92 It has been shown that in clinically unstable patients with ACS, surgical decompression can be safely performed at the bedside in the ICU.93 Temporary containment of the abdominal contents is achieved using a mesh (silastic, polypropylene or polygalactin), plastic bag (intravenous fluid container or bowel bag) or vacuum systems (Figure 19.9a). Alternatively the skin can be closed leaving the fascia un-apposed. Following formation of the laparostomy consideration has to be given as to the method used to close the resulting defect. After the patient has recovered from the organ dysfunction associated with ACS delayed primary closure can be considered. The rate of unsuccessful closure of the abdomen has been reported between 20% and 78%.94,95 Coverage of the bowel, prevention and management of enterocutaneous fistulas, and management of large ventral hernias are some of the potential problems of an open abdomen. The incidence of failed closure and additional complications increases with delay and therefore efforts should be made to close the abdomen as soon as the underlying cause of the ACS has been dealt with. This window of opportunity is in general about 7 days but varies greatly from patient to patient, and actual timing should be tailored to the individual assessment of the abdomen. After this time the body’s attempt to heal secondarily will lead to development of adhesions and early granulation tissue culminating in a ‘frozen abdomen’ that prevents fascial closure. In cases where primary closure of the fascia is not possible, the abdominal contents should be left to granulate over and then either heal by secondary intention or skin grafts applied (Figure 19.9b). The resultant ventral hernia will then need to be repaired at a later date.
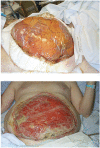
FIGURE 9
a) Immediate containment of abdominal contents using plastic bowel bag after decompressive laparostomy following ruptured abdominal aortic aneurysm repair. b) The same wound after 4 weeks, the viscera having granulated over.
Normal intra-abdominal pressure is less than 10.5mmHg in men and 8.8mmHg in women.96 Historical studies35,50 that proposed threshold pressures above 25mmHg to start definitive decompressive treatment have largely been disregarded. In patients with ruptured AAA, Papavassiliou et al41 demonstrated that an IAP threshold of 15mmHg was associated with significant physiological dysfunction, lower than ACS of other aetiology. The algorithm for the assessment and management of patients after abdominal aortic aneurysm surgery as proposed by Loftus et al,58 suggested immediate bladder pressure measurement of IAP in patients at risk after surgery and if the pressure is greater than 20mmHg, abdominal closure should be delayed. Following surgery patients should be monitored, if the IAP rises above 30mmHg urgent decompression should be mandatorily performed. In those with IAP between 21 to 30mmHg, urgent decompression should be considered. In those with IAP between 16 and 20mmHg urgent decompression should be considered in the presence of organ dysfunction. In those patients with IAP less than 15mmHg physiological support should be continued. Ganeshanatham et al60 proposed a lower threshold of IAP more than or equal to 25mmHg for which to perform mandatory decompression in post-operative vascular patients. At IAP of between 16 and 25mmHg, these authors suggested decompressive laparotomy if there has been no recovery of deranged physiological parameters despite optimisation.
Complications of surgical decompression
Much of the morbidity of ACS is due to the decompression surgery itself. Once healed the patient is frequently left with a large ventral hernia. With mesh closures, entero-laparostomy fistulas are not uncommon (7%) and the rate of mesh dehiscence from the fascia or skin was 22%.97 Enterocutaneous fistula arose in 5% of patients with negative pressure vacuum dressings.89 Infection is a potential complication of the temporary abdominal wound closures and this risk increases if more than one dressing application is needed.89 The temporary open wounds are not only an obvious point of entry for pathogens but are also a potential source of excessive fluid loss. In hypovolaemic patients, decompression may result in haemodynamic instability. Rebleeding can occur if coagulopathies are not reversed prior to the decompression. The increased pulmonary compliance from rapid reduction in IAP can lead to increased minute ventilation resulting in a respiratory alkalosis. The toxic metabolites entering the systemic circulation can precipitate a cardiac event as with any reperfusion injury,60
Outcome
ACS is associated with an overall high mortality of between 60% and 70%, not solely due to the development of ACS itself but in addition to the insult that caused it.98,99 A combined analysis of 18 papers with total of 250 patients who underwent decompressive surgery demonstrated that surgical decompression significantly reduced the mean IAP from 34.6mmHg to 15.5mmHg with associated improvements in respiratory function and cardiac output.100 With an average mortality of 49.2%, it has not been definitively demonstrated whether surgical decompression for ACS confers any beneficial overall effect on survival although in many studies, ACS without intervention results in mortality approaching 100%. Given this consideration and the potential risks of decompressive surgery, the best treatment for ACS is therefore prevention where possible.
References
- 1.
- Volkmann R. Die ischaemischen Muskellahmungen und kontrakturen. Centralbl Chir 1881; 8: 801–3.
- 2.
- Scott DJ, Allen MJ, Bell PR, McShane M, Barnes MR. Does oedema following lower limb revascularisation cause compartment syndromes? Ann R Coll Surg Engl 1988; 70: 372–6. [PMC free article: PMC2498632] [PubMed: 3207329]
- 3.
- Patman RD,. Thompson JE. Fasciotomy in peripheral vascular surgery. Report of 164 patients. Arch Surg 1970; 101: 663–72. [PubMed: 5489289]
- 4.
- Allenberg JR,. Meybier H. [The compartment syndrome from the vascular surgery viewpoint]. Chirurg 1988; 59: 722–7. [PubMed: 3234086]
- 5.
- Jensen SL,. Sandermann J. Compartment syndrome and fasciotomy in vascular surgery. A review of 57 cases. Eur J Vasc Endovasc Surg 1997; 13: 48–53. [PubMed: 9046914]
- 6.
- Papalambros EL, Panayiotopoulos YP, Bastounis E, Zavos G, Balas P. Prophylactic fasciotomy of the legs following acute arterial occlusion procedures. Int.Angiol. 1989; 8: 120–4. [PubMed: 2592793]
- 7.
- Abouezzi Z, Nassoura Z, Ivatury RR, Porter JM, Stahl WM. A critical reappraisal of indications for fasciotomy after extremity vascular trauma. Arch.Surg 1998; 133: 547–51. [PubMed: 9605919]
- 8.
- Hoffmeyer P, Cox JN, Fritschy D. Ultrastructural modifications of muscle in three types of compartment syndrome. Int Orthop 1987; 11: 53–9. [PubMed: 3557756]
- 9.
- Bown MJ, Nicholson ML, Bell PR, Sayers RD. Cytokines and inflammatory pathways in the pathogenesis of multiple organ failure following abdominal aortic aneurysm repair. Eur J Vasc Endovasc Surg 2001; 22: 485–95. [PubMed: 11735196]
- 10.
- Mubarak SJ, Hargens AR,Owen CA, Garetto LP, Akeson WH. The wick catheter technique for measurement of intramuscular pressure. A new research and clinical tool. J Bone Joint Surg Am 1976; 58: 1016–20. [PubMed: 977611]
- 11.
- Rorabeck CH, Castle GS, Hardie R, Logan J. Compartmental pressure measurements: an experimental investigation using the slit catheter. J Trauma 1981; 21: 446–9. [PubMed: 7230297]
- 12.
- Whitesides TE, Jr., Haney TC, Harada H, Holmes HE, Morimoto K. A simple method for tissue pressure determination. Arch Surg 1975; 110: 1311–3. [PubMed: 1191023]
- 13.
- Giannotti G, Cohn SM, Brown M, Varela JE, Mckenney MG, Wiseberg JA. Utility of near-infrared spectroscopy in the diagnosis of lower extremity compartment syndrome. J Trauma 2000; 48: 396–9. [PubMed: 10744275]
- 14.
- Qvarfordt P, Christenson JT, Eklof B, Ohlin P. Intramuscular pressure after revascularization of the popliteal artery in severe ischaemia. Br J Surg 1983; 70: 539–41. [PubMed: 6616159]
- 15.
- Holden CE. The pathology and prevention of Volkmann’s ischaemic contracture. J Bone Joint Surg Br 1979; 61–B: 296–300. [PubMed: 479252]
- 16.
- Mubarak SJ, Owen CA, Hargens AR, Garetto LP, Akeson WH. Acute compartment syndromes: diagnosis and treatment with the aid of the wick catheter. J Bone Joint Surg Am 1978; 60: 1091–5. [PubMed: 721856]
- 17.
- Allen MJ, Stirling AJ, Crawshaw CV, Barnes MR. Intracompartmental pressure monitoring of leg injuries. An aid to management. J Bone Joint Surg Br 1985; 67: 53–7. [PubMed: 3968144]
- 18.
- Whitesides TE, Haney TC, Morimoto K, Harada H. Tissue pressure measurements as a determinant for the need of fasciotomy. Clin Orthop Relat Res 1975; 43–51. [PubMed: 1192674]
- 19.
- McQueen MM,. Court-Brown CM. Compartment monitoring in tibial fractures. The pressure threshold for decompression. J Bone Joint Surg Br 1996; 78: 99–104. [PubMed: 8898137]
- 20.
- Moyer RA, Boden BP, Marchetto PA, Kleinbart F, Kelly JD. Acute compartment syndrome of the lower extremity secondary to noncontact injury. Foot Ankle 1993; 14: 534–7. [PubMed: 8314190]
- 21.
- Sheridan GW,. Matsen FA III, Fasciotomy in the treatment of the acute compartment syndrome. J Bone Joint Surg Am 1976; 58: 112–5. [PubMed: 1249096]
- 22.
- Williams AB, Luchette FA, Papaconstantinou HT, Lim E, Hurst JM, Johannigman JA et al. The effect of early versus late fasciotomy in the management of extremity trauma. Surgery 1997; 122: 861–6. [PubMed: 9347868]
- 23.
- Finkelstein JA, Hunter GA, Hu RW. Lower limb compartment syndrome: course after delayed fasciotomy. J Trauma 1996; 40: 342–4. [PubMed: 8601846]
- 24.
- Cooper GG. A method of singleincision, four compartment fasciotomy of the leg. Eur.J Vasc.Surg 1992; 6: 659–61. [PubMed: 1451825]
- 25.
- Mubarak SJ,. Owen CA. Doubleincision fasciotomy of the leg for decompression in compartment syndromes. J Bone Joint Surg Am 1977; 59: 184–7. [PubMed: 15455478]
- 26.
- Johnson SB, Weaver FA, Yellin AE, Kelly R, Bauer M. Clinical results of decompressive dermotomy-fasciotomy. Am J Surg 1992; 164: 286–90. [PubMed: 1415931]
- 27.
- Oredsson S, Arlock P, Plate G, Qvarfordt P. Metabolic and electrophysiological changes in rabbit skeletal muscle during ischaemia and reperfusion. Eur J Surg 1993; 159: 3–8. [PubMed: 8095803]
- 28.
- Perler BA, Tohmeh AG, Bulkley GB. Inhibition of the compartment syndrome by the ablation of free radical-mediated reperfusion injury. Surgery 1990; 108: 40–7. [PubMed: 2360189]
- 29.
- Shah DM, Bock DE, Darling RC, III, Chang BB, Kupinski AM, Leather RP. Beneficial effects of hypertonic mannitol in acute ischemia – reperfusion injuries in humans. Cardiovasc Surg 1996; 4: 97–100. [PubMed: 8634857]
- 30.
- Dabby D, Greif F, Yaniv M, Rubin M, Dekel S, Lelcuk S. Thromboxane A2 in postischemic acute compartmental syndrome. Arch Surg 1998; 133: 953–6. [PubMed: 9749846]
- 31.
- Bouachour G, Cronier P, Gouello JP, Toulemonde JL, Talha A, Alquier P. Hyperbaric oxygen therapy in the management of crush injuries: a randomized double-blind placebo-controlled clinical trial. J Trauma 1996; 41: 333–9. [PubMed: 8760546]
- 32.
- Ris HB, Furrer M, Stronsky S, Walpoth B, Nachbur B. Four-compartment fasciotomy and venous calf pump function: long-term results. Surgery 1993; 113: 55–8. [PubMed: 8417489]
- 33.
- McQueen MM, Christie J, Court-Brown CM. Acute compartment syndrome in tibial diaphyseal fractures. J Bone Joint Surg Br 1996; 78: 95–8. [PubMed: 8898136]
- 34.
- Matsen, FA III, Winquist RA, Krugmire RB, Jr. Diagnosis and management of compartmental syndromes. J Bone Joint Surg Am 1980; 62: 286–91. [PubMed: 7358759]
- 35.
- Kron IL, Harman PK, Nolan SP. The measurement of intra-abdominal pressure as a criterion for abdominal re-exploration. Ann Surg 1984; 199: 28–30. [PMC free article: PMC1353253] [PubMed: 6691728]
- 36.
- Cheatham ML, Malbrain ML, Kirkpatrick A, Sugrue M, Parr M, De Waele J et al. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. II. Recommendations. Intensive Care Med 2007; 33: 951–62. [PubMed: 17377769]
- 37.
- Malbrain ML, Cheatham ML, Kirkpatrick A, Sugrue M, Parr M, De Waele J et al.. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. I. Definitions. Intensive Care Med 2006; 32: 1722–32. [PubMed: 16967294]
- 38.
- Offner PJ, de Souza AL, Moore EE, Biffl WL, Franciose RJ, Johnson JL et al. Avoidance of abdominal compartment syndrome in damage-control laparotomy after trauma. Arch Surg 2001; 136: 676–81. [PubMed: 11387007]
- 39.
- Cheatham ML, White MW, Sagraves SG, Johnson JL, Block EF. Abdominal perfusion pressure: a superior parameter in the assessment of intra-abdominal hypertension. J Trauma 2000; 49: 621–6. [PubMed: 11038078]
- 40.
- Sugerman H, Windsor A, Bessos M, Wolfe L. Intra-abdominal pressure, sagittal abdominal diameter and obesity comorbidity. J Intern Med 1997; 241: 71–9. [PubMed: 9042096]
- 41.
- Papavassiliou V, Anderton M, Loftus IM, Turner DA, Naylor AR, London NJ et al.. The physiological effects of elevated intra-abdominal pressure following aneurysm repair. Eur J Vasc Endovasc Surg 2003; 26: 293–8. [PubMed: 14509893]
- 42.
- Djavani K, Wanhainen A, Bjorck M. Intra-abdominal hypertension and abdominal compartment syndrome following surgery for ruptured abdominal aortic aneurysm. Eur J Vasc Endovasc Surg 2006; 31: 581–4. [PubMed: 16458547]
- 43.
- Mehta M, Darling RC, III, Roddy SP, Fecteau S, Ozsvath KJ, Kreienberg PB et al. Factors associated with abdominal compartment syndrome complicating endovascular repair of ruptured abdominal aortic aneurysms. J Vasc Surg 2005; 42: 1047–51. [PubMed: 16376190]
- 44.
- Platell CF, Hall J, Clarke G, Lawrence-Brown M. Intra-abdominal pressure and renal function after surgery to the abdominal aorta. Aust NZ J Surg 1990; 60: 213–6. [PubMed: 2327926]
- 45.
- Fietsam R, Jr., Villalba M, Glover JL, Clark K. Intra abdominal compartment syndrome as a complication of ruptured abdominal aortic aneurysm repair. Am Surg 1989; 55: 396–402. [PubMed: 2729780]
- 46.
- Jacques T,. Lee R. Improvement of renal function after relief of raised intra-abdominal pressure due to traumatic retroperitoneal haematoma. Anaesth Intensive Care 1988; 16: 478–82. [PubMed: 3232805]
- 47.
- Ertel W, Oberholzer A, Platz A, Stocker R, Trentz O. Incidence and clinical pattern of the abdominal compartment syndrome after ‘damagecontrol’ laparotomy in 311 patients with severe abdominal and/or pelvic trauma. Crit Care Med 2000; 28: 1747–53. [PubMed: 10890613]
- 48.
- Offenbartl K,. Bengmark S. Intra-abdominal infections and gut origin sepsis. World J Surg 1990; 14: 191–5. [PubMed: 2183481]
- 49.
- Saggi BH, Sugerman HJ, Ivatury RR, Bloomfield GL. Abdominal compartment syndrome. J Trauma 1998; 45: 597–609. [PubMed: 9751558]
- 50.
- Sharp KW,. Locicero RJ. Abdominal packing for surgically uncontrollable hemorrhage. Ann Surg 1992; 215: 467–74. [PMC free article: PMC1242477] [PubMed: 1616383]
- 51.
- Smith PC, Tweddell JS, Bessey PQ. Alternative approaches to abdominal wound closure in severely injured patients with massive visceral edema. J Trauma 1992; 32: 16–20. [PubMed: 1732567]
- 52.
- Maxwell RA, Fabian TC, Croce MA, Davis KA. Secondary abdominal compartment syndrome: an underappreciated manifestation of severe hemorrhagic shock. J Trauma 1999; 47: 995–9. [PubMed: 10608523]
- 53.
- Greenhalgh DG,. Warden GD. The importance of intra-abdominal pressure measurements in burned children. J Trauma 1994; 36: 685–90. [PubMed: 8189471]
- 54.
- McSwain NE, Jr. Pneumatic anti-shock garment: state of the art 1988. Ann Emerg Med 1988; 17: 506–25. [PubMed: 3284422]
- 55.
- Pierri A, Munegato G, Carraro L, Zaccaria F, Tiso E, Zotti EF. Hemodynamic alterations during massive incisional hernioplasty. J Am.Coll.Surg 1995; 181: 299–302. [PubMed: 7551322]
- 56.
- Kopelman T, Harris C, Miller R, Arrillaga A. Abdominal compartment syndrome in patients with isolated extraperitoneal injuries. J Trauma 2000; 49: 744–7. [PubMed: 11038095]
- 57.
- Cullen DJ, Coyle JP, Teplick R, Long MC. Cardiovascular, pulmonary, and renal effects of massively increased intra-abdominal pressure in critically ill patients. Crit Care Med 1989; 17: 118–21. [PubMed: 2914444]
- 58.
- Loftus IM,. Thompson MM. The abdominal compartment syndrome following aortic surgery. Eur J Vasc Endovasc Surg 2003; 25: 97–109. [PubMed: 12552469]
- 59.
- Rasmussen TE, Hallett JW, Jr., Noel AA, Jenkins G, Bower TC, Cherry KJ, Jr. et al. Early abdominal closure with mesh reduces multiple organ failure after ruptured abdominal aortic aneurysm repair: guidelines from a 10–year case-control study. J Vasc Surg 2002; 35: 246–53. [PubMed: 11854721]
- 60.
- Ganeshanantham G, Walsh SR, Varty K. Abdominal compartment syndrome in vascular surgery –A review. Int J Surg 2010; 8: 181–5. [PubMed: 20074677]
- 61.
- Diebel LN, Dulchavsky SA, Wilson RF. Effect of increased intra-abdominal pressure on mesenteric arterial and intestinal mucosal blood flow. J Trauma 1992; 33: 45–8. [PubMed: 1635105]
- 62.
- Diebel LN, Wilson RF, Dulchavsky SA, Saxe J. Effect of increased intra-abdominal pressure on hepatic arterial, portal venous, and hepatic microcirculatory blood flow. J Trauma 1992; 33: 279–82. [PubMed: 1507294]
- 63.
- Ivatury RR, Porter JM, Simon RJ, Islam S, John R, Stahl WM. Intra-abdominal hypertension after lifethreatening penetrating abdominal trauma: prophylaxis, incidence, and clinical relevance to gastric mucosal pH and abdominal compartment syndrome. J Trauma 1998; 44: 1016–21. [PubMed: 9637157]
- 64.
- Gudmundsson FF, Gislason HG, Dicko A, Horn A, Viste A, Grong K et al. Effects of prolonged increased intra-abdominal pressure on gastrointestinal blood flow in pigs. Surg Endosc 2001; 15: 854–60. [PubMed: 11443466]
- 65.
- Diebel LN, Dulchavsky SA, Brown WJ. Splanchnic ischemia and bacterial translocation in the abdominal compartment syndrome. J Trauma 1997; 43: 852–5. [PubMed: 9390500]
- 66.
- Harman PK, Kron IL, McLachlan HD, Freedlender AE, Nolan SP. Elevated intra-abdominal pressure and renal function. Ann Surg 1982; 196: 594–7. [PMC free article: PMC1352794] [PubMed: 7125746]
- 67.
- Kirsch AJ, Hensle TW, Chang DT, Kayton ML, Olsson CA, Sawczuk IS. Renal effects of CO2 insufflation: oliguria and acute renal dysfunction in a rat pneumoperitoneum model. Urology 1994; 43: 453–9. [PubMed: 8154067]
- 68.
- Paramore RH. The Intra-abdominal Pressure in Pregnancy. Proc R Soc Med 1913; 6: 291–334. [PMC free article: PMC2006436] [PubMed: 19977092]
- 69.
- Ridings PC, Bloomfield GL, Blocher CR, Sugerman HJ. Cardiopulmonary effects of raised intra-abdominal pressure before and after intravascular volume expansion. J Trauma 1995; 39: 1071–5. [PubMed: 7500396]
- 70.
- Mutoh T, Lamm WJ, Embree LJ, Hildebrandt J, Albert RK. Abdominal distension alters regional pleural pressures and chest wall mechanics in pigs in vivo. J Appl Physiol 1991; 70: 2611–8. [PubMed: 1885456]
- 71.
- Bloomfield GL, Ridings PC, Blocher CR, Marmarou A, Sugerman HJ. Effects of increased intra-abdominal pressure upon intracranial and cerebral perfusion pressure before and after volume expansion. J Trauma 1996; 40: 936–41. [PubMed: 8656480]
- 72.
- Bloomfield GL, Ridings PC, Blocher CR, Marmarou A, Sugerman HJ. A proposed relationship between increased intra-abdominal, intrathoracic, and intracranial pressure. Crit Care Med 1997; 25: 496–503. [PubMed: 9118668]
- 73.
- Diebel L, Saxe J, Dulchavsky S. Effect of intra-abdominal pressure on abdominal wall blood flow. Am Surg 1992; 58: 573–5. [PubMed: 1388005]
- 74.
- Mutoh T, Lamm WJ, Embree LJ, Hildebrandt J, Albert RK. Volume infusion produces abdominal distension, lung compression, and chest wall stiffening in pigs. J Appl Physiol 1992; 72: 575–82. [PubMed: 1559935]
- 75.
- Kirkpatrick AW, Brenneman FD, McLean RF, Rapanos T, Boulanger BR. Is clinical examination an accurate indicator of raised intra-abdominal pressure in critically injured patients? Can J Surg 2000; 43: 207–11. [PMC free article: PMC3695163] [PubMed: 10851415]
- 76.
- Dry SM, Bechard KM, Milford EL, Churchill WH, Benjamin RJ. The pathology of transfusion-related acute lung injury. Am J Clin Pathol 1999; 112: 216–21. [PubMed: 10439802]
- 77.
- Iberti TJ, Kelly KM, Gentili DR, Hirsch S, Benjamin E. A simple technique to accurately determine intra-abdominal pressure. Crit Care Med 1987; 15: 1140–2. [PubMed: 3677766]
- 78.
- Cheatham ML, Safcsak K. Intra-abdominal pressure: a revised method for measurement. J Am, Coll Surg 1998; 186: 594–5. [PubMed: 9583702]
- 79.
- Johna S, Taylor E, Brown C, Zimmerman G. Abdominal compartment syndrome: does intracystic pressure reflect actual intra-abdominal pressure? A prospective study in surgical patients. Crit Care 1999; 3: 135–8. [PMC free article: PMC29028] [PubMed: 11056737]
- 80.
- Sugrue M, Buist MD, Lee A, Sanchez DJ, Hillman KM. Intra-abdominal pressure measurement using a modified nasogastric tube: description and validation of a new technique. Intensive Care Med 1994; 20: 588–90. [PubMed: 7706574]
- 81.
- Lacey SR, Bruce J, Brooks SP, Griswald J, Ferguson W, Allen JE et al. The relative merits of various methods of indirect measurement of intra-abdominal pressure as a guide to closure of abdominal wall defects. J Pediatr Surg 1987; 22: 1207–11 [PubMed: 2964519]
- 82.
- Collee GG, Lomax DM, Ferguson C, Hanson GC. Bedside measurement of intra-abdominal pressure (IAP) via an indwelling nasogastric tube: clinical validation of the technique. Intensive Care Med 1993; 19: 478–80. [PubMed: 8294633]
- 83.
- An G,. West MA. Abdominal compartment syndrome: a concise clinical review. Crit Care Med 2008; 36: 1304–10. [PubMed: 18379259]
- 84.
- De L, I, Hoste E, Verholen E, De Waele JJ. The effect of neuromuscular blockers in patients with intra-abdominal hypertension. Intensive Care Med 2007; 33: 1811–4. [PubMed: 17594072]
- 85.
- De Waele JJ, Benoit D, Hoste E, Colardyn F. A role for muscle relaxation in patients with abdominal compartment syndrome? Intensive Care Med 2003; 29: 332. [PubMed: 12675044]
- 86.
- Goodrich C. Endpoints of resuscitation: what should we be monitoring? AACN Adv Crit Care 2006; 17: 306–16. [PubMed: 16931926]
- 87.
- Oelschlager BK, Boyle EM, Jr., Johansen K, Meissner MH. Delayed abdominal closure in the management of ruptured abdominal aortic aneurysms. Am.J Surg 1997; 173: 411–5. [PubMed: 9168078]
- 88.
- Benninger E, Labler L, Seifert B, Trentz O, Menger MD, Meier C. In vitro comparison of intra-abdominal hypertension development after different temporary abdominal closure techniques. J Surg Res 2008; 144: 102–6. [PubMed: 17764694]
- 89.
- Barker DE, Green JM, Maxwell RA, Smith PW, Mejia VA, Dart BW et al. Experience with vacuum-pack temporary abdominal wound closure in 258 trauma and general and vascular surgical patients. J Am Coll Surg 2007; 204: 784–92. [PubMed: 17481484]
- 90.
- Irgau I, Koyfman Y, Tikellis JI. Elective intraoperative intracranial pressure monitoring during laparoscopic cholecystectomy. Arch Surg 1995; 130: 1011–3. [PubMed: 7661661]
- 91.
- Shelly MP, Robinson AA, Hesford JW, Park GR. Haemodynamic effects following surgical release of increased intra-abdominal pressure. Br J Anaesth 1987; 59: 800–5. [PubMed: 3300756]
- 92.
- Bloomfield GL, Dalton JM, Sugerman HJ, Ridings PC, DeMaria EJ, Bullock R. Treatment of increasing intracranial pressure secondary to the acute abdominal compartment syndrome in a patient with combined abdominal and head trauma. J Trauma 1995; 39: 1168–70. [PubMed: 7500414]
- 93.
- Diaz JJ, Jr., Mejia V, Subhawong AP, Subhawong T, Miller RS, O’Neill PJ et al.. Protocol for bedside laparotomy in trauma and emergency general surgery: a low return to the operating room. Am.Surg 2005; 71: 986–91. [PubMed: 16372620]
- 94.
- Fabian TC. Damage control in trauma: laparotomy wound management acute to chronic. Surg Clin.North Am 2007; 87: 73–93, vi. [PubMed: 17127124]
- 95.
- Joels CS, Vanderveer AS, Newcomb WL, Lincourt AE, Polhill JL, Jacobs DG et al. Abdominal wall reconstruction after temporary abdominal closure: A ten-year review. Surg Innov 2006; 13: 223–30. [PubMed: 17227920]
- 96.
- Sanchez NC, Tenofsky PL, Dort JM, Shen LY, Helmer SD, Smith RS. What is normal intra-abdominal pressure? Am.Surg 2001; 67: 243–8. [PubMed: 11270882]
- 97.
- Rasmussen TE, Hallett JW, Jr., Noel AA, Jenkins G, Bower TC, Cherry KJ, Jr. et al. Early abdominal closure with mesh reduces multiple organ failure after ruptured abdominal aortic aneurysm repair: guidelines from a 10-year case-control study. J Vase Surg 2002; 35: 246–53. [PubMed: 11854721]
- 98.
- Eddy V, Nunn C, Morris JA, Jr. Abdominal compartment syndrome. The Nashville experience. Surg Clin. North Am 1997; 77: 801–12. [PubMed: 9291982]
- 99.
- Meldrum DR, Moore FA, Moore EE, Franciose RJ, Sauaia A, Burch JM. Prospective characterization and selective management of the abdominal compartment syndrome. Am J Surg 1997; 174: 667–72. [PubMed: 9409594]
- 100.
- De Waele JJ, Hoste EA, Malbrain ML. Decompressive laparotomy for abdominal compartment syndrome – a critical analysis. Crit Care 2006; 10: R51. [PMC free article: PMC1550894] [PubMed: 16569255]
- Compartment syndromes from head to toe.[Crit Care Med. 2010]Compartment syndromes from head to toe.Balogh ZJ, Butcher NE. Crit Care Med. 2010 Sep; 38(9 Suppl):S445-51.
- Diagnosis and treatment of less common compartment syndromes of the upper and lower extremities: current evidence and best practices.[Instr Course Lect. 2011]Diagnosis and treatment of less common compartment syndromes of the upper and lower extremities: current evidence and best practices.Roberts CS, Gorczyca JT, Ring D, Pugh KJ. Instr Course Lect. 2011; 60:43-50.
- Review Common compartment syndromes in athletes. Treatment and rehabilitation.[Sports Med. 1994]Review Common compartment syndromes in athletes. Treatment and rehabilitation.Hutchinson MR, Ireland ML. Sports Med. 1994 Mar; 17(3):200-8.
- Endoscopically assisted release for exertional compartment syndromes of the lower leg.[Arch Orthop Trauma Surg. 2007]Endoscopically assisted release for exertional compartment syndromes of the lower leg.Lohrer H, Nauck T. Arch Orthop Trauma Surg. 2007 Nov; 127(9):827-34. Epub 2007 Feb 6.
- Review Current concepts in the treatment of common compartment syndromes in athletes.[Sports Med. 1993]Review Current concepts in the treatment of common compartment syndromes in athletes.Black KP, Taylor DE. Sports Med. 1993 Jun; 15(6):408-18.
- Compartment Syndromes - Mechanisms of Vascular DiseaseCompartment Syndromes - Mechanisms of Vascular Disease
- Pathophysiology of Vascular Graft Infections - Mechanisms of Vascular DiseasePathophysiology of Vascular Graft Infections - Mechanisms of Vascular Disease
Your browsing activity is empty.
Activity recording is turned off.
See more...