By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2001, Garland Science.
Bookshelf ID: NBK27100

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Janeway CA Jr, Travers P, Walport M, et al. Immunobiology: The Immune System in Health and Disease. 5th edition. New York: Garland Science; 2001.

Complement was discovered many years ago as a heat-labile component of normal plasma that augments the opsonization of bacteria by antibodies and allows antibodies to kill some bacteria. This activity was said to ‘complement’ the antibacterial activity of antibody, hence the name. Although first discovered as an effector arm of the antibody response, complement can also be activated early in infection in the absence of antibodies. Indeed, it now seems clear that complement first evolved as part of the innate immune system, where it still plays an important role.
The complement system is made up of a large number of distinct plasma proteins that react with one another to opsonize pathogens and induce a series of inflammatory responses that help to fight infection. A number of complement proteins are proteases that are themselves activated by proteolytic cleavage. Such enzymes are called zymogens and were first found in the gut. The digestive enzyme pepsin, for example, is stored inside cells and secreted as an inactive precursor enzyme, pepsinogen, which is only cleaved to pepsin in the acid environment of the stomach. The advantage to the host of not being autodigested is obvious.
In the case of the complement system, the precursor zymogens are widely distributed throughout body fluids and tissues without adverse effect. At sites of infection, however, they are activated locally and trigger a series of potent inflammatory events. The complement system activates through a triggered-enzyme cascade. In such a cascade, an active complement enzyme generated by cleavage of its zymogen precursor then cleaves its substrate, another complement zymogen, to its active enzymatic form. This in turn cleaves and activates the next zymogen in the complement pathway. In this way, the activation of a small number of complement proteins at the start of the pathway is hugely amplified by each successive enzymatic reaction, resulting in the rapid generation of a disproportionately large complement response. As might be expected, there are many regulatory mechanisms to prevent uncontrolled complement activation. The blood coagulation system is another example of a triggered-enzyme cascade. In this case, a small injury to a blood vessel wall can lead to the development of a large thrombus.
There are three distinct pathways through which complement can be activated on pathogen surfaces. These pathways depend on different molecules for their initiation, but they converge to generate the same set of effector molecules (Fig. 2.7). There are three ways in which the complement system protects against infection. First, it generates large numbers of activated complement proteins that bind covalently to pathogens, opsonizing them for engulfment by phagocytes bearing receptors for complement. Second, the small fragments of some complement proteins act as chemoattractants to recruit more phagocytes to the site of complement activation, and also to activate these phagocytes. Third, the terminal complement components damage certain bacteria by creating pores in the bacterial membrane.
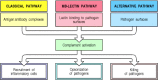
Schematic overview of the complement cascade. There are three pathways of complement activation: the classical pathway, which is triggered by antibody or by direct binding of complement component C1q to the pathogen surface; the MB-lectin pathway, which (more...)
In the early phases of an infection, the complement cascade can be activated on the surface of a pathogen through any one, or more, of the three pathways shown in Fig. 2.8. The classical pathway can be initiated by the binding of C1q, the first protein in the complement cascade, directly to the pathogen surface. It can also be activated during an adaptive immune response by the binding of C1q to antibody:antigen complexes, and is thus a key link between the effector mechanisms of innate and adaptive immunity. The mannan-binding lectin pathway (MB-lectin pathway) is initiated by binding of the mannan-binding lectin, a serum protein, to mannose-containing carbohydrates on bacteria or viruses. Finally, the alternative pathway can be initiated when a spontaneously activated complement component binds to the surface of a pathogen. Each pathway follows a sequence of reactions to generate a protease called a C3 convertase. These reactions are known as the ‘early’ events of complement activation, and consist of triggered-enzyme cascades in which inactive complement zymogens are successively cleaved to yield two fragments, the larger of which is an active serine protease. The active protease is retained at the pathogen surface, and this ensures that the next complement zymogen in the pathway is also cleaved and activated at the pathogen surface. By contrast, the small peptide fragment is released from the site of the reaction and can act as a soluble mediator.

Overview of the main components and effector actions of complement. The early events of all three pathways of complement activation involve a series of cleavage reactions that culminate in the formation of an enzymatic activity called a C3 convertase, (more...)
The C3 convertases formed by these early events of complement activation are bound covalently to the pathogen surface. Here they cleave C3 to generate large amounts of C3b, the main effector molecule of the complement system, and C3a, a peptide mediator of inflammation. The C3b molecules act as opsonins; they bind covalently to the pathogen and thereby target it for destruction by phagocytes equipped with receptors for C3b. C3b also binds the C3 convertase to form a C5 convertase that produces the most important small peptide mediator of inflammation, C5a, as well as a large active fragment, C5b, that initiates the ‘late’ events of complement activation. These comprise a sequence of polymerization reactions in which the terminal complement components interact to form a membrane-attack complex, which creates a pore in the cell membranes of some pathogens that can lead to their death.
The nomenclature of complement proteins is often a significant obstacle to
understanding this system, and before discussing the complement cascade in more
detail, we will explain the conventions, and the nomenclature used in this book.
All components of the classical complement pathway and the membrane-attack
complex are designated by the letter C followed by a number. The native
components have a simple number designation, for example, C1 and C2, but
unfortunately, the components were numbered in the order of their discovery
rather than the sequence of reactions, which is C1, C4, C2, C3, C5, C6, C7, C8,
and C9. The products of the cleavage reactions are designated by added
lower-case letters, the larger fragment being designated b and the smaller a;
thus, for example, C4 is cleaved to C4b, the large fragment of C4 that binds
covalently to the surface of the pathogen, and C4a, a small fragment with weak
pro-inflammatory properties. The components of the alternative pathway, instead
of being numbered, are designated by different capital letters, for example
factor B and factor D. As with the classical pathway, their cleavage products
are designated by the addition of lower-case a and b: thus, the large fragment
of B is called Bb and the small fragment Ba. Finally, in the mannose-binding
lectin pathway, the first enzymes to be activated are known as the
mannan-binding lectin-associated
serine proteases MASP-1 and MASP-2, after which the
pathway is essentially the same as the classical pathway. Activated complement
components are often designated by a horizontal line, for example,
 ; however, we will not use this
convention. It is also useful to be aware that the large active fragment of C2
was originally designated C2a, and is still called that in some texts and
research papers. Here, for consistency, we will call all large fragments of
complement b, so the large active fragment of C2 will be designated C2b.
; however, we will not use this
convention. It is also useful to be aware that the large active fragment of C2
was originally designated C2a, and is still called that in some texts and
research papers. Here, for consistency, we will call all large fragments of
complement b, so the large active fragment of C2 will be designated C2b.
The formation of C3 convertase activity is pivotal in complement activation, leading to the production of the principal effector molecules, and initiating the late events. In the classical and MB-lectin pathways, the C3 convertase is formed from membrane-bound C4b complexed with C2b. In the alternative pathway, a homologous C3 convertase is formed from membrane-bound C3b complexed with Bb. The alternative pathway can act as an amplification loop for all three pathways, as it is initiated by the binding of C3b.
It is clear that a pathway leading to such potent inflammatory and destructive effects, and which, moreover, has a series of built-in amplification steps, is potentially dangerous and must be subject to tight regulation. One important safeguard is that key activated complement components are rapidly inactivated unless they bind to the pathogen surface on which their activation was initiated. There are also several points in the pathway at which regulatory proteins act on complement components to prevent the inadvertent activation of complement on host cell surfaces, hence protecting them from accidental damage. We will return to these regulatory mechanisms later.
We have now introduced all the relevant components of complement and are ready for a more detailed account of their functions. To help distinguish the different components according to their functions, we will use a color code in the figures in this part of the chapter. This is introduced in Fig. 2.9, where all the components of complement are grouped by function.

Functional protein classes in the complement system.
The classical pathway plays a role in both innate and adaptive immunity. As we will see in Chapter 9, the first component of this pathway, C1q, links the adaptive humoral immune response to the complement system by binding to antibodies complexed with antigens. C1q can, however, also bind directly to the surface of certain pathogens and thus trigger complement activation in the absence of antibody. C1q is part of the C1 complex, which comprises a single C1q molecule bound to two molecules each of the zymogens C1r and C1s. C1q is a calcium-dependent sugar-binding protein, a lectin, belonging to the collectin family of proteins, which contains both collagen-like and lectin domains hence the name collectin. It has six globular heads, linked together by a collagen-like tail, which surround the (C1r:C1s)2 complex (Fig. 2.10). Binding of more than one of the C1q heads to a pathogen surface causes a conformational change in the (C1r:C1s)2 complex, which leads to activation of an autocatalytic enzymatic activity in C1r; the active form of C1r then cleaves its associated C1s to generate an active serine protease.
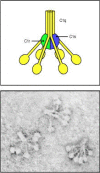
The first protein in the classical pathway of complement activation is C1, which is a complex of C1q, C1r, and C1s. C1q is composed of six identical subunits with globular heads and long collagen-like tails. The tails combine to bind to two molecules each (more...)
Once activated, the C1s enzyme acts on the next two components of the classical pathway, cleaving C4 and then C2 to generate two large fragments, C4b and C2b, which together form the C3 convertase of the classical pathway. In the first step, C1s cleaves C4 to produce C4b, which binds covalently to the surface of the pathogen. The covalently attached C4b then binds one molecule of C2, making it susceptible, in turn, to cleavage by C1s. C1s cleaves C2 to produce the large fragment C2b, which is itself a serine protease. The complex of C4b with the active serine protease C2b remains on the surface of the pathogen as the C3 convertase of the classical pathway. Its most important activity is to cleave large numbers of C3 molecules to produce C3b molecules that coat the pathogen surface. At the same time, the other cleavage product, C3a, initiates a local inflammatory response. These reactions, which comprise the classical pathway of complement activation, are shown in schematic form in Fig. 2.11; the proteins involved, and their active forms, are listed in Fig. 2.12.

The classical pathway of complement activation generates a C3 convertase that deposits large numbers of C3b molecules on the pathogen surface. The steps in the reaction are outlined here and detailed in the text. The cleavage of C4 by C1s exposes a reactive (more...)

The proteins of the classical pathway of complement activation.
The MB-lectin pathway uses a protein very similar to C1q to trigger the complement cascade. This protein, called the mannan-binding lectin (MBL), is a collectin, like C1q. Mannan-binding lectin binds specifically to mannose residues, and to certain other sugars, which are accessible and arranged in a pattern that allows binding on many pathogens. On vertebrate cells, however, these are covered by other sugar groups, especially sialic acid. Thus, mannan-binding lectin is able to initiate complement activation by binding to pathogen surfaces. It is present at low concentrations in normal plasma of most individuals, and, as we will see in the last part of this chapter, its production by the liver is increased during the acute-phase reaction of the innate immune response.
Mannan-binding lectin, like C1q, is a six-headed molecule that forms a complex with two protease zymogens, which in the case of the mannanbinding lectin complex (MBL complex) are MASP-1 and MASP-2 (Fig. 2.13). MASP-1 and MASP-2 are closely homologous to C1r and C1s, and all four enzymes are likely to have evolved from gene duplication of a common precursor. When the MBL complex binds to a pathogen surface, MASP-1 and MASP-2 are activated to cleave C4 and C2. Thus the MB-lectin pathway initiates complement activation in the same way as the classical pathway, forming a C3 convertase from C2b bound to C4b. People deficient in mannan-binding lectin experience a substantial increase in infections during early childhood, indicating the importance of the MB-lectin pathway for host defense. The age window of susceptibility to infections associated with mannan-binding lectin deficiency illustrates the particular importance of innate host defense mechanisms in childhood, before the child’s adaptive immune responses are fully matured and after maternal antibodies transferred across the placenta and in colostrum have been lost.

Mannan-binding lectin forms a complex with serine proteases that resembles the complement C1 complex. MBL forms clusters of two to six carbohydrate-binding heads around a central collagen-like stalk. This structure, easily discernible under the electron (more...)
We have seen that the classical and MB-lectin pathways of complement activation are initiated by proteins that bind to pathogen surfaces. During the triggered-enzyme cascade that follows, it is important that activating events are confined to this same site, so that C3 activation also occurs on the surface of the pathogen, and not in the plasma or on host cell surfaces. This is achieved principally by the covalent binding of C4b to the pathogen surface. Cleavage of C4 exposes a highly reactive thioester bond on the C4b molecule that allows it to bind covalently to molecules in the immediate vicinity of its site of activation. In innate immunity, C4 cleavage is catalyzed by a C1 or MBL complex bound to the pathogen surface, and C4b can bind adjacent proteins or carbohydrates on the pathogen surface. If C4b does not rapidly form this bond, the thioester bond is cleaved by reaction with water and this hydrolysis reaction irreversibly inactivates C4b (Fig. 2.14). This helps to prevent C4b from diffusing from its site of activation on the microbial surface and becoming coupled to host cells.

Cleavage of C4 exposes an active thioester bond that causes the large fragment, C4b, to bind covalently to nearby molecules on the bacterial cell surface. Intact C4 consists of an α, a β, and a γ chain with a shielded thioester (more...)
C2 becomes susceptible to cleavage by C1s only when it is bound by C4b, and the C2b serine protease is thereby also confined to the pathogen surface, where it remains associated with C4b, forming a C3 convertase. The activation of C3 molecules thus also occurs at the surface of the pathogen. Furthermore, the C3b cleavage product is also rapidly inactivated unless it binds covalently by the same mechanism as C4b, and it therefore opsonizes only the surface on which complement activation has taken place.
The third pathway of complement activation is called the alternative pathway because it was discovered as a second, or ‘alternative,’ pathway for complement activation after the classical pathway had been defined. This pathway can proceed on many microbial surfaces in the absence of specific antibody, and it leads to the generation of a distinct C3 convertase designated C3b,Bb. In contrast to the classical and MB-lectin pathways of complement activation, the alternative pathway does not depend on a pathogen-binding protein for its initiation; instead it is initiated through the spontaneous hydrolysis of C3, as shown in the top three panels of Fig. 2.15. The distinctive components of the pathway are listed in Fig. 2.16. A number of mechanisms ensure that the activation pathway will only proceed on the surface of a pathogen.

Complement activated by the alternative pathway attacks pathogens while sparing host cells, which are protected by complement regulatory proteins. The complement component C3 is cleaved spontaneously in plasma to give C3(H2O), which binds factor B and (more...)

The proteins of the alternative pathway of complement activation.
C3 is abundant in plasma, and C3b is produced at a significant rate by spontaneous cleavage (also known as ‘tickover’). This occurs through the spontaneous hydrolysis of the thioester bond in C3 to form C3(H2O) which has an altered conformation, allowing binding of the plasma protein factor B. The binding of B by C3(H2O) then allows a plasma protease called factor D to cleave factor B to Ba and Bb, the latter remaining associated with C3(H2O) to form the C3(H2O)Bb complex. This complex is a fluid-phase C3 convertase, and although it is only formed in small amounts it can cleave many molecules of C3 to C3a and C3b. Much of this C3b is inactivated by hydrolysis, but some attaches covalently, through its reactive thioester group, to the surfaces of host cells or to pathogens. C3b bound in this way is able to bind factor B, allowing its cleavage by factor D to yield the small fragment Ba and the active protease Bb. This results in formation of the alternative pathway C3 convertase, C3b,Bb (see Fig. 2.15).
When C3b binds to host cells, a number of complement-regulatory proteins, present in the plasma and on host cell membranes combine to prevent complement activation from proceeding. These proteins interact with C3b and either prevent the convertase from forming, or promote its rapid dissociation (see Fig. 2.15). Thus, the complement receptor 1 (CR1) and a membrane-attached protein known as decay-accelerating factor (DAF or CD55) compete with factor B for binding to C3b on the cell surface, and can displace Bb from a convertase that has already formed. Convertase formation can also be prevented by cleaving C3b to its inactive derivative iC3b. This is achieved by a plasma protease, factor I, in conjunction with C3b-binding proteins that can act as cofactors, such as CR1 and membrane cofactor of proteolysis (MCP or CD46), another host cell membrane protein. Factor H is another complement-regulatory protein in plasma that binds C3b and, like CR1, it is able to compete with factor B and displace Bb from the convertase in addition to acting as a cofactor for factor I. Factor H binds preferentially to C3b bound to vertebrate cells as it has an affinity for the sialic acid residues present on these cells.
By contrast, because pathogen surfaces lack these regulatory proteins and sialic acid residues, the C3b,Bb convertase can form and persist. Indeed, this process may be favored by a positive regulatory factor, known as properdin or factor P, which binds to many microbial surfaces and stabilizes the convertase. Deficiencies in factor P are associated with a heightened susceptibility to infection with Neisseria species. Once formed, the C3b,Bb convertase rapidly cleaves yet more C3 to C3b, which can bind to the pathogen and either act as an opsonin or reinitiate the pathway to form another molecule of C3b,Bb convertase. Thus, the alternative pathway activates through an amplification loop that can proceed on the surface of a pathogen, but not on a host cell. This same amplification loop enables the alternative pathway to contribute to complement activation initially triggered through the classical or MB-lectin pathways (Fig. 2.17).

The alternative pathway of complement activation can amplify the classical or the MB-lectin pathway by forming an alternative C3 convertase and depositing more C3b molecules on the pathogen. C3b deposited by the classical or MB-lectin pathways can bind (more...)
The C3 convertases resulting from activation of the classical and MB-lectin pathways (C4b,2b) and from the alternative pathway (C3b,Bb) are apparently distinct. However, understanding of the complement system is simplified somewhat by recognition of the close evolutionary relationships between the different complement proteins. Thus the complement zymogens, factor B and C2, are closely related proteins encoded by homologous genes located in tandem in the major histocompatibility complex (MHC) on human chromosome 6. Furthermore, their respective binding partners, C3 and C4, both contain thioester bonds that provide the means of covalently attaching the C3 convertases to a pathogen surface. Only one component of the alternative pathway appears entirely unrelated to its functional equivalents in the classical and MB-lectin pathways; this is the initiating serine protease, factor D. Factor D can also be singled out as the only activating protease of the complement system to circulate as an active enzyme rather than a zymogen. This is both necessary for the initiation of the alternative pathway through spontaneous C3 cleavage, and safe for the host because factor D has no other substrate than factor B when bound to C3b. This means that factor D only finds its substrate at a very low level in plasma, and at pathogen surfaces where the alternative pathway of complement activation is allowed to proceed.
Comparison of the different pathways of complement activation illustrates the general principle that most of the immune effector mechanisms that can be activated in a nonclonal fashion as part of the early nonadaptive host response against infection have been harnessed during evolution to be used as effector mechanisms of adaptive immunity. It is almost certain that the adaptive response evolved by adding specific recognition to the original nonadaptive system. This is illustrated particularly clearly in the complement system, because here the components are defined, and the functional homologues can be seen to be evolutionarily related (Fig. 2.18).
There is a close relationship between the factors of the alternative, MB-lectin, and classical pathways of complement activation. Most of the factors are either identical or the products of genes that have duplicated and then diverged in sequence. The (more...)
The formation of C3 convertases is the point at which the three pathways of complement activation converge, because both the classical pathway and MB-lectin pathway convertases C4b,2b, and the alternative pathway convertase C3b,Bb have the same activity, and they initiate the same subsequent events. They both cleave C3 to C3b and C3a. C3b binds covalently through its thioester bond to adjacent molecules on the pathogen surface; otherwise it is inactivated by hydrolysis. C3 is the most abundant complement protein in plasma, occurring at a concentration of 1.2 mg ml–1, and up to 1000 molecules of C3b can bind in the vicinity of a single active C3 convertase (see Fig. 2.11). Thus, the main effect of complement activation is to deposit large quantities of C3b on the surface of the infecting pathogen, where it forms a covalently bonded coat that, as we will see, can signal the ultimate destruction of the pathogen by phagocytes.
The next step in the cascade is the generation of the C5 convertases. In the classical and the MB-lectin pathways, a C5 convertase is formed by the binding of C3b to C4b,2b to yield C4b,2b,3b. By the same token, the C5 convertase of the alternative pathway is formed by the binding of C3b to the C3 convertase to form C3b2,Bb. C5 is captured by these C5 convertase complexes through binding to an acceptor site on C3b, and is then rendered susceptible to cleavage by the serine protease activity of C2b or Bb. This reaction, which generates C5b and C5a, is much more limited than cleavage of C3, as C5 can be cleaved only when it binds to C3b that is part of the C5 convertase complex. Thus, complement activation by both the alternative, MB-lectin and classical pathways leads to the binding of large numbers of C3b molecules on the surface of the pathogen, the generation of a more limited number of C5b molecules, and the release of C3a and C5a (Fig. 2.19).

Complement component C5 is cleaved when captured by a C3b molecule that is part of a C5 convertase complex. As shown in the top panel, C5 convertases are formed when C3b binds either the classical or MB-lectin pathway C3 convertase C4b,2b to form C4b,2b,3b, (more...)
The most important action of complement is to facilitate the uptake and destruction of pathogens by phagocytic cells. This occurs by the specific recognition of bound complement components by complement receptors (CRs) on phagocytes. These complement receptors bind pathogens opsonized with complement components: opsonization of pathogens is a major function of C3b and its proteolytic derivatives. C4b also acts as an opsonin but has a relatively minor role, largely because so much more C3b than C4b is generated.
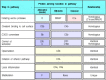
The five known types of receptor for bound complement components are listed, with their functions and distributions, in Fig. 2.20. The best-characterized is the C3b receptor CR1 (CD35), which is expressed on both macrophages and polymorphonuclear leukocytes. Binding of C3b to CR1 cannot by itself stimulate phagocytosis, but it can lead to phagocytosis in the presence of other immune mediators that activate macrophages. For example, the small complement fragment C5a can activate macrophages to ingest bacteria bound to their CR1 receptors (Fig. 2.21). C5a binds to another receptor expressed by macrophages, the C5a receptor, which has seven membrane-spanning domains. Receptors of this type couple with intracellular guanine-nucleotide-binding proteins called G proteins, and the C5a receptor signals in this way. Proteins associated with the extracellular matrix, such as fibronectin, can also contribute to phagocyte activation; these are encountered when phagocytes are recruited to connective tissue and activated there. C3a, which has inflammatory activities similar to those of C5a, although it is a less potent chemoattractant, binds to its own specific receptor, the C3a receptor, which is homologous in structure to the C5a receptor.

Distribution and function of receptors for complement proteins on the surfaces of cells. There are several different receptors specific for different bound complement components and their fragments. CR1 and CR3 are especially important in inducing phagocytosis (more...)

The anaphylotoxin C5a can enhance phagocytosis of opsonized microorganisms. Activation of complement, either by the alternative or the MB-lectin pathway, leads to the deposition of C3b on the surface of the microorganism (left panel). C3b can be bound (more...)
Three other complement receptors—CR2 (also known as CD21), CR3 (CD11b:CD18), and CR4 (CD11c:CD18)—bind to inactivated forms of C3b that remain attached to the pathogen surface. Like several other key components of complement, C3b is subject to regulatory mechanisms and can be cleaved into derivatives that cannot form an active convertase. One of the inactive derivatives of C3b, known as iC3b (see Section 2-9) acts as an opsonin in its own right when bound by the complement receptors CR2 or CR3. Unlike the binding of iC3b to CR1, the binding of iC3b to CR3 is sufficient on its own to stimulate phagocytosis. A second breakdown product of C3b, called C3dg, binds only to CR2. CR2 is found on B cells as part of a co-receptor complex that can augment the signal received through the antigen-specific immunoglobulin receptor. Thus a B cell whose antigen receptor is specific for a given pathogen will receive a strongly augmented signal on binding this pathogen if it is also coated with C3dg. The activation of complement can therefore contribute to producing a strong antibody response (see Chapters 6 and 9). This example of how an innate humoral immune response can contribute to activating adaptive humoral immunity parallels the contribution made by the innate cellular response of macrophages and dendritic cells to the initiation of a T-cell response, which we will discuss later in this chapter.
The central role of opsonization by C3b and its inactive fragments in the destruction of extracellular pathogens can be seen in the effects of various complement deficiency diseases. Whereas individuals deficient in any of the late components of complement are relatively unaffected, individuals deficient in C3 or in molecules that catalyze C3b deposition show increased susceptibility to infection by a wide range of extracellular bacteria, as we will see in Chapter 11.
The small complement fragments C3a, C4a, and C5a act on specific receptors (see Fig. 2.20) to produce local inflammatory responses. When produced in large amounts or injected systemically, they induce a generalized circulatory collapse, producing a shocklike syndrome similar to that seen in a systemic allergic reaction involving IgE antibodies (see Chapter 12). Such a reaction is termed anaphylactic shock and these small fragments of complement are therefore often referred to as anaphylotoxins. Of the three, C5a is the most stable and has the highest specific biological activity. All three induce smooth muscle contraction and increase vascular permeability, but C5a and C3a also act on the endothelial cells lining blood vessels to induce adhesion molecules. In addition, C3a and C5a can activate the mast cells that populate submucosal tissues to release mediators such as histamine and TNF-α that cause similar effects. The changes induced by C5a and C3a recruit antibody, complement, and phagocytic cells to the site of an infection (Fig. 2.22), and the increased fluid in the tissues hastens the movement of pathogen-bearing antigen- presenting cells to the local lymph nodes, contributing to the prompt initiation of the adaptive immune response.
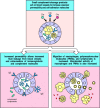
Local inflammatory responses can be induced by small complement fragments, especially C5a. The small complement fragments are differentially active: C5a is more active than C3a, which is more active than C4a. They cause local inflammatory responses by (more...)
C5a also acts directly on neutrophils and monocytes to increase their adherence to vessel walls, their migration toward sites of antigen deposition, and their ability to ingest particles, as well as increasing the expression of CR1 and CR3 on the surfaces of these cells. In this way C5a and, to a smaller extent, C3a and C4a, act in concert with other complement components to hasten the destruction of pathogens by phagocytes. C5a and C3a signal through transmembrane receptors that activate G proteins; thus the action of C5a in attracting neutrophils and monocytes is analogous to that of chemokines, which also act via G proteins to control cell migration.
One of the important effects of complement activation is the assembly of the terminal components of complement (Fig. 2.23) to form a membrane-attack complex. The reactions leading to the formation of this complex are shown schematically in Fig. 2.24. The end result is a pore in the lipid bilayer membrane that destroys membrane integrity. This is thought to kill the pathogen by destroying the proton gradient across the pathogen cell membrane.

The terminal complement components assemble to form the membrane-attack complex.

Assembly of the membrane-attack complex generates a pore in the lipid bilayer membrane. The sequence of steps and their approximate appearance are shown here in schematic form. C5b triggers the assembly of a complex of one molecule each of C6, C7, and (more...)
The first step in the formation of the membrane-attack complex is the cleavage of C5 by a C5 convertase to release C5b (see Fig. 2.19). In the next stages, shown in Fig. 2.24, C5b initiates the assembly of the later complement components and their insertion into the cell membrane. First, one molecule of C5b binds one molecule of C6, and the C5b,6 complex then binds one molecule of C7. This reaction leads to a conformational change in the constituent molecules, with the exposure of a hydrophobic site on C7, which inserts into the lipid bilayer. Similar hydrophobic sites are exposed on the later components C8 and C9 when they are bound to the complex, allowing these proteins also to insert into the lipid bilayer. C8 is a complex of two proteins, C8β and C8α-γ. The C8β protein binds to C5b, and the binding of C8β to the membrane-associated C5b,6,7 complex allows the hydrophobic domain of C8α-γ to insert into the lipid bilayer. Finally, C8α-γ induces the polymerization of 10 to 16 molecules of C9 into a pore-forming structure called the membrane-attack complex. The membrane-attack complex, shown schematically and by electron microscopy in Fig. 2.24, has a hydrophobic external face, allowing it to associate with the lipid bilayer, but a hydrophilic internal channel. The diameter of this channel is about 100 Å, allowing the free passage of solutes and water across the lipid bilayer. The disruption of the lipid bilayer leads to the loss of cellular homeostasis, the disruption of the proton gradient across the membrane, the penetration of enzymes such as lysozyme into the cell, and the eventual destruction of the pathogen.
Although the effect of the membrane-attack complex is very dramatic, particularly in experimental demonstrations in which antibodies against red blood cell membranes are used to trigger the complement cascade, the significance of these components in host defense seems to be quite limited. To date, deficiencies in complement components C5–C9 have been associated with susceptibility only to Neisseria species, the bacteria that cause the sexually transmitted disease gonorrhea and a common form of bacterial meningitis. Thus, the opsonizing and inflammatory actions of the earlier components of the complement cascade are clearly most important for host defense against infection. Formation of the membrane-attack complex seems to be important only for the killing of a few pathogens, although, as we will see in Chapter 13, it might have a major role in immunopathology.
Given the destructive effects of complement, and the way in which its activation is rapidly amplified through a triggered-enzyme cascade, it is not surprising that there are several mechanisms to prevent its uncontrolled activation. As we have seen, the effector molecules of complement are generated through the sequential activation of zymogens, which are present in plasma in an inactive form. The activation of these zymogens usually occurs on a pathogen surface, and the activated complement fragments produced in the ensuing cascade of reactions usually bind nearby or are rapidly inactivated by hydrolysis. These two features of complement activation act as safeguards against uncontrolled activation. Even so, all complement components are activated spontaneously at a low rate in plasma, and activated complement components will sometimes bind proteins on host cells. The potentially damaging consequences are prevented by a series of complement control proteins, summarized in Fig. 2.25, which regulate the complement cascade at different points. As we saw in discussing the alternative pathway of complement activation (see Section 2-9) many of these control proteins specifically protect host cells while allowing complement activation to proceed on pathogen surfaces. The complement control proteins therefore allow complement to distinguish self from nonself.

The proteins that regulate the activity of complement.
The reactions that regulate the complement cascade are shown in Fig. 2.26. The top two panels show how the
activation of C1 is controlled by a plasma serine proteinase inhibitor or
serpin, the C1 inhibitor (C1INH). C1INH binds the active enzyme
C1r:C1s, and causes it to dissociate from C1q, which remains bound to the
pathogen. In this way, C1INH limits the time during which active C1s is able to
cleave C4 and C2. By the same means, C1INH limits the spontaneous activation of
C1 in plasma. Its importance can be seen in the C1INH deficiency disease, hereditary angioneurotic edema , in
which chronic spontaneous complement activation leads to the production of
excess cleaved fragments of C4 and C2. The small fragment of C2, C2a, is further
cleaved into a peptide, the C2 kinin, which causes extensive swelling—the most
dangerous being local swelling in the trachea, which can lead to suffocation.
Bradykinin, which has similar actions to C2 kinin, is also produced in an
uncontrolled fashion in this disease, as a result of the lack of inhibition of
another plasma protease, kallikrein, which is activated by tissue damage and
also regulated by C1INH. This disease is fully corrected by replacing C1INH. The
large activated fragments of C4 and C2, which normally combine to form the C3
convertase, do not damage host cells in such patients because C4b is rapidly
inactivated in plasma (see Fig. 2.14) and
the convertase does not form. Furthermore, any convertase that accidentally
forms on a host cell is inactivated by the mechanisms described below.
, in
which chronic spontaneous complement activation leads to the production of
excess cleaved fragments of C4 and C2. The small fragment of C2, C2a, is further
cleaved into a peptide, the C2 kinin, which causes extensive swelling—the most
dangerous being local swelling in the trachea, which can lead to suffocation.
Bradykinin, which has similar actions to C2 kinin, is also produced in an
uncontrolled fashion in this disease, as a result of the lack of inhibition of
another plasma protease, kallikrein, which is activated by tissue damage and
also regulated by C1INH. This disease is fully corrected by replacing C1INH. The
large activated fragments of C4 and C2, which normally combine to form the C3
convertase, do not damage host cells in such patients because C4b is rapidly
inactivated in plasma (see Fig. 2.14) and
the convertase does not form. Furthermore, any convertase that accidentally
forms on a host cell is inactivated by the mechanisms described below.

Complement activation is regulated by a series of proteins that serve to protect host cells from accidental damage. These act on different stages of the complement cascade, dissociating complexes or catalyzing the enzymatic degradation of covalently bound (more...)
The thioester bond of activated C3 and C4 is extremely reactive and has no
mechanism for distinguishing an acceptor hydroxyl or amine group on a host cell
from a similar group on the surface of a pathogen. A series of protective
mechanisms, mediated by other proteins, has evolved to ensure that the binding
of a small number of C3 or C4 molecules to host cell membranes results in
minimal formation of C3 convertase and little amplification of complement
activation. We have already encountered most of these mechanisms in the
description of the alternative pathway (see Fig.
2.15), but we will consider them again here, as they are important
regulators of the classical pathway convertase as well (see Fig. 2.26, second and third rows). The mechanisms can be
divided into three categories. The first catalyze the cleavage of any C3b or C4b
that does bind to host cells into inactive products. The complement-regulatory
enzyme responsible is the plasma serine protease factor I; it circulates in
active form but can only cleave C3b and C4b when they are bound to a cofactor
protein. In these circumstances, factor I cleaves C3b, first into iC3b and then
further to C3dg, thus permanently inactivating it. C4b is similarly inactivated
by cleavage into C4c and C4d. There are two cell-membrane proteins that bind C3b
and C4b and possess cofactor activity for factor I; these are CR1 and MCP (see
Section 2-9). Microbial cell walls
lack these protective proteins and cannot promote the breakdown of C3b and C4b.
Instead, these proteins act as binding sites for factor B and C2, promoting
complement activation. The importance of factor I can be seen in people with
genetic factor I deficiency. Because of uncontrolled complement activation,
complement proteins rapidly become depleted and such people suffer repeated
bacterial infections, especially with ubiquitous pyogenic bacteria.
There are also plasma proteins with cofactor activity for factor I. C4b is bound by a cofactor known as the C4b-binding protein (C4BP), which mainly acts as a regulator of the classical pathway in the fluid phase. C3b is bound in both the fluid phase and at cell membranes by a cofactor protein called factor H (see Section 2-9). Factor H is an important complement regulator at cell membranes, and at first sight it is not obvious how factor H can distinguish C3b bound to host cells or to a pathogen. However, the carbohydrate content of the cell membranes of bacterial pathogens differs from that of their hosts and this is the basis for the protective effect of factor H. Factor H has affinity for the terminal sialic acids of host cell membrane glycoproteins and this increases the binding of factor H to any C3b deposited on host cells. In contrast, factor H has a much lower affinity for C3b deposited on the cell walls of many bacteria, and factor B binds in preference, resulting in amplification of complement activation on bacterial cell surfaces. In effect, factor H and factor B compete for binding to C3b bound to cells. If factor B ‘wins,’ as is typically the case on a pathogen surface, then more C3b,Bb C3 convertase forms and complement activation is amplified. If factor H ‘wins,’ as is the case on cells of the host, then the bound C3b is catabolized by factor I to iC3b and C3dg and complement activation is inhibited.
The competition between factor H and factor B for binding to surface-bound C3b is an example of the second mechanism for inhibiting complement activation on host cell membranes. A number of proteins competitively inhibit the binding of C2 to cell-bound C4b and of factor B to cell-bound C3b, thereby inhibiting convertase formation. These proteins bind to C3b and C4b on the cell surface, and also mediate protection against complement through the third mechanism, which is to augment the dissociation of C4b,2b and C3b,Bb convertases that have already formed. Host cell membrane molecules that regulate complement through both these mechanisms include DAF (see Section 2-9) and CR1, which promotes dissociation of convertase in addition to its cofactor activity. All the proteins that bind the homologous C4b and C3b molecules share one or more copies of a structural element called the short consensus repeat (SCR), complement control protein (CCP) repeat, or (especially in Japan) the sushi domain.
In addition to the mechanisms for preventing C3 convertase formation and C4 and C3 deposition on cell membranes, there are also inhibitory mechanisms that prevent the inappropriate insertion of the membrane-attack complex into membranes. We saw in Section 2-13 that the membrane-attack complex polymerizes onto C5b molecules released from C5 convertase. This complex mainly inserts into cell membranes adjacent to the site of the C5 convertase, that is, close to the site of complement activation on a pathogen. However, some newly formed membrane-attack complexes may diffuse from the site of complement activation and insert into adjacent host cell membranes. Several plasma proteins bind to the C5b,6,7 complex and thereby inhibit its random insertion into cell membranes. The most important is probably C8β itself, when it binds to C5b,6,7 in the fluid phase. Host cell membranes also contain an intrinsic protein, CD59 or protectin, which inhibits the binding of C9 to the C5b,6,7,8 complex (see Fig. 2.26, bottom row). CD59 and DAF are both linked to the cell surface by a phosphoinositol glycolipid (PIG) tail, like many other membrane proteins. One of the enzymes involved in the synthesis of PIG tails is encoded on chromosome X. In people with a somatic mutation in this gene in a clone of hematopoietic cells, both CD59 and DAF fail to function. This causes the disease paroxysmal nocturnal hemoglobinuria, which is characterized by episodes of intravascular red blood cell lysis by complement. Red blood cells that lack CD59 only are also susceptible to destruction as a result of spontaneous activation of the complement cascade.
The complement system is one of the major mechanisms by which pathogen recognition is converted into an effective host defense against initial infection. Complement is a system of plasma proteins that can be activated directly by pathogens or indirectly by pathogen-bound antibody, leading to a cascade of reactions that occurs on the surface of pathogens and generates active components with various effector functions. There are three pathways of complement activation: the classical pathway, which is triggered directly by pathogen or indirectly by antibody binding to the pathogen surface; the MB-lectin pathway; and the alternative pathway, which also provides an amplification loop for the other two pathways. All three pathways can be initiated independently of antibody as part of innate immunity. The early events in all pathways consist of a sequence of cleavage reactions in which the larger cleavage product binds covalently to the pathogen surface and contributes to the activation of the next component. The pathways converge with the formation of a C3 convertase enzyme, which cleaves C3 to produce the active complement component C3b. The binding of large numbers of C3b molecules to the pathogen is the central event in complement activation. Bound complement components, especially bound C3b and its inactive fragments, are recognized by specific complement receptors on phagocytic cells, which engulf pathogens opsonized by C3b and its inactive fragments. The small cleavage fragments of C3, C4, and especially C5, recruit phagocytes to sites of infection and activate them by binding to specific trimeric G protein-coupled receptors. Together, these activities promote the uptake and destruction of pathogens by phagocytes. The molecules of C3b that bind the C3 convertase itself initiate the late events, binding C5 to make it susceptible to cleavage by C2b or Bb. The larger C5b fragment triggers the assembly of a membrane-attack complex, which can result in the lysis of certain pathogens. The activity of complement components is modulated by a system of regulatory proteins that prevent tissue damage as a result of inadvertent binding of activated complement components to host cells or spontaneous activation of complement components in plasma.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...