- 1.
All the specific reagents for gene editing (crRNA, tracrRNA-ATTO550, ssDNA template, Cas9 Nuclease) explained in this protocol were obtained from IDT (Integrated DNA Technology). However, it is important to note that there are several other companies that sell these same products performing equally well. It is the researcher’s decision to decide which company he wants to work with.
- 2.
The method of delivery and/or the transfection reagent will depend on our cell line or on our preferences. Lipofection with Lipofectamine™ CRISPRMAX™ Cas9 and its Transfection Reagent (Thermofisher) has been the method and the reagent chosen is this protocol.
- 3.
Before starting the gene editing experiment, it is important to check that the chosen cell line expresses the gene of interest (mRNA and protein) and corresponds to a tissue relevant for your studies, i.e., splicing defect was observed in this type of cells, as splicing outcomes may depend on tissue-specific splice factors. It is also essential to take into account the organism from which the cell line is derived. For example, intronic sequences are not well conserved among species, and this is crucial when, for example, the aim is to study intronic splicing mutations.
- 4.
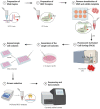
It is necessary to verify the karyotype of the chosen cell line, to confirm it is normal, at least in relation to the pair of chromosomes where the gene that is going to be edited is located. Most established cell lines show aneuplodies and structural chromosomal alterations that will hinder the desired gene edition if the corresponding chromosome is affected. In our case, we tested a battery of human hepatoma cell lines, Hep3B, HepG2, Huh7 among others and selected an HepG2 cell line with two chromosomes 12 where the PAH gene is located. Karyotype analysis is a routine service offered by many human genetic diagnosis laboratories.
- 5.
The chosen cell line should have the ability to form “single-cell colonies.” This is necessary to isolate individual cells after transfection that will be subsequently expanded for genetic characterization to confirm and select correctly gene edited clones. There are different procedures for the generation of “single-cell colonies”: (a) cell sorting: 1 cell/96-well-plate well using a cell sorter, (b) serial dilutions, and (c) seeding the cells at a high dilution (approximately 100 cells in one 150 mm plate).
The election of one method or another will depend on the cell line, so it is advisable to test this before generating the colonies with the edited cells. In our hands, for example, HepG2 cells exhibited high mortality after sorting and plating in 96-well plates, so we selected option c. For some cell lines the use of conditioned medium (filtered culture medium collected from control cells) can aid the growth in the form of a colony derived from a single cell. The time of growth and appearance of single-cell colonies will depend on the type of cells you are working with. With HepG2 cells, colonies emerged and reached the correct size after circa 20 days.
- 6.
It is advisable to have the region sequenced before starting the editing experiment to identify single-nucleotide polymorphisms in the specific cell line used which may affect the design of RNA guides and DNA templates, as well as result in erroneous interpretation of the sequencing analysis of the edited clones (concluding there has been an extra change introduced during DNA repair after Cas9 reaction when it was already present in the sequence prior to editing).
- 7.
There are multiple softwares for designing RNA guides for CRISPR assays. In our case we have used Breaking Cas software (http://bioinfogp.cnb.csic.es/tools/breakingcas) [19], which we find user-friendly, and the one offered by the company IDT, obtaining nearly identical results. In this sense, it is advisable to use and compare the results from at least two different softwares, to be sure that the selected guides are the most suitable.
- 8.
It is advisable to test at least two RNA guides in a parallel and independent way. Generally, according to IDT, in 2/3 of the cases, sense sequence guides will work better than antisense guides. As we cannot predict which ones will do best for a given locus, we recommend testing both orientations.
- 9.
As an optional step, you can pretest your RNA guides with an in vitro digestion after PCR amplification of the target region to confirm their efficiency (following IDT protocol).
- 10.
SnapGene Viewer (https://www.snapgene.com/) has been the software used for visualization of sequences used in this project, location of crRNA, DNA templates, restriction sites, etc. and for sequence analysis of the individual edited clones. However, other programs and software can be used.
- 11.
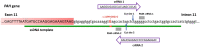
Cas9 nuclease cut site should be as close as possible to the sequence (nucleotide) which is to be edited. This requirement limits the region where we will design the RNA guides, especially if we want to introduce a point mutation as is the case here. It should be noted that this does not generally apply for the generation of a knock-out model, or in general, if we are not focused on introducing a mutation in a specific DNA position; in those cases the cut site can be in any position, so the design and choice of the RNA guide is much easier.
- 12.
If possible, it is recommended to choose an RNA guide targeting the region that includes the nucleotide we intend to edit. Once the edition of that locus has occurred, the affinity of our RNA guide is reduced (because of a mismatch due to the mutation introduced), thus hindering possible reediting.
- 13.
A ssDNA oligonucleotide containing the desired point mutation to be introduced is used as a template by the cell to repair the double strand break induced by Cas9 through HDR. The mutation of interest included in the ssDNA template should be in the middle of the sequence flanked by the homology arms. The length of the homology arms should be 35–40 nucleotides if it is a single-nucleotide change. Using longer homology arms does not increase the homologous recombination success rate. However, for longer edits (e.g. insertion/deletion of several nucleotides), the length of the homology arms must also be increased.
- 14.
For small insertions or single-nucleotide changes, ssDNA template is recommended. In other experimental situations (introduction of >100 nucleotide sequences) it may be advisable to use double stranded DNA templates.
- 15.
In most gene editing protocols, introducing translationally silent sequence changes in the DNA template eliminating the PAM sequence is recommended, to avoid reediting of our target which may introduce unwanted changes. However, when dealing with intronic or exonic splice mutations, any extra change may alter the final splicing outcome so this should be avoided.
- 16.
Standard desalting or HLPC are the purification methods recommended when ordering the ssDNA template. Also, especially in rich nucleases environments, phosphorothioate bonds (PS Bonds) at the extremes of the oligonucleotide are advisable, ideally putting at least two for each end of the template.
- 17.
This protocol is written to use separate crRNA and tracrRNA. There is also the possibility of working with single guide RNA, where both are linked together, so this step will be different, refer to manufacturer’s recommendations.
- 18.
The resuspension volumes depend on the amount of purchased crRNA and tracrRNA. A table of equivalences for different quantities is available in the IDT protocols. It is important to keep in mind that the resuspended RNA oligonucleotides can be stored at −20 °C. The volumes and quantities referred to in this protocol are calculated for a 6-well plate, which has been the format used by the authors. Refer to the protocols available on the IDT website for other formats (e.g. 96-well plate).
- 19.
The use of tracrRNA fused to the ATTO550 fluorophore is not strictly necessary but, in our hands, it was very useful for measuring transfection efficiency and to select transfected cells by fluorescence activated cell sorting (FACS) before clone generation. However, in cell types where transfection efficiency is known to be high/very high this step may be waived. In addition, there are certain cell types that are more prone to damage during the sorting process, so it would not be advisable to use this procedure to avoid increasing cell mortality. The protocol described can also be used for tracrRNA without ATTO550. In addition, it is important not to confuse transfection efficiency rate with editing efficiency, since a cell may have been transfected, but not edited. It is important to keep in mind that the success rate of the gene editing will not only depend on the quality of the guide, but also on the transfection method, the locus we are editing, the cell type, etc.
- 20.
The RNA Duplex can be prepared at a final concentration >1 μM and stored at −20 °C during, at least, 6 months. Before use, it should be diluted in Nuclease-Free Duplex Buffer to a working concentration of 1 μM.
- 21.
IDT provides Cas9 nuclease at a stock concentration of 62 μM. It can be diluted in different buffers, such as PBS or Cas9 Working Buffer (20 mM HEPES, 150 mM KCl, pH 7.5). This will depend on our cell type. In our case we have used OptiMEM to dilute the Cas9 enzyme. It will be important to take these details into account when purchasing Cas9 nuclease from other companies.
- 22.
The final concentration of the ssDNA template is variable depending on the cell type, delivery method, etc. In this case (transfection of HepG2 cells with Lipofectamine (CRISPRMax)), a final concentration of 3 nM ssDNA template was used, following the manufacturer’s recommendations. Transfecting higher amounts of ssDNA template does not ensure a higher rate of editing success. In addition, large amounts of DNA oligonucleotide can become toxic for the cells and increase cell mortality.
- 23.
In initial experiments, it is advisable to perform the reverse transfection of each crRNA guide in triplicate (three 6-well plate wells/crRNA).
- 24.
As with any transfection assay, it is advisable to split and pass the cells at least once after defrosting before starting the test.
- 25.
Before generating colonies derived from a single cell, it is important to freeze the remaining total pool of cells transfected with each crRNA. In the event of any problem we could defrost those cells to generate the colonies again without the need to repeat the transfection.
- 26.
Once the colonies have grown to a size allowing us to handle them efficiently, they must be expanded for analysis . You can select as many colonies as you can manage. You must consider that expansion, cultivation, and analysis of individual colonies require considerable effort and dedication. Normally we grow around 50–70 colonies for each crRNA used.
- 27.
The system used to select colonies and pick them can be very variable. For example, cloning cylinders can be used or other methods of choice of the researcher.
- 28.
We expanded the single-cell colonies in 24-well plates, but this can be modified according to the researcher’s preferences and/or cell line characteristics using plates with different formats. In our case, once the cells are confluent, we divide each well of the 24-well plate into two wells of a 12-well plate. It is important to keep accurate record of each duplicate, since one of them will be used to extract DNA for analysis , and the other will be used to freeze the colony and, in case it is the one selected, expand it for further characterization and use.
- 29.
The analysis of the colonies derived from a single cell is necessary to identify edited ones. In our case, the point mutation that we are introducing generates a new restriction site for the PshAI enzyme. This is very useful to rapidly and easily screen by RFLP analysis for the presence of the introduced mutation , although the edited region must be verified by sequence analysis . In some applications, translationally silent changes are introduced in the donor template near the mutation to create/destroy a restriction site, thus allowing RFLP screening. However, this is not recommended for splicing mutations as any nearby change may alter the splicing outcome. Alternative approaches to evaluate edition efficiency include next-generation sequencing approaches or digital droplet PCR.
- 30.
Other commercial kits or in-house methods can be used for DNA extraction.
- 31.
Other alternative software and resources can be used with the same objective. Primers are designed to amplify the region with the desired change, which should ideally be in the middle of the amplicon, so after digesting with the corresponding enzyme and running the products in an agarose gel we can easily distinguish digested and undigested DNA bands, which will facilitate the identification of the positive clone. Care should be taken during primer design to ensure that there are no other restriction sites for the corresponding enzyme (in our case PshAI) within the amplicon.
- 32.
Due to the high number of colonies, it is very laborious to analyze all of them individually. Therefore, it is advisable to make pools with DNA extracted from 4 or 5 colonies, mixing them to obtain 200 ng of total DNA. Once edition is observed in the RFLP analysis , colonies will then be analyzed individually.
- 33.
It is not necessary to purify the PCR products before restriction enzyme digestion. Purification of PCR products does not improve digestion efficiency, as the PCR product is diluted enough so that the different components of the PCR reaction do not interfere with the enzymatic activity.
- 34.
The conditions, temperatures, and times of the restriction reaction may depend on the enzyme and/or the trademark.
- 35.
Usually, amplification and subsequent sequencing of the three possible off-targets with the highest scores identified by the software used is enough. Based on our experience we can conclude that off-targets, although it is important to sequence and validate them, are not the biggest problem. However, we frequently found extra changes in the area near the edited nucleotide (on-target). In this sense, these errors have been the main problem and the cause of having had to discard many clones before finding the final positive one.
- 36.
There are many commercially available kits for purification of PCR products. We routinely use Cycle Pure Kit (Omega).
- 37.
Sequencing is necessary for the validation of the positive clone. And to discard off-target effects in the correctly edited clone. We should confirm that no extra changes have been made in the edited region. IMPORTANT: Do not confuse these random changes that CRISPR introduces when repairing the DSB in the DNA (on-target effects) with potential off-targets, which are locus to which our crRNAs can bind and induce a DSB in the DNA.
- 38.
It is important, once the positive clone is selected and genetically analyzed, to carry out the phenotypic characterization as cellular model of the disease phenotype, to confirm that it accurately recapitulates the splicing defect, resulting (in our case) in the absence of protein and activity. To that aim, RT-PCR and cDNA sequencing, followed by Western blot analysis of PAH protein and PAH activity assay were performed. The specific analyses to be performed will depend on each case according to the aim of the study, but, in the case of splicing mutations they should include at least RT-PCR and subsequent cDNA sequencing analysis .



 1.
1.