Copyright Notice
This is a work of the US government and distributed under the terms of the Public Domain
Bookshelf ID: NBK576210

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Leakey JEA, Allaben WT, Dunnick JK, et al. NTP Genetically Modified Model Report on the Toxicology and Carcinogenicity Studies of 3’-Azido-3’-Deoxythymidine (CASRN 30516-87-1) in Genetically Modified C3B6.129F1-Trp53tm1Brd N12 Haploinsufficient Mice (In Utero and Postnatal Gavage Study): NTP GMM 14 [Internet]. Research Triangle Park (NC): National Toxicology Program; 2013 Oct.

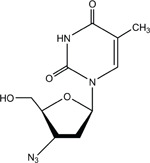
CAS No. 30516-87-1
Chemical Formula: C10H13N5O4 Molecular Weight: 267.24
Synonyms: AZT; 3′-azido-2′,3′-dideoxythymidine; azidodeoxythymidine; azidothymidine; 3′-azidothymidine; 3′-deoxy-3′-azidothymidine; 3′-deoxy-(8CI) (9CI); BW A509U; Compound S; ZDV; zidovudine
Trade Name: Retrovir®
Antiviral therapy is essential for treatment and prevention of human immunodeficiency virus (HIV) disease in adults and children and to prevent mother-to-child transmission of HIV (DHHS, 2009). The studies described in this report were designed to determine possible long-term sequelae from 3′-azido-3′-deoxythymidine (AZT) treatment, often used in combination with other antivirals, in preventing mother-to-child transmission of HIV.
AZT is a dideoxynucleoside of thymine and a structural analogue of 2′-deoxythymidine and is the most widely used and evaluated chemotherapeutic agent for the treatment of persons with acquired immune deficiency syndrome (AIDS) and persons seropositive for HIV.
AZT is a white to off-white, odorless, crystalline solid and is moderately soluble in water (20 mg/mL) and alcohol (71 mg/mL) at 25° C. Aqueous solutions of AZT are clear to pale yellow and are mildly acidic (e.g., pH 5.5 for 10 mg AZT/mL solution) (NTP, 2006).
AZT was first synthesized by Horowitz et al. (1964), and it was subsequently reported by Mitsuya et al. (1985) to inhibit HIV replication in vitro at concentrations ranging from 50 to 500 nmol/L. Clinical activity for the treatment of AIDS was first reported by Yarchoan et al. (1986), and the drug was commercially developed by Burroughs Wellcome Company (Research Triangle Park, NC) under the trade name Retrovir®, which was approved by the Food and Drug Administration (FDA) in March 1987, as the first nucleoside analogue reverse transcriptase inhibitor (NRTI) for the treatment of adult patients with AIDS or advanced AIDS-related complex (Anonymous, 1987; Brook, 1987). By May 2007, patent restrictions on all forms of AZT had expired, allowing the FDA to complete approval of a full range of generic alternatives for patented AZT formulations (Anonymous, 2007). Currently, AZT is available in capsules or syrup for oral administration and in formulations suitable for intravenous infusion. These include: solutions containing 10 mg AZT/mL for oral or intravenous administration, capsules of either 100 or 300 mg of AZT alone, combination capsules containing either 300 mg AZT and 150 mg of the NRTI 3TC (lamivudine, 2′-deoxy-3′-thiacytidine), and combination capsules containing 300 mg AZT and 150 mg of 3TC and 300 mg of the NRTI ABC [abacavir, (1S,4R)-4-(2-amino-6-(cyclopropylamino)-9H-purin-9-yl)-2-cyclopentene-1-methanol] (DHHS, 2008a).
Antiviral therapy is essential for treatment and prevention of AIDS in adults and children infected with HIV and to prevent mother-to-child transmission of HIV during pregnancy and labor (DHHS, 2008a,b). AZT is a primary drug of choice for pediatric prophylactic monotherapy in infants at risk for contracting HIV and as part of the triple drug combination Highly Active Antiretroviral Therapy (HAART) for infants and children infected with HIV (DHHS, 2008b). Typical pediatric doses range from 1.5 mg/kg in intravenous infusion or oral doses of 2 mg/kg every 12 hours for term neonates to 100 mg three times daily for children weighing more than 30 kg (DHHS, 2008b). AZT is also included as part of a therapeutic regimen to prevent mother-to-child transmission of HIV and has been shown to prevent the vertical transmission of HIV by nearly 70% (7.2% in treated patients versus 21.9% in a placebo control group; Connor et al., 1994). AZT is a primary drug of choice for combination HAART or prophylactic treatment in pregnant women who are HIV positive and is the primary drug of choice for intravenous infusion during labor (PHS, 2008).
In 2007, it was estimated that 30 to 36 million people worldwide were living with HIV infections, of whom approximately half were women (UNAIDS, 2008). During 2007, 2.2 to 3.2 million new infections occurred, while 1.8 to 2.3 million people died. The 2007 estimates suggest that there are 0.69 to 1.9 million people living with HIV in the United States, of whom 140,000 to 400,000 are women (UNAIDS, 2008). Earlier estimates (Steinbrook, 2004; WHO, 2004) have suggested that worldwide, some 2.2 million HIV-infected women give birth each year and that approximately 700,000 of their neonates become infected.
The antiviral activity of AZT depends on its conversion to a nucleotide triphosphate (3′-azido-2′,3′-dideoxythymidine 5′-triphosphate; AZTTP). AZT enters mammalian cells by non-facilitated diffusion (Zimmerman et al., 1987), and it is then phosphorylated in successive reactions catalyzed in proliferating cells primarily by thymidine kinase 1, thymidylate kinase, and nucleoside diphosphokinase present in cell cytosol (Avramis et al., 1989; Törnevik et al., 1995; Bradshaw et al., 2005). The resulting nucleoside triphosphate, AZTTP, is a substrate for HIV reverse transcriptase and a competitive inhibitor of deoxythymidine triphosphate. Because the 3′ position of AZT is blocked with an azido group, incorporation of AZTTP into a growing polynucleotide chain (e.g., viral DNA) terminates elongation at that position. Thus, AZT intervenes at a relatively early stage of the viral replication cycle. AZTTP is also a substrate for cellular DNA polymerases; however, the Ki and Km of AZTTP for HIV reverse transcriptase are considerably lower than for cellular DNA polymerases. Accordingly, AZTTP inhibits viral replication at doses lower than those at which it is an efficient substrate for the cellular DNA polymerases (Furman et al., 1986; Huang et al., 1990; Parker et al., 1991).
Following oral administration, AZT is rapidly absorbed, and after oral or intravenous administration, it is rapidly distributed (NTP, 2006). Elimination is also rapid, with essentially all parent drug and its metabolites being completely excreted within 24 hours (Singlas et al., 1989; Taburet et al., 1990; Child et al., 1991; Stagg et al., 1992). However, there are significant interspecies differences in the extent to which the parent compound is metabolized (NTP, 2006). In humans and monkeys, the majority of an administered dose is converted to the 5′-O-glucuronide and eliminated in urine along with unmetabolized parent drug and a minor metabolite, 3′-amino-2′,3′dideoxythymidine (AMT), formed by reduction of the 3′-azido group of AZT.
However, in rodents, the majority of absorbed AZT is eliminated in urine as the parent compound with relatively little conversion to the glucuronide or to AMT (Singlas et al., 1989; de Miranda et al., 1990; Good et al., 1990; Cretton et al., 1991; Mays et al., 1991; Stagg et al., 1992; Trang et al., 1993). The reduced metabolism in rodents results in equivalent doses producing greater Cmax values than for humans, but plasma half-life values are similar to humans (Doshi et al., 1989; Patel et al., 1989; Trang et al., 1993). The glucuronide of AMT has been reported to be a minor urinary metabolite in monkeys and a minor biliary metabolite in humans, but it has not been identified in rats (de Miranda et al., 1990).
In animals, AZT causes hematologic toxicities (Thompson et al., 1991) and impaired function of the electron transport chain in cardiac (Walker et al., 2004) and skeletal (Lamperth et al., 1991) muscle mitochondria. The latter are associated with morphological damage including enlarged mitochondria with disorganized or absent cristae (Lamperth et al., 1991; Lewis et al., 1992). In Sprague-Dawley rats administered 1 mg AZT/mL in drinking water for 35 days, decreases in mitochondrial DNA, RNA, and protein synthesis were observed in skeletal muscle mitochondria (Lewis et al., 1992). AZTTP is an inhibitor and alternate substrate for mitochondrial DNA polymerase γ from both skeletal and cardiac muscle (Simpson et al., 1989; Lewis et al., 1994), and therefore, it is possible that AZT, via AZTTP, is acting as an inhibitor and chain terminator, disrupting mitochondrial DNA synthesis. However, more recent evidence suggests that AZT is also a potent inhibitor of thymidine phosphorylation in mitochondria as a result of its inhibition of mitochondrial thymidine kinase 2 (Lynx and McKee 2006; Lynx et al., 2006; Susan-Resiga et al., 2007). This enzyme plays a secondary role in the conversion of thymidine into thymidine monophosphate in rapidly dividing cells, which express cytosolic thymidine kinase 1, but in non-mitotic cells, such as mature hepatocytes or cardiac myocytes, it provides the primary source of thymidine triphosphate that is required for mitochondrial DNA replication and nuclear DNA repair (Pérez-Pérez et al., 2005; Samuels, 2006). AZT inhibits thymidine kinase 2 at lower concentrations (7 to 14 μM) than AZTTP inhibits DNA polymerase γ (Lynx and McKee, 2006), and pharmacological levels of AZT would be expected to result in accumulative mitochondrial damage and increased observed mutation frequencies through this mechanism.
Heart toxicities associated with AZT treatment have been reported in rats, mice, and monkeys. For example, rats exposed to approximately 29 to 102 mg AZT/kg body weight per day in drinking water for up to 49 days developed cardiac mitochondrial swelling with fractured and disrupted cristae (Lewis et al., 1991). These ultrastructural defects did not reverse after a 14-day recovery period. Ultrastructural examination of cardiomyocytes of Sprague-Dawley rats exposed to
AZT in drinking water at approximately 90 mg/kg per day showed disruption of cristae and increased size of mitochondria after 30 or 60 days of exposure; no alterations were seen in rats after 120 days of exposure (Corcuera Pindado et al., 1994). Sprague-Dawley rats given intraperitoneal injections of approximately 17 to 51 mg AZT/kg body weight for 3 months developed enlarged cardiomyocytic mitochondria with disorganized or absent cristae and increased serum concentrations of creatine kinase, lactate, and glucose (Lamperth et al., 1991).
Transgenic mice (that express replication incompetent HIV) or FVB mice exposed to AZT in drinking water at doses of approximately 180 to 200 mg AZT/kg body weight for 35 days developed cardiac toxicity characterized by mitochondrial destruction (Lewis et al., 2000). Treatment-related histopathologic changes were described as numerous cardiomyocytes with granular cytoplasm in normal and transgenic mice. The lesions were generally more severe in transgenic mice. Neither interstitial inflammation nor fibrosis was found. The National Toxicology Program (NTP) (1999) did not report cardiac toxicity in B6C3F1/N mice administered AZT by oral gavage in corn oil at doses of 0, 50, 100, 200, 800, or 2,000 mg/kg for 14 days, or at doses of 0, 30, 60, or 120 mg/kg for 2 years. However, in an NTP study where Swiss (CD-1®) mouse pups were exposed in utero, via lactation, and by direct gavage on postnatal days (PND) 4 through 28 with twice-daily doses of 75/37.5 mg/kg AZT/3TC or the vehicle control mixture of 0.1% polysorbate 80 and 0.2% methylcellulose, the hearts of PND 28 pups treated with AZT/3TC showed significant increases in the mean area and decreases in the mean number of cardiomyocytic mitochondria compared to vehicle controls (Bishop et al., 2004a; NTP, 2006). AZT has also been shown to cause alteration in fat metabolism in rats; male Wistar rats exposed to AZT (0.6 mg/mL in drinking water) for 4 weeks exhibited increased serum triglyceride levels and decreased cytochrome c oxidase and fatty acid synthase activities in their inguinal fat (Deveaud et al., 2007).
Studies in monkeys at the National Cancer Institute showed that daily doses of AZT during the second half of gestation at approximately 86% of the recommended human daily dose caused mitochondrial abnormalities (Gerschenson et al., 2000), which were similar to those observed in human neonates exposed to antiretroviral drugs (Divi et al., 2007). In skeletal muscle, these abnormalities were characterized as abnormally shaped mitochondria with disrupted cristae (Gerschenson et al., 2000). In heart muscle, small mitochondria in myocytes with myofibrillar loss and abnormal alignment of sarcomeres were observed.
Exposure to AZT has resulted in myelosuppression and anemia in some human patients as in experimental animals. In humans, this toxicity limits the useful therapeutic dose range of AZT (Fischl, 1989; Pluda et al., 1991; Balzarini, 1994). The primary target of AZT toxicity is the hematopoietic system of the bone marrow; in vitro coculture studies have demonstrated that AZT is cytotoxic to human and murine hematopoietic progenitor cells (Sommadossi and Carlisle, 1987; Dainiak et al., 1988; Gallicchio et al., 1989). In cultures of human bone marrow cells, the extent of incorporation of AZTTP into cellular DNA and the growth inhibition of human clonal peripheral blood mononu-clear cells have been correlated (Sommadossi and Carlisle, 1987; Sommadossi et al., 1989). In human erythroid K-562 leukemia cells induced to differentiate by butyric acid treatment, AZT selectively reduced the steady-state level of globin mRNA (Weidner and Sommadossi, 1990). Neither the kinetics of induction nor the steady-state mRNA levels of other components of the heme biosynthetic pathway were altered, including erythroid-specific isozymes of aminolevulinate synthase and porphobilinogen deaminase (Fowler et al., 1995). These results suggest a specific effect on transcription of the globin gene in erythroid cells.
A few patients receiving long-term AZT therapy have been reported to have toxic mitochondrial myopathy (Dalakas et al., 1990). Clinical symptoms include myalgia, muscle weakness, and elevated levels of creatinine kinase in serum. These symptoms correlate with the presence in muscle biopsies of abnormal mitochondria containing paracrystalline inclusions. Human muscle myotubes grown in tissue culture exposed to AZT for 9 days exhibited increased numbers of mitochondria as well as enlarged mitochondria with abnormal cristae and electron-dense deposits in the matrix (Lamperth et al., 1991).
The United States Department of Health and Human Services updates information on current treatment regimens for HIV and observed toxicities on an ongoing basis (PHS, 2008). Common adverse effects noted from AZT use in humans include bone marrow suppression, anemia and/or neutropenia, and subjective complaints including gastrointestinal intolerance, headache, insomnia, and asthenia. In addition, lactic acidosis with hepatic steatosis has been reported as a rare side effect from the NRTI components used in HAART, including AZT.
NRTIs such as AZT have been reported to produce mitochondrial dysfunction in human patients (Dalakas et al., 1990; Arnaudo et al., 1991; Lamperth et al., 1991; Brinkman et al., 1999; DHHS, 2008a), possibly resulting from inhibition of human mitochondrial DNA polymerase γ or thymidine kinase 2. Mitochondrial DNA dysfunction may result in pancreatitis, peripheral neuropathy, myopathy, and cardiomyopathy (Lim and Copeland 2001; DHHS, 2008a). It is thought that combinations of NRTIs will act synergistically to induce mitochondrial dysfunction. Protease inhibitors may also aggravate this mechanism (DHHS, 2008a). Lipodystrophy (fat redistribution syndrome) may be seen in patients receiving NRTIs and is related to mitochondrial toxicities (Brinkman et al., 1999; Kakuda et al., 1999; Mallal et al., 2000; DHHS, 2008a). Metabolic complications of HAART include vascular necrosis, decreased bone density, and skin rashes (DHHS, 2008a). A recent study suggests that mitochondrial damage in children of mothers taking AZT may persist for up to 2 years after birth; however, HIV infection by itself may lead to cardiac toxicity (Artandi et al., 2000; Raidel et al., 2002).
AZT has been shown to cross the placenta of mice (Child et al., 1991) and of monkeys (Ewings et al., 2000). In a series of studies in rats, mice, and rabbits, AZT has been shown to cause adverse reproductive effects but no overt teratogenic effects. AZT was evaluated for adverse effects on reproductive and fetal development in CD (Sprague-Dawley) rats and New Zealand White rabbits (Greene et al., 1996). Male and female CD rats were given twice-daily oral AZT doses of 0, 25, 75, or 225 mg/kg, approximately 6 hours apart. Males were dosed for 85 days prior to mating and continued on dosing throughout two mating cycles for a total of 175 dosing days. Treated males were mated to females (F0) dosed for 26 days prior to mating and throughout gestation and lactation. Early resorptions and decreased litter size were noted following parental dosing with 75 or 225 mg/kg. In a second mating, treated males were mated to untreated females and the pups were monitored for growth, survival, and developmental characteristics. All reproductive parameters were normal. The authors concluded that the embryotoxicity of AZT noted with the first mating (treated males with treated females) was not mediated by a genotoxic effect in the males. The live-born off-spring showed no developmental abnormalities or teratogenic effects. Also in these studies, pregnant New Zealand White rabbits given an oral dose of 250 mg AZT/kg body weight per day from gestational day (GD) 6 until 18 had reduced body weight gain, anemia, and increased late fetal deaths. The live-born offspring showed no developmental abnormalities or teratogenic effects.
When pregnant CD-1® mice were administered daily intragastric doses of 25 mg AZT/kg body weight per day from GD 12 to 18, no developmental toxicity was seen in the F1 generation (Diwan et al., 2000). When these treated offspring were mated to untreated off-spring, the live-born F2 pups showed no adverse effects on reproductive parameters. Other studies have shown that AZT can cause cytotoxic effects in preimplantation mouse embryos by inhibition of blastocyst and postblastocyst development at doses similar to human therapeutic doses (Toltzis et al., 1991; DHHS, 2008b).
There have been no reported increases in congenital abnormalities in infants born to women with antepartum AZT exposure (Connor et al., 1994; Sperling et al., 1998). The National Institutes of Health (NIH) panel cautions that definitive conclusions regarding teratogenic risk cannot be thoroughly evaluated because of limited numbers of children evaluated (Corcuera Pindado et al., 1994; DHHS, 2008a; PHS, 2008). When AZT crosses the human placenta, it is incorporated into the DNA of cord blood leukocytes (Olivero et al., 1999).
Preclinical studies in rodents were conducted by GlaxoSmithKline to determine the potential for toxicity and/or cancer from exposure to AZT. AZT was administered to CD rats by oral gavage once a day at 0, 80, 220, or 600 mg AZT/kg body weight per day for up to 2 years (Ayers et al., 1996a). Because of anemia, the high dose was reduced to 450 mg/kg per day at day 91; on day 278, the high dose was again reduced to 300 mg/kg per day. Squamous cell carcinoma of the vagina occurred in two females receiving 300 mg/kg; no vaginal neoplasms/hyperplasia occurred in any other group of female rats. These investigators also administered AZT to CD-1® mice by oral gavage at 0, 30, 60, or 120 mg/kg per day. Because of anemia, the doses were reduced to 0, 20, 30, or 40 mg/kg per day at day 90, where they remained for the rest of the 22-month study of AZT.
The only neoplasms associated with administration of AZT in the mice were vaginal squamous cell carcinoma in five females receiving 40 mg/kg, vaginal squamous cell papilloma in one female receiving 30 mg/kg and in one female receiving 40 mg/kg, and one vaginal squamous polyp in a female receiving 40 mg/kg. Although the incidences of hyperplasia of the vaginal epithelium were not increased above that in the controls, the severity of this lesion increased with increasing dose. In order to clarify the role of AZT in producing vaginal neoplasms, AZT was administered intravaginally to CD-1® mice for 22 months (Ayers et al., 1996b). Higher incidences of vaginal neoplasms occurred than were seen in the AZT oral gavage study in CD-1® mice. There was a retrograde flow of urine from the discharge point at the base of the vulva into the region of the vagina where the vaginal neoplasms occurred. In mice, 90% of AZT is eliminated in the urine as the parent compound following oral administration. Because there is a high rate of cell turnover in the vaginal epithelium as a consequence of the short estrous cycle in mice (4 to 5 days), the investigators concluded that prolonged exposure of the vaginal epithelium to the relatively high concentrations of AZT in the urine could explain the observed vaginal neoplasms. In humans, the concentration of free AZT in the urine is low, and the authors concluded that the vaginal neoplasms seen in mice would not necessarily be predictive of vaginal neoplasms in humans.
The NTP’s 2-year chronic studies of AZT and AZT/interferon were conducted in B6C3F1/N mice (NTP, 1999). AZT was administered to male and female mice by oral gavage at doses of 0, 30, 60, or 120 mg/kg per day in two equal doses, at least 6 hours apart, 5 days per week for 105 weeks. In the AZT/interferon studies, male and female mice received AZT by oral gavage at daily doses of 0, 30, 60, or 120 mg/kg body weight, given in two equal doses, 5 days per week for 105 weeks; the groups receiving AZT also received subcutaneous injections of 500 or 5,000 U α-interferon A/D three times per week for 105 weeks. Additional groups of 80 male and 80 female mice received subcutaneous injections of the vehicle, 500 U α-interferon A/D, 5,000 U α-interferon A/D, or 5,000 U α-interferon A (all without AZT), three times per week for 105 weeks. There was equivocal evidence of carcinogenic activity of AZT in male mice based on marginally increased incidences of renal tubule and Harderian gland neoplasms in groups receiving AZT alone. There was clear evidence of carcinogenic activity of AZT in female mice based on increased incidences of squamous cell neoplasms of the vagina in groups that received AZT alone (2/197, 0/49, 5/45, 11/49) or in combination with α-interferon A/D (0/49, 0/44, 5/48, 6/48). Hematotoxicity occurred in all groups that received AZT. Treatment with AZT alone and AZT in combination with α-interferon A/D resulted in increased incidences of epithelial hyperplasia of the vagina in all dosed groups of females.
In a follow-up transplacental exposure study (NTP, 2006), Swiss (CD-1®) mice were administered 0, 50, 100, 200, or 300 mg AZT/kg body weight per day by oral gavage from 10 to 14 days prior to conception until GD 19, and up to 4 pups (F1 generation) from each litter were followed for 2 years and evaluated for carcinogenicity. Under the conditions of this study, there was clear evidence of carcinogenic activity in F1 male mice exposed transplacentally to AZT based on increased incidences of alveolar/bronchiolar neoplasms (14/50, 20/50, 13/50, 7/37, 8/32; Poly-3 survival-adjusted rates of 40.2%, 54.5%, 40.5%, 66.3%, 72.9%). There was no evidence of carcinogenic activity in F1 female mice exposed transplacentally to AZT at 50, 100, 200, or 300 mg/kg.
Studies by the National Cancer Institute suggest that AZT, when given at relatively high doses, is a moderately effective perinatal carcinogen in mice, targeting several tissue types (Olivero et al., 1997; Diwan et al., 1999). In these studies, AZT was given to CD-1® mice at doses of 12.5 or 25 mg (equivalent to up to 1,000 mg AZT/kg nonpregnant body weight or 450 mg AZT/kg of terminal body weight) orally from GD 12 through GD 18. AZT was incorporated into nuclear and mitochondrial DNA of the fetuses. A dose-dependent increase in tumor multiplicity in the lung, liver, and female reproductive organs occurred. However, in a transplacental carcinogenicity study using lower doses, CD-1® mice were given 20 or 40 mg AZT/kg body weight per day in the drinking water from gestation day 10 through lactation day 21 (Ayers et al., 1997). Some of the pups from these litters were then continued on AZT treatment by daily gavage at doses of 20 or 40 mg/kg per day for 24 months. AZT tumor findings were limited to the vaginal epithelium.
A more recent study exposed B6C3F1 mice and Fischer 344 rats to relatively high doses (80, 240, 480 mg/kg) of AZT during gestation and evaluated the animals for tumor incidence at 24 months of age (Walker et al., 2007). In male mice, there were statistically significant increases in the incidences of hemangiosarcoma in all three dosed groups and of hepatocellular carcinoma in the highest dose group. In female mice there was a statistically significant increase in the incidence of neoplasms of the uterus in the 480 mg/kg group. In female rats, the incidences of mononuclear cell leukemia were increased in all three dosed groups.
There have been no studies reported in the literature on any association between AZT and/or HAART and cancer. However, a recent epidemiology study reported that patients with HIV or AIDS do have increased risk of developing lung cancer (Kirk et al., 2007). Since the increased risk was not significantly correlated with either HAART or low CD4 cell count, it is not yet known whether AZT exposure contributes to this increased cancer risk. Cancer often takes many years to develop, and follow-up of patients is continuing (Antiretroviral Pregnancy Registry, 2003). An NIH panel has recommended long-term follow-up in children receiving in utero exposure to AZT and other antiretroviral drugs (Corcuera Pindado et al., 1994).
AZT is a DNA-reactive chemical that is positive in the Salmonella mutation assay (NTP, 1999) and has been shown to increase mutation frequencies and induce chromosomal damage in mammalian cells in vivo and in vitro. Its genotoxic effects have been extensively reviewed in previous NTP technical reports (NTP, 1999, 2006). A brief summary of the extensive genetic toxicity literature follows.
AZT was reported to be weakly positive in the mouse lymphoma cell mutagenicity test (Ayers, 1988; Olin and Kastrup, 1995) and to induce transformation in cultured mammalian cells (Olin and Kastrup, 1995). Results of in vitro cytogenetic assays with mammalian cells showed that AZT induced sister chromatid exchanges, chromosomal aberrations, and micronuclei in human lymphocytes, as well as chromosomal aberrations and sister chromatid exchanges in cultured Chinese hamster ovary cells (Gonzalez Cid and Larripa, 1994). In cyto-genetic studies in Chinese hamster ovary cells conducted by the NTP, sister chromatid exchanges were remarkably elevated by AZT, particularly in the absence of S9 activation, but no induction of chromosomal aberrations was observed (NTP, 1999).
In vivo, AZT has been shown in several studies to be an effective inducer of micronucleated erythrocytes (markers of chromosomal damage) in rats and mice exposed through various combinations of routes and exposure durations (Phillips et al., 1991; Dertinger et al., 1996; Von Tungeln et al., 2002; Bishop et al., 2004b; Witt et al., 2004). For example, significantly increased micronucleus frequencies (6 to 27 times the frequency in concurrent controls) were noted in peripheral blood and bone marrow erythrocytes of mice after multiple treatments with 100 to 2,000 mg AZT/kg body weight per day for periods of 72 hours, 96 hours, or 90 days. However, other studies using lower doses have reported no increase in micronucleus frequency (Motimaya et al., 1994). A recent study by Dobrovolsky et al. (2007) using C3B6F1-Trp53tm1Brd p53+/− haploinsufficient mice reported that transplacental followed by neonatal exposure to AZT increased the reticulocyte micronucleus frequency 4.8-, 7.1-, and 11.3-fold when measured on PND 1 with maternal doses of 48, 80, and 160 mg AZT/kg body weight per day respectively, and 10.3-, 10.6-, and 26.5-fold when measured on PND 10 with maternal/neonatal doses of 40/20, 80/40, and 160/80 mg AZT/kg body weight per day, respectively.
Incorporation of AZT into the DNA of leukocytes and multiple organs of cynomolgus monkeys was demonstrated following a 30-day treatment period (40 mg/day by nasogastric intubation) (Olivero et al., 2001). Organ specific differences in the amount of AZT incorporation were noted, and the average levels of incorporation were similar to what had been reported for human leukocytes (Olivero et al., 2000).
Pregnant CD-1® mice and Erythrocebus patas monkeys were treated with AZT (mice, 12.5 or 25 mg/day; monkeys, 10 mg/day) during critical periods of gestation, and AZT incorporation into both nuclear and mitochondrial DNA, along with telomere length of chromosomes, was measured in the newborns (Comstock et al., 1993). The transplacentally exposed animals showed significant AZT incorporation into nuclear as well as mitochondrial DNA of several organs, and decreased telomere lengths were seen in chromosomes from liver and brain cells of mice but not monkeys. Similarly, a human-equivalent dose of AZT (8 mg/kg) administered continuously over 4 hours to pregnant rhesus macaques just prior to hysterotomy at the end of gestation resulted in AZT incorporation into DNA extracted from cells of several fetal organs (Poirier et al., 1999).
A study conducted in neonatal CD-1® mice reported that treatment with AZT (150 mg/kg per day) for 4 weeks resulted in reduced mtDNA copy number and increased mtDNA lesions in heart mitochondria evaluated on PND 28 (Chan et al., 2007). Females appeared to be more susceptible to damage than males.
In humans, HAART designed to stop mother-to-child HIV transmission has been reported to increase frequencies of micronuleated reticulocytes in blood samples from mothers and their babies taken from the cord blood and during the first postnatal week. Percent micronucleated reticulocyte frequencies were up to 10-fold greater in blood from infants exposed to HAART incorporating AZT than from those exposed to HAART that did not include AZT (Witt et al., 2007).
The relevance of the positive results from animal mutation studies to humans is not yet clear, but numerous investigations have yielded data supporting a potential for genetic damage in humans exposed to AZT and other nucleoside analogues (Olivero, 2007, 2008). Several studies have demonstrated increased mutation frequencies in cultured human lymphoblastoid cells following AZT exposure. For example, there are a number of studies showing incorporation of AZT into DNA of human lymphoblastoid cells, followed by loss of heterozygosity at loci for the thymidine kinase 1, hypoxanthine phosphoribosyltransferase, and adenine phosphoribosyltransferase genes, resulting in significant increases in mutant frequencies (Sussman et al., 1999; Meng et al., 2000a,b,c). Analysis of the AZT-induced mutational spectra in cultured human lymphoblastoid cells showed an increase in complete gene deletions, a result consistent with DNA chain termination, and loss of heterozygosity (Meng et al., 2002). In vivo, anti-AZT radioimmunoassays were used to demonstrate that AZT is incorporated into lymphocyte DNA of HIV-infected adults taking AZT (Olivero et al., 2000).
Currently there is some evidence that AZT exposure in conjunction with HAART can result in chromosomal damage in humans. An early paper by Shafik et al. (1991) reported significantly increased chromosomal aberration frequencies in lymphocytes of AIDS patients treated with AZT alone when compared to a healthy control group. In a more recent study, children born to HIV-infected mothers who received treatment with AZT and other NRTIs were evaluated periodically for up to 9 years after birth (Senda et al., 2007). Heterochromatin analysis of blood leukocytes showed an increased frequency (P<0.001) of chromatin dispersal in samples from children exposed to NTRIs (predominantly AZT) as compared to frequencies from children of HIV-infected mothers who were not exposed to NTRIs. The heterochromatin defects persisted long after the end of the exposure period and were present in leukocytes of both myeloid and lymphoid lineages, suggesting that hematopoietic stem cells were affected.
The p53 tumor suppressor gene suppresses cancer in both humans and mice. The p53 protein is critical to cell cycle control, DNA repair and apoptosis, etc., and is often mutated or lost in human and rodent cancers. The haploinsufficient Trp53 tumor suppressor gene mouse model heterozygous for wildtype and null (+/–) Trp53 alleles (Donehower et al., 1992, 1995) was used in these studies. In this model, a Trp53 null mutation was introduced by homologous recombination in AB1 murine embryonic stem cells that were derived from a black agouti 129Sv inbred mouse. By targeted insertion of a polII neo cassette, an engineered null mutation was induced as a result of the deletion of a 450-base pair gene fragment from the Trp53 gene that included 106 nucleotides of exon 5 and approximately 350 nucleotides of intron 4 that eliminated both mRNA and p53 protein expression from this allele. This Trp53 protein haploinsufficient mouse model has been extensively tested as a short-term cancer bioassay mouse model (Tennant et al., 1995; Dunnick et al., 1997; French et al., 2001a,b; Pritchard et al., 2003; French, 2004) based upon the observation that mice with only a single wildtype Trp53 allele show a significant decrease in the time required for genotoxic carcinogen-induced tumors to develop. These tumors are often associated with either a mutation and/or a loss of heterozygosity of the remaining wildtype Trp53 allele. Few to no sporadic tumors occur in concurrent or historical study control groups in this GMM model, which allows tests to be conducted with fewer animals and direct analysis of the target wildtype Trp53 allele to test for genotoxicity in vivo as a mode of action.
For these studies, an outcross between C3H/HeNTac (C3) female mice homozygous for the wildtype (+) Trp53 allele and the C57BL/6.129Sv-Trp53tm1Brd N12 congenic (abbreviated B6.129-Trp53tm1Brd) N12 back-cross generation males homozygous for the Trp53 null (−) allele produced C3B6.129F1/Tac-Trp53tm1Brd N12 progeny heterozygous for a Trp53 wildtype (+) and null allele (−) inbred mouse progeny [hereafter referred to in the abbreviated form as the heterozygous F1 p53+/− mouse, Taconic Laboratory Animals and Services (Germantown, NY)]. The heterozygous F1 p53+/− mouse was selected for the 30- and 45-week studies of AZT because the B6.129-Trp53tm1Brd (N5) haplo-insufficient male and female mice (backcrossed to C57BL/6, subline unspecified, for two generations and then to C57BL/6NTac females for an additional three generations) were not sufficiently inbred. The N5 generation of this line retained both C57BL/6 and 129Sv strain allele heterozygosity at both the Trp53 locus and the flanking region on chromosome 11 and at unknown loci throughout the genome of this line. This residual heterozygosity in the B6.129-Trp53tm1Brd N5 backcross generation mice was one covariate that may have been responsible for large variations in the p-cresidine-induced urinary bladder tumors (0% to 80%, 10 of 11 studies were positive) in males, which was used as a positive control genotoxic carcinogen in the ILSI/HESI Alternatives to Carcinogenicity Testing initiative (Storer et al., 2001). Therefore, additional inbreeding to the N12 generation was anticipated to decrease the variance in tumor incidence and stabilize the penetrance of tumor phenotypes in NTP studies.
The majority of B6.129-Trp53tm1Brd homozygous null females die in utero and only a few are born alive and most die early. Thus, the B6.129-Trp53tm1Brd N12 line is maintained by intercross of the B6.129-Trp53tm1Brd female heterozygote with the B6.129-Trp53tm1Brd homo-zygous null male to produce a 1:2 population of homo-zygous null males and heterozygous null males and females. Therefore it is necessary to select the B6.129-Trp53tm1Brd homozygous null male as the carrier of the null allele. However, the selection of the C3H/HeNTac female as the wildtype Trp53 allele carrier provides 1) increased fecundity and maternal instincts, 2) increased hybrid vigor of an F1 outcross that increases the number of progeny, 3) the advantage of expanding the pattern of tumor susceptibility associated with this genetic background, and 4) a genetic background similar to the B6C3F1/N mouse used in NTP studies (NTP, 2013). Together, these factors provided a rational basis for selection of this GMM test model. In addition, the NTP study reported on senna also used the C3B6.129F1/Tac-Trp53tm1Brd N12 haploinsufficient GMM model (NTP, 2012), and the background rate for spontaneous tumors in the control group C3B6.129F1-Trp53tm1Brd haploinsufficient mice in both studies (AZT and senna) was not statistically different from the background rates for spontaneous tumors observed in control B6.129-Trp53tm1Brd (N5) haploinsufficient mice used in previous NTP GMM studies (NTP, 2005a,b, 2007a,b,c,d,e, 2008).
The development of HAART to combat the AIDS pandemic was a major public health triumph of the late 20th century. For countries where HAART drugs are widely available, they have transformed HIV infection from a death sentence into a manageable chronic disease. In the United States, the FDA has made a significant contribution to this triumph by rapidly evaluating and approving new antiretroviral drugs. An estimated 7,000 infants are born to HIV-infected women in the United States every year, and due to the implementation of HAART, the vast majority of these infants escape infection (Steinbrook, 2004; UNAIDS, 2008).
In the United States, AZT is a primary drug of choice for treating pregnant women who are HIV positive to prevent transmission of the virus to their children. AZT is either used alone or in combination with other antiretroviral drugs. Because AZT has only been in use for less than 25 years, its long-term toxicological impact on children exposed in utero or in infancy is currently unknown. As outlined in the previous sections, AZT has been shown to be both genotoxic and a rodent carcinogen.
The studies described in this report were designed to determine possible long-term sequelae from AZT treatment, often used in combination with other antiviral drugs, in preventing mother-to-child transmission of HIV. The C3B6.129F1-Trp53tm1Brd N12 haplo-insufficient mouse was selected for these studies because previous NTP studies had shown that p53 mutations were associated with AZT-induced carcinogenesis processes (NTP, 2006). It was hypothesized that when a p53 gene change is involved in the multiple genetic steps to cancer, a mouse deficient in this gene will develop cancer in a shorter time period than in the traditional 2-year mouse carcinogenesis studies. The studies were therefore designed to determine whether genetically modified mice that are haploinsufficient for the p53 gene would provide a suitable model for (1) further evaluating the potential carcinogenicity of AZT and (2) determining whether current and future HAART drug combinations incorporating AZT may potentiate the harmful side effects of AZT. The current study was conducted in conjunction with ongoing NTP-sponsored 2-year studies of AZT, alone or in combination with other HAART drugs, in B6C3F1 mice exposed either during gestation, via maternal gavage, or during gestational exposure followed by neonatal exposure via direct oral gavage to the pups (NTP, 2013).
Doses of AZT were selected based on range-finding studies conducted in B6C3F1 mice performed in conjunction with the previously mentioned 2-year studies. The heterozygous F1 p53+/− mice were exposed to AZT transplacentally via maternal dosing at the same doses that were used for the 2-year studies (NTP, 2013). These transplacental AZT doses were 80, 160, or 240 mg/kg per day from GD 12 though 18 given to the dams as a single daily gavage. These doses fall within the range of doses used in other animal studies that varied from 20 mg/kg per day (Ayers et al., 1997) to 800 mg/kg (Olivero et al., 1997). Pregnant women receiving HAART typically receive doses of 8 to 9 mg/kg per day (NTP, 2013). According to traditional dose scaling calculations (Freireich et al., 1966), this is approximately equivalent to 100 mg/kg per day for adult mice and by the same scaling formulas the selected dam doses of 80, 160, and 240 mg AZT/kg body weight per day were estimated as equivalent to daily AZT doses of 6.5, 13.0, and 19.5 mg/kg, respectively, for a pregnant woman. In the current studies, dosing was continued by daily oral gavage of the pups throughout postnatal development until scheduled evaluation at either 30 or 45 weeks of age. Pup dosing was initiated on PND 1, but the doses and dose volumes were reduced by half from PND 1 to PND 10 to compensate for immaturity of detoxification systems. Continuous dosing until evaluation was used to maximize exposure. A 45-week evaluation time was selected so that the age of the mice would match that of other GMM studies that utilized p53 haploinsufficient mice (e.g., NTP, 2007e) where dosing was initiated at 6 to 7 weeks of age and continued for 39 weeks. Additional mice from the 0/0/0 mg/kg and 240/120/240 mg/kg groups were evaluated at 30 weeks of age (30-week study) to help establish the rate of tumor progression in heterozygous F1 p53+/− mice. Additional mice that were dosed only up to PND 8 (45-week stop-study) were also incorporated into the current studies. This dosing regime (0/0 and 240/40 mg/kg per day) was identical to that used in the concurrent ongoing 2-year studies in B6C3F1 mice and allowed a direct comparison between the studies.
This is a work of the US government and distributed under the terms of the Public Domain
Your browsing activity is empty.
Activity recording is turned off.
See more...