

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Mobley HLT, Mendz GL, Hazell SL, editors. Helicobacter pylori: Physiology and Genetics. Washington (DC): ASM Press; 2001.

When Warren and Marshall first identified spiral organisms closely applied to the gastric epithelium in active chronic gastritis (84), they brought to light an etiological explanation for a whole series of pathological changes that had been long-recognized but not understood. It was widely appreciated that chronic gastritis was a common denominator linking peptic ulceration, gastric carcinoma, and lymphoma and that the histological picture encompassed chronic inflammation, atrophy, and intestinal metaplasia. But our state of ignorance concerning etiology led to a plethora of possible causes including stress, excessive alcohol consumption, bile reflux, and even the ingestion of hot beverages and spicy food. The importance of Helicobacter pylori infection to the pathologist is threefold. We now have an etiology for chronic gastritis that can be readily identified in histological sections using simple techniques. The accumulated knowledge of bacterial pathogenicity and the host response now offers plausible explanations for the tissue changes observed. Finally, the ecology of the organism with regard to tissue colonization and local environment affords explanations for hitherto inexplicable differences in the patterns of inflammation seen in different clinical phenotypes. These relatively recent advances in our understanding of gastritis and peptic ulceration form the substance of this chapter.
General Features of H. pylori Gastritis
The Acute Phase
The initial, acute phase of infection is subclinical in the great majority of subjects. Following ingestion, organisms penetrate through the viscid mucous layer and multiply in close proximity to the surface epithelial cells. The epithelium responds to infection by mucin depletion, cellular exfoliation, and compensatory regenerative changes. Polymorph infiltration into foveolar and surface epithelium, and lamina propria edema are conspicuous. Collections of polymorphs in the foveolae and adherent neutrophil exudate on the surface may also be present. In the few cases of acute helicobacter gastritis in the literature that have been studied histologically, there appears to be relatively equal involvement of antrum and body (46, 51, 69). This acute phase is accompanied by profound hypochlorhydria and a failure of ascorbic acid secretion into gastric juice (71). It may take several weeks for acid output to return to preinfection levels, and in a proportion of patients output remains low. However, ascorbic acid secretion remains lower than normal for the duration of chronic gastritis, indicating that low secretion is related to persisting inflammation rather than hypochlorhydria (71).
The acute response is mediated by release of bacterial lipopolysaccharide (67) and a number of directly acting chemotactic moieties, which penetrate through the damaged surface epithelium and induce polymorph emigration into the lamina propria and epithelium (8). Bacterial products also activate mast cells, and subsequent degranulation releases other acute inflammatory mediators that increase vascular permeability, up-regulate expression of leukocyte adhesion molecules on endothelial cells, and increase polymorph emigration (26). H. pylori stimulates the gastric epithelium to produce a potent neutrophil chemokine interleukin (IL) 8, whose production is up-regulated by tumor necrosis factor alpha (TNF-α) and IL-1 released by macrophages in response to bacterial lipopolysaccharide (8). Additional IL-8 is released by the polymorphs themselves in response to soluble H. pylori proteins (35).
The acute phase is short lived. In a small minority of people, and particularly in childhood, the organisms may be spontaneously cleared, the polymorph infiltrate resolves, and appearances return to normal. In the majority, however, the host immune response fails to eliminate the infection and over the next 3 or 4 weeks there is a gradual accumulation of chronic inflammatory cells that come to dominate the histological picture. As a consequence, the diagnosis of an acute neutrophilic gastritis gives way to that of an active chronic gastritis (69).
Active Chronic Gastritis
The arrival of lymphocytes and plasma cells in the mucosa signals augmentation of the acute inflammatory response by the production of cytokines and specific anti-H. pylori antibodies. B-cell proliferation and subsequent plasma cell differentiation result in the synthesis of IgM-opsonizing and complement-fixing antibodies, which amplify the inflammatory reaction (Fig. 1 and 2). However, this vigorous response fails to eliminate infection, and the continued presence of H. pylori leads to the development of a second arm of the immune response more specifically aimed at preventing the damaging effects of intraluminal pathogens. This second-line response involves the recruitment of primed B cells into lymphoid follicles, with the production of plasma cells largely committed to the synthesis of "mucosally protective" IgA antibodies. The fact that, even when augmented by IgA, the response is insufficient to eradicate H. pylori in the great majority of cases means that antigenic stimulation persists and the formation of follicles becomes a consistent feature of chronic H. pylori gastritis. Indeed, it is claimed that if enough biopsies are examined, follicles will always be found in H. pylori-infected stomachs (23). This acquisition of "organized" lymphoid tissue in the gastric mucosa constitutes a mucosa-associated lymphoid tissue (MALT). As such it provides the background tissue in which gastric marginal zone (B-cell) lymphoma (so-called MALToma) arises, and this underlines the crucial role of H. pylori in lymphomagenesis in the stomach.

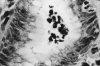
Figure 2
Polymorphs infiltrate the foveloar epithelium, which is colonized by helicobacters. Degenerate polymorphs have accumulated in the lumen. Modified Giemsa stain; magnification, ×400.
The two arms of the reaction to H. pylori, the acute "inflammatory" and the chronic "immune," are thought to be directed by two subsets of T-helper cells, Th1 cells that promote inflammation and by activating CD8+ T cells lead to autoantibody formation and cell-mediated epithelial damage, and Th2 cells that are responsible for the secretory immune response, which has the potential to reduce the bacterial load (17, 50). While these two arms are both interrelated and to a large degree inseparable, the response leans heavily toward the Th1 arm, although its strength will be influenced by bacterial and host factors. The typical histological picture of active chronic gastritis with lymphoid follicles reflects the overlap of these two processes, but down-regulation of the acute inflammatory component can result in a picture of inactive chronic gastritis, i.e., one in which there is no polymorph component. In human H. pylori infection this disparity between Th1 and Th2 responses is most evident in children in whom the lymphofollicular pattern is dominant and is sufficient to give rise to a characteristic nodularity of the mucosa, yet polymorph activity is minimal or absent. The dissociation between inflammatory and immune (follicular) lymphoid infiltration reaches its ultimate expression in some animal models. For example, in the BALB/c mouse infected with Helicobacter felis, no gastritis is observed for most of the animal's life span. However, aged infected mice may develop a pronounced follicular lymphoid infiltrate in the corpus mucosa, which in some animals progresses to lymphoma (16). These findings indicate the importance of host factors in determining the balance between inflammation and immunity, but bacterial (strain) factors should not be overlooked. Thus, while genotypic differences in cagA and vacA appear to be important in determining inflammatory activity (1, 83), follicle formation seems to be a universal response to H. pylori irrespective of the infecting strain (23).
Atrophy
Atrophy in the stomach is conventionally defined as loss of glandular tissue from repeated or continuing mucosal injury and is a common denominator in all pathological processes causing progressive mucosal damage, including long-standing H. pylori infection (Fig. 3). Thus, loss of glands may follow erosion or ulceration of the mucosa, with destruction of the glandular layer, or as a result of a prolonged inflammatory process in which individual glands undergo destruction. When such loss occurs, it is followed by fibrous replacement. However, atrophy can also be thought of as simply "a loss of specialized or functional cells." Under this broader definition it is possible to include situations in which there is loss or replacement of parietal and chief cells without glandular destruction. Such partial or "preatrophy" has been described in human autoimmune gastritis (76) and is frequently encountered in animal models of both autoimmune gastritis (5) and chronic helicobacter infection (62). In these latter situations, specialized oxyntic cells within intact glandular tubules are replaced by mucous cells ("mucous metaplasia"). It is likely that downward proliferation of mucous neck cells is the explanation, but whether this gives rise to so-called pyloric gland metaplasia has yet to be determined.
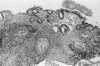
Figure 3
Chronic H. pylori gastritis. This low-power view shows marked glandular atrophy, lymphoid follicles, and centrally a focus of intestinal metaplasia. H&E ×25.
The prevalence and severity of atrophy among patients with chronic H. pylori gastritis increase with time (41, 45). However, atrophy is not an effect of aging per se; there is no evidence that atrophy occurs as a physiological aging phenomenon and elderly subjects without gastritis have a normal acid output (34).
The prevalence of H. pylori positivity declines with increasing glandular atrophy. There are two main reasons for the loss of organisms. First, H. pylori only colonizes gastric epithelium; thus, the organisms are absent from areas of complete intestinal metaplasia. Second, the organism only thrives within the narrow pH range provided by a partially acidic environment and the hypochlorhydric stomach is inimical to H. pylori (6). Therefore, the failure to demonstrate H. pylori in the atrophic stomach does not deny a role for infection in the causation of the underlying gastritis.
Atrophy in H. pylori gastritis could result from direct bacterial effects or alternatively as a consequence of the host inflammatory or immune response. Direct injury by cytotoxins and ammonia products can bring about epithelial cell destruction, but this is unlikely to be an important mechanism in the glandular layer as bacterial colonization is restricted to surface and foveolar epithelium. While infection with more virulent strains of H. pylori, that is, cytotoxin-positive and CagA-positive strains, is more likely to be associated with atrophy (3, 21, 32), this does not necessarily argue for a direct mechanism. Such strains are "proinflammatory" and could also lead to increased release of potentially autodestructive agents such as proteases and free radicals from inflammatory cells. Interestingly, active chronic H. pylori gastritis is characterized by accumulations of neutrophil polymorphs around and within the stem-cell compartment in the gastric pit, a phenomenon possibly related to the rich vascular supply of this segment (29). Release of injurious products at this site could destroy stem cells, arrest the renewal of glandular epithelial cells, and lead to complete loss of the pit-gland unit.
Animal models of helicobacter infection (62) show a divergence between active chronic inflammation in the antrum, which is strongly linked to bacterial colonization density, and the development of atrophy in the corpus, which is not. Such experiments raise the possibility that atrophy is governed largely by host-related factors, a view that has gathered strength with the demonstration that antibodies produced during H. pylori infection react with antigens on the luminal aspect of foveolar cells and the secretory canaliculi of parietal cells (53) and the occasional finding of lymphocytic infiltration of glandular epithelium showing apoptotic degeneration (personal observation). The presence of anticanalicular antibodies in H. pylori infection is significantly related to the severity of corpus inflammation and the presence of atrophy (18). Once initiated, it is possible that these autoimmune effects could become self-perpetuating.
Intestinal Metaplasia
Metaplasia is defined as a potentially reversible change in which a fully differentiated cell type is replaced by another differentiated cell type, and usually represents a change to cells better able to withstand an adverse environment. Thus, intestinal metaplasia represents a change from a gastric epithelial phenotype to a small- or large-intestinal phenotype. Metaplasia is always associated with some abnormal stimulation of growth, for example, during regeneration following mucosal injury (52, 54, 58) and may be transient. However, persistence of metaplasia indicates that the change must either be heritable on cell division, indicating an alteration at the level of stem cells, i.e., somatic mutation, or is a consequence of sustained epigenetic modulation of the normal sequence of gene expression, which occurs during differentiation and which determines the ultimate cellular phenotype. The pattern of gene expression is under the control of a complex heirarchy of transcription factors of which homeodomain proteins are important members. These proteins are themselves regulated by homeobox genes whose expression is pivotal to cellular differentiation and organogenesis (20, 33). Under normal circumstances the homeobox genes cdx-1 and cdx-2 are expressed only in the intestine, but the corresponding transcription factor (CDX-1 protein) has been demonstrated in intestinal metaplasia in the stomach (66). However, whether such expression results from a mutation in stem cells or epigenetic changes affecting progeny cells has not been determined. While the frequent coexistence of glandular loss and intestinal metaplasia implies survival of stem cells with subsequent formation of a neocrypt exhibiting intestinal differentiation, it is also possible that intestinalized crypts could arise by fission from preexisting pit-gland units adjacent to the foci of atrophy.
Intestinal metaplasia is a common finding in chronic gastritis of all causes and is found in around 20% of symptomatic European subjects undergoing endoscopy (20, 33). Its extent usually, but not invariably, parallels the development of atrophy. It is rare in Europeans below the age of 30 (15, 68). Intestinal metaplasia is found more frequently in H. pylori-positive than -negative cases, despite the tendency for stomachs with extensive atrophy and metaplasia to become negative (7). In H. pylori gastritis, intestinal metaplasia is significantly correlated with age, is much more frequently observed in the antrum than in the body, and is more commonly associated with gastric ulcer than pyloric or duodenal ulcers (14). In addition to an increased prevalence in H. pylori-infected subjects, intestinal metaplasia is significantly associated with bile reflux (70) and is a feature of autoimmune gastritis (13).
Insofar as H. pylori cannot adhere to intestinal epithelium, it is possible to view intestinal metaplasia as a defense response against infection. This view is in accord with current opinion on the histogenesis of gastric metaplasia in the duodenum and columnar metaplasia in the esophagus as defense responses, in these instances to excess luminal acid, but nevertheless, this interpretation remains speculative.
Patterns of H. pylori Gastritis
Antral Predominant Gastritis and Local Acid Production
In the majority of infected individuals, at least in developed countries, H. pylori gastritis is to some degree more pronounced in the antrum than in the corpus. When there is a substantial difference between the two compartments, such that there is minimal inflammation in the corpus and marked involvement of the antrum, the gastritis is designated "antral predominant." This pattern is found in patients with duodenal (and prepyloric) ulceration and is a marker of the duodenal ulcer diathesis. The relative resistance of the corpus mucosa to H. pylori colonization and inflammation and the susceptibility of the duodenal mucosa to ulceration are best explained by high local acid concentrations in the corpus and increased acid output from stomach to duodenum in subjects at risk of duodenal ulceration.
H. pylori has evolved mechanisms that ensure its survival in the acidic environment of the stomach. In the pH range of 3.5 to 5.0 in the presence of urea, the organism can maintain the proton motive force (PMF) across its periplasmic membrane, ensuring a continued supply of energy through ATP synthesis (48). Urea entering the bacterium is acted on by its cytoplasmic urease to produce ammonia, which neutralizes excess hydrogen ions to maintain a pH of 6.2 in the periplasmic space and thereby preserve the PMF (63, 64). However, when there is a high local acid concentration, the protective mechanism fails to keep up with hydrogen ion influx, ATP synthesis declines, and the bacterium dies, or at least loses virulence (19). Thus, in the presence of high acid output, the corpus becomes an even more hostile environment, colonization density is greatly diminished, and infection and inflammation are concentrated in the antrum. Conversely, when the pH in its microenvironment rises above 5.0, the organism produces ammonia in excess of that needed to neutralize the much-diminished influx of hydrogen ions, and the environment becomes increasingly alkaline. Above a pH of 8 the cell ceases to function (6, 27). Thus, in achlorhydric states gastric helicobacters will self-destruct and infection will be spontaneously eliminated.
Corpus Gastritis
In a minority of infected subjects in Western populations, a corpus-predominant pattern of gastritis is observed. Such individuals generally have a low acid output. It is thought that the inflammatory infiltrate itself influences parietal cell function by the release of acid-inhibitory cytokines such as IL-1.
The influence of local acid production on corpus colonization and virulence is clearly demonstrated by observations in humans and experimental animals. Acid reduction by vagotomy in duodenal ulcer patients resulted in a substantial increase in corpus inflammation over the ensuing 3 months (61), while sustained acid suppression with proton-pump inhibitors in patients with reflux esophagitis led to much-increased H. pylori colonization over the corpus relative to antrum and a swing from antral- to corpus-predominant gastritis (49, 77). Similarly in animals infected with H. felis, acid suppression led to colonization of previously uninfected oxyntic mucosa (10). On the other hand, patients with the Zollinger-Ellison syndrome have a low prevalence of H. pylori infection because their entire gastric mucosa is hostile to colonization (25).
Duodenal Ulceration
Gastric Metaplasia
It has long been recognized that patients with duodenal ulcer (DU) have, on average, higher acid secretion than do normal, healthy individuals. We now know that H. pylori-infected individuals with the DU diathesis have hypergastrinemia and increased sensitivity to gastrin. The arrival of unneutralized acid in the first part of the duodenum has certain consequences. Notable among these is gastric metaplasia, the presence of gastric-type mucus-secreting cells in the surface epithelium of the duodenum (81). The pathogenesis of gastric metaplasia is controversial; recent work suggests local goblet-cell transformation to cells of gastric phenotype in response to inflammatory signals (65). However, it is a common observation that gastric metaplasia exists without inflammation. The change probably develops as an adaptive response to acid, a suggestion that is supported by animal experiments with induced hyperchlorhydria (22, 78), and in human subjects by the correlation between the presence and extent of metaplasia and maximal acid output (40). Furthermore, gastric metaplasia is extensive in patients with the Zollinger-Ellison syndrome (59), has a lower prevalence following truncal and proximal selective vagotomy, and is not seen in patients with autoimmune gastritis and achlorhydria (86).
Active Chronic Duodenitis
While the coexistence of gastric metaplasia and chronic inflammation in duodenitis has been long recognized, it was Marshall et al. (47) who first suggested that H. pylori was responsible for colonizing "antral type" mucosa in the duodenal bulb and giving rise to chronic inflammation. However, these authors considered the presence of gastric epithelium as a primary, presumably congenital, phenomenon rather than accepting its metaplastic origin. Even before Warren and Marshall's rediscovery of bacterial infection of the human stomach (84), Steer (73) had described the migration of polymorphs through gastric epithelium at the sites of bacterial attachment, and later established the presence of a bacterium and the associated polymorph response in gastric metaplasia in the duodenum (74, 75). However, H. pylori is much more difficult to recognize in duodenal than gastric biopsies. The bacteria are usually scanty and may adopt a coccoid form, which then renders recognition based purely on morphology impossible. Thus, prevalence rates for H. pylori in duodenitis are very variable and depend on the number of biopsies examined and the methods of detection used. With the modified Giemsa stain, organisms were observed in 57% of anterior duodenal biopsies with active chronic duodenitis in one study (86), but three methods of microscopy detected H. pylori in 75% in another study (82). Similar prevalence rates for colonization of gastric metaplasia in H. pylori-infected patients with duodenal ulcer and duodenitis have been obtained when multiple tests have been employed (i.e., histology, culture, and rapid urease tests) (81). The conclusion to be drawn from these studies is that intraduodenal infection cannot be demonstrated in a substantial proportion of patients who have all the components of the duodenal ulcer phenotype, namely, gastric metaplasia, chronic duodenitis, and H. pylori gastritis. However, even in histologically H. pylori-negative chronic duodenitis it can be shown that the mucosa contains plasma cells that secrete specific IgA anti-H. pylori antibodies and that this response is restricted to the first part of the duodenum, i.e., the area most likely to exhibit gastric metaplasia and infection (9).
Ulceration—pH and Hp
The close relationship between acid-induced gastric metaplasia, H. pylori gastritis, and active chronic duodenitis was first emphasized by Wyatt et al. (86). A sequence of events was proposed that embraced acid-induced metaplasia in the first part of the duodenum, spread of H. pylori infection from the stomach, and following colonization of the gastric-type epithelium, the development of an acute and chronic inflammatory cell response. Chronic inflammation and direct bacterial effects on epithelial structure and bicarbonate production render the duodenal mucosa more susceptible to acid/peptic attack and, in a proportion of subjects, frank duodenal ulceration ensues. Thus, in this model two risk factors, acid-induced gastric metaplasia and H. pylori infection, are essential prerequisites for the development of chronic duodenitis and, by extrapolation, duodenal ulceration. Thus the prevalence of gastric metaplasia needs to be taken into account when considering the overall relationship between the incidence of DU and H. pylori infection. It seems likely that in countries where there is a high rate of infection but a low incidence of DU there will also be a low frequency of gastric metaplasia as a consequence of the lower acid output prevailing in these populations. These considerations are pertinent in many developing countries and may go some way toward explaining the so-called "African enigma," where there is a low peptic ulcer frequency in populations with high H. pylori infection rates (12).
Gastric Ulceration
The Antral-Body Transitional Zone
Transitional zones are the junctional areas between two contiguous mucosae. The antral-corpus zone is a narrow area in which acid-secreting oxyntic glands give way to simple mucous glands of the antrum. The site of the boundary between antrum and corpus will be governed by the relative size of these compartments. Individuals who have a constitutionally enlarged parietal cell mass will have a relatively large corpus and the transitional zone (TZ) will be shifted caudally. On the other hand, subjects with low acid output will have a relatively larger antrum and proximal shift of the antral-corpus TZ. However, proximal displacement of this zone is usually an acquired change consequent to inflammation, atrophy, and metaplasia encroaching on the corpus mucosa.
It has been appreciated for many years that the boundary between nonatrophic corpus and atrophic antral mucosa can be endoscopically visible. The boundary has been called the "atrophic border," and Japanese investigators have demonstrated that it moves proximally with advancing age (36, 37). The dynamics of this migrating border are interesting. Inflammation in the TZ results in glandular atrophy. Loss of corpus glands at the interface with the TZ is followed by mucous cell and pyloric gland metaplasia. Thus, the original corpus-type mucosa close to the boundary takes on the appearance of antrum. Interestingly, the degree of inflammation found in the antral-corpus TZ is often greater than that seen in the adjacent mucosa of either the antrum or corpus proper. The increased inflammation could be a reflection of changes in H. pylori colonization density or the virulence of the organisms. Certainly there is increased colonization immediately proximal to the atrophic border, and there is greater polymorph activity when compared to mucosa immediately distal to the border (88). Conversely, and in keeping with our concepts of causation, the mucosa distal (antral) to the border shows significantly more atrophy and intestinal metaplasia. It is likely that H. pylori colonization and virulence will be affected by the local acid levels prevailing across the transitional zones.
H. pylori and the TZ
There are two possible explanations for the apparent ability of H. pylori to induce a more intense inflammatory response at the acid-gradient TZs that border the oxyntic (corpus) mucosa (i.e., the antral-corpus and the corpus-cardia zones). The bacteria are either metabolizing and proliferating maximally because the local environment is at their pH optimum, or they are generating more inflammatory products due to induction of stress proteins. Bacteria tend to find their growth optimum when confronted with an environmental gradient. Thus, it is likely that at some point across the gradient of local acid found through the TZs, H. pylori finds optimal conditions and can maximize adhesion, growth, and release of inflammatory products and induce maximal cytokine production via signal transduction at the surface. Alternatively, it is known that bacteria have a series of well-developed physiological mechanisms for dealing with environmental stress, among which is the acid-tolerance response (80). Various genes are switched on that encode proteins that allow the bacterium to survive (57). Examples of such molecules are the acid (heat) shock proteins (24, 38). In the TZ, there will be a "watershed" area where the local acid environment is such that the acid-tolerance response of the bacterium becomes further stressed. It is conceivable that at this site novel proteins could be produced that are more inflammatory than those produced elsewhere. Whatever the explanation, these areas of peak inflammatory response are the sites of maximal development of atrophy and intestinal metaplasia and are at greatest risk of ulceration.
Gastric Ulceration—a Moving Experience
A distinction is made between distal ulcers occurring within the antrum, i.e., prepyloric ulcers, and more proximal ulcers in the stomach. The former share epidemiological characteristics with duodenal ulcer, and the patients usually have raised acid levels. In such cases, the site of ulceration may be determined by patches of atrophy in the antrum, making the mucosa more susceptible to acid attack. However, the TZs play a pivotal role in the pathogenesis of more proximal gastric ulcers. Oi et al. in 1959 (55, 56) and Stadelmann et al. in 1971 (72) were the first to draw attention to the close topographical relationship between gastric ulcers and the TZ, but it required knowledge of the behavior of H. pylori at the TZs to advance a plausible explanation for the finding of peptic ulcers at these sites. Colonization, particularly by the more virulent CagA-positive strains, leads to surface epithelial degeneration and increased exfoliation of surface epithelial cells (83). Accelerated cell exfoliation results in compensatory cell proliferation so that immature cells populate the foveolae and surface. Mucin and bicarbonate production is impaired, and the integrity of the mucous barrier may be compromised. In addition, activation of complement via the alternate pathway and the release of chemical mediators by mast cells and activated polymorphs may lead to microvascular disturbances and focal ischemic damage to the surface epithelium (2, 11). Apart from these inflammatory factors, ulcerogenesis may be promoted by the greater degree of atrophy and intestinal metaplasia found immediately distal to the TZ. Differences in mucus composition and bicarbonate production in metaplastic mucosa may lower the protection afforded by the mucous barrier. Indeed, metaplastic and atrophic mucosa also differs from normal mucosa in the local production of epithelial growth factors and regulatory (trefoil) peptides (28), and there may also be differences in the pattern of receptors for luminal growth factors (e.g., epidermal growth factor) (39). Diminished growth factor or regulatory peptide stimulation will adversely affect mucosal regeneration and exaggerate the effects of injury. However, there may also be bacterial factors at work. H. pylori infection down-regulates E-cadherin expression in gastric epithelial cells (79). E-cadherin is involved in cell-to-cell adhesion and in epithelial cell proliferation so that depressed production could adversely affect the resistance of the mucosa to acid attack. All these consequences of H. pylori infection conspire to make the antrum-corpus TZ peculiarly susceptible to acid-peptic attack and, therefore, the principal site of peptic ulceration. The proximal migration of the TZ with time explains the endoscopic observation that gastric ulcers are found progressively higher up the lesser curve with increasing age.
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) and Synergy with H. pylori
Given that NSAIDs (including aspirin) are gastric irritants, it could be predicted that such drugs would act synergistically with H. pylori gastritis to exacerbate mucosal damage. However, a role for H. pylori in increasing the risk of acute gastroduodenal injury has been difficult to establish or refute. Only one study has looked, in a controlled fashion, at the influence of H. pylori status on the frequency and severity of mucosal hemorrhage, erosions, and ulcers after 1 week of treatment with NSAIDs (44). No significant differences between H. pylori-positive and -negative subjects were detected. Although stronger claims have been made for a synergistic link with H. pylori in the causation of chronic gastric and duodenal ulcers, the evidence is controversial. While the point prevalence of ulcers appears higher in H. pylori-positive versus-negative NSAID users, the differences have not achieved statistical significance, even after amalgamation in a meta-analysis (80). Furthermore, two large randomized controlled trials on the prevention of peptic ulcers by prior eradication of H. pylori arrived at opposite conclusions, one (4) claiming that eradication reduced the occurrence of ulcers and another (85) concluding that there was no effect. With regard to bleeding peptic ulcers, it seems quite clear that NSAID use and H. pylori infection are independent risk factors (31, 42, 43). Indeed, one authority on the effects of NSAIDs on the gastrointestinal tract has concluded that under some circumstances patients who are infected are less prone to NSAID-induced ulcers than are uninfected individuals because, at least in part, of opposing effects on mucosal prostaglandin synthesis (30).
Conclusion
H. pylori infection is now accepted as the cause of the most common form of chronic gastritis. It is also widely accepted that the infection is at least the triggering factor for, if not the direct cause of, atrophy and intestinal metaplasia. These alterations in the gastric mucosa predispose to peptic ulceration. Given that they are maximal in the antrum-corpus TZ, this is the site of predilection for gastric ulcers. Different patterns of H. pylori gastritis are associated with profound alterations in acid output. In antral-predominant gastritis, acid production from the largely unaffected corpus is enhanced whereas corpus inflammation is associated with hypochlorhydria. These changes have an important bearing on peptic ulcer pathogenesis. Eradication of H. pylori infection is followed by resolution of gastritis, and this greatly reduces, if not eliminates, the risk of further peptic ulceration. Whether such intervention can remove the increased risk of progression to gastric cancer has yet to be determined.
References
- 1.
- Atherton J. C., Peek R. M., Tham K. T., Cover T. L., Blaser M. J. Clinical and pathological importance of heterogeneity in vacA, the vacuolating cytotoxin gene of Helicobacter pylori. Gastroenterology. 1997;112:92–99. [PubMed: 8978347]
- 2.
- Atuma C., Engstrand L., Holm L. Extracts of Helicobacter pylori reduce gastric mucosal blood flow through a VacA- and CagA-independent pathway in rats. Scand. J. Gastroenterol. 1998;33:1256–1261. [PubMed: 9930388]
- 3.
- Beales I. L., Crabtree J. E., Scunes D., Covacci A., Calam J. Antibodies to CagA protein are associated with gastric atrophy in Helicobacter pylori infection. Eur. J. Gastroenterol. Hepatol. 1996;8:645–649. [PubMed: 8853252]
- 4.
- Chan F. K., Sung J. J., Chung S. C., To K. F., Yung M. Y., Leung V. K., Lee Y. T., Chan C. S., Li E. K., Woo J. Randomised trial of eradication of Helicobacter pylori before non-steroidal anti-inflammatory drug therapy to prevent peptic ulcers. Lancet. 1997;350:975–979. [PubMed: 9329511]
- 5.
- Claeys D., Karapetian O., Saraga E., Schreyer M., Louis J., Acha-Orbea H., Blum A. L., Kraehenbuhl J.-P. Mouse mammary tumor virus superantigens and murine auto-immune gastritis. Gastroenterology. 1994;107:924–933. [PubMed: 7926482]
- 6.
- Clyne M., Labigne A., Drumm B. Helicobacter pylori requires an acidic environment to survive in the presence of urea. Infect. Immun. 1995;63:1669–1673. [PMC free article: PMC173208] [PubMed: 7729871]
- 7.
- Craanen M. E., Dekker W., Blok P., Ferwerda J., Tytgat G. N. J. Intestinal metaplasia and Helicobacter pylori: an endoscopic bioptic study of the gastric antrum. Gut. 1992;33:16–20. [PMC free article: PMC1373858] [PubMed: 1740271]
- 8.
- Crabtree J. Immunopathological aspects of Helicobacter pylori associated injury of the gastric mucosa. Mol. Med. 1994;31:1340–1348.
- 9.
- Crabtree J. E., Shallcross T. M., Wyatt J. I., Taylor J. D., Heatley R. V., Rathbone B. J., Lowsowsky M. S. Mucosal humoral immune response to Helicobacter pylori in patients with duodenitis. Dig. Dis. Sci. 1991;36:1266–1273. [PubMed: 1893811]
- 10.
- Danon S. J., O'Rourke J. L., Moss N. D., Lee A. The importance of local acid production in the distribution of Helicobacter felis in the mouse stomach. Gastroenterology. 1995;108:1386–1395. [PubMed: 7729630]
- 11.
- Dixon M. F. Pathophysiology of Helicobacter pylori infection. Scand. J. Gastroenterol. 1994;29(Suppl. 201):7–10. [PubMed: 8047828]
- 12.
- Dixon M. F., Wyatt J. I. Helicobacter pylori infection and duodenal ulcer. Br. Med. J. 1991;302:1535.
- 13.
- Eidt S., Oberhuber G., Schneider A., Stolte M. The histopathological spectrum of type A gastritis. Path. Res. Pract. 1996;192:101–106. [PubMed: 8692709]
- 14.
- Eidt S., Stolte M. Antral intestinal metaplasia in Helicobacter pylori gastritis. Digestion. 1994;55:13–18. [PubMed: 8112491]
- 15.
- Eidt S., Stolte M. Prevalence of intestinal metaplasia in Helicobacter pylori gastritis. Scand. J. Gastroenterol. 1994;29:607–610. [PubMed: 7939396]
- 16.
- Enno A., O'Rourke J. L., Howlett C. R., Jack A., Dixon M. F., Lee A. MALToma-like lesions in the murine gastric mucosa after long-term infection with Helicobacter felis. A mouse model of Helicobacter pylori-induced gastric lymphoma. Am. J. Pathol. 1995;147:217–222. [PMC free article: PMC1869885] [PubMed: 7604881]
- 17.
- Ernst, P. B., Y. Jin, J. Navarro, V. Reyes, and S. Crowe. 1994. Overview of the immune response to H. pylori, p. 295–305. In R. H. Hunt and G. N. J. Tytgat (ed.), Helicobacter pylori: Basic Mechanisms to Clinical Cure. Kluwer Academic Publishers, London, United Kingdom.
- 18.
- Faller G., Steininger H., Kränzlein J., Maul H., Kerkau T., Hensen J., Hahn E. G., Kirchner T. Antigastric autoantibodies in Helicobacter pylori infection: implications of histological and clinical parameters of gastritis. Gut. 1997;41:619–623. [PMC free article: PMC1891571] [PubMed: 9414967]
- 19.
- Ferrero R. L., Lee A. 1991The importance of urease in acid protection for the gastric-colonising bacteria H. pylori and H. felis sp. nov. Microbiol. Ecol. Health Dis 4121–134.
- 20.
- Filipe M. I., Potet F., Bogomoletz W. V., Dawson P. A., Fabiani B., Chauveinc P., Fenzy A., Gazzard B., Goldfain D., Zeegen R. Incomplete sulphomucin-secreting intestinal metaplasia for gastric cancer: preliminary data from a prospective study from three centres. Gut. 1985;26:1319–1326. [PMC free article: PMC1433103] [PubMed: 4085908]
- 21.
- Fox J. G., Correa P., Taylor N. S., Thompson N., Fontham E., Janney F., Sobhan M., Ruiz B., Hunter F. High prevalence and persistence of cytotoxin-positive Helicobacter pylori strains in a population with high prevalence of atrophic gastritis. Am. J. Gastroenterol. 1992;87:1554–1560. [PubMed: 1442673]
- 22.
- Gaskin R. J., Gad A., Barros A. A. J., D'Sa A. D., Joffe S. N., Baron J. H. Natural history and morphology of secretagogue-induced ulcers in rats. Gastroenterology. 1975;69:903–910. [PubMed: 1175885]
- 23.
- Genta R. M., Hamner H. W., Graham D. Y. Gastric lymphoid follicles in Helicobacter pylori infection: frequency, distribution, and response to triple therapy. Hum. Pathol. 1993;24:577–583. [PubMed: 8505036]
- 24.
- Goodwin S. Helicobacters shed new light on chaperonins. Lancet. 1995;346:653–655. [PubMed: 7658815]
- 25.
- Graham D. Y. Campylobacter pylori and peptic ulcer disease. Gastroenterology. 1989;96:615–625. [PubMed: 2642447]
- 26.
- Graham D. Y. Pathogenic mechanisms leading to Helicobacter pylori-induced inflammation. Eur. J. Gastroenterol. Hepatol. 1992;4(Suppl. 2):9–16.
- 27.
- Greig M. A., Neithercut W. D., Hossack M., McColl K. E. L. Harnessing of urease activity of H. pylori to induce self-destruction of the bacterium. J. Clin. Pathol. 1991;44:157–159. [PMC free article: PMC496980] [PubMed: 1864988]
- 28.
- Hanby A. M., Poulsom R., Singh S. et al. Spasmolytic polypeptide is a major antral peptide: distribution of the trefoil peptides human spasmolytic polypeptide and pS2 in the stomach. Gastroenterology. 1993;105:1110–1116. [PubMed: 8405856]
- 29.
- Hattori T. On cell proliferation and differentiation of the fundic mucosa of the golden hamster. Cell Tiss. Res. 1974;148:213–226. [PubMed: 4836489]
- 30.
- Hawkey C. J. Personal review: Helicobacter pylori, NSAIDs and cognitive dissonance. Aliment. Pharmacol. Ther. 1999;13:695–702. [PubMed: 10383497]
- 31.
- Hawkey C. J., Tulassay Z., Szczepanski L., van Rensburg C. J., Filipowicz-Sosnowska A., Lanas A., Wason C. M., Peacock R. A., Gillon K. R. Randomised controlled trial of Helicobacter pylori eradication in patients on non-steroidal anti-inflammatory drugs: HELP NSAIDs study. Helicobacter Eradication for Lesion Prevention. Lancet. 1998;352:1016–1021. [PubMed: 9759744]
- 32.
- Ito S., Azuma T., Murakita H., Hirai M., Miyaji H., Ito Y., Ohtaki Y., Yamazaki Y., Kuriyama M., Keida Y., Kohli Y. Profile of Helicobacter pylori cytotoxin derived from two areas of Japan with different prevalence of atrophic gastritis. Gut. 1996;39:800–806. [PMC free article: PMC1383450] [PubMed: 9038660]
- 33.
- James R. J. Homeodomain proteins and cell phenotype. Gastroenterology. 1997;113:680–686. [PubMed: 9247492]
- 34.
- Katelaris P. H., Seow F., Lin B., Napoli J., Ngu M. C., Jones D. B. The effect of age, Helicobacter pylori infection and gastric atrophy on serum gastrin and gastric acid secretion in healthy men. Gut. 1993;34:1032–1037. [PMC free article: PMC1374348] [PubMed: 8174948]
- 35.
- Kim J. S., Jung H. C., Kim J. M., Song I. S., Kim C. Y. Interleukin-8 expression by human neutrophils activated by Helicobacter pylori soluble proteins. Scand. J. Gastroenterol. 1998;33:1249–1255. [PubMed: 9930387]
- 36.
- Kimura K. Chronological transition of the fundic-pyloric border determined by stepwise biopsy of the lesser and greater curvatures of the stomach. Gastroenterology. 1972;63:584–592. [PubMed: 5077145]
- 37.
- Kimura K., Takemoto T. Endoscopic recognition of the atrophic border and its significance in chronic gastritis. Endoscopy. 1969;8:87–97.
- 38.
- Kjelleberg, S. (ed.). 1993. Starvation in Bacteria. Plenum Press, New York, N. Y.
- 39.
- Konturek P. C., Ernst H., Konturek S. J., Bobrzynski A. J., Faller G., Klingler C., Hahn E. G. Mucosal expression and luminal release of epidermal and transforming growth factors in patients with duodenal ulcer before and after eradication of Helicobacter pylori. Gut. 1997;40:463–469. [PMC free article: PMC1027119] [PubMed: 9176072]
- 40.
- Kreuning J., Wal A. M. V. D., Kuiper G., Lindeman J. Chronic nonspecific duodenitis: a multiple biopsy study of the duodenal bulb in health and disease. Scand. J. Gastroenterol. 1989;24(Suppl. 167):16–20. [PubMed: 2617162]
- 41.
- Kuipers E. J., Uyterlinde A. M., Pena A. S., Roosendaal R., Pals G., Nelis G. F., Festen H. P., Meuwissen S. G. Long term follow-up of Helicobacter pylori associated gastritis. Lancet. 1995;345:1525–1528. [PubMed: 7791437]
- 42.
- Kuyvenhoven J. P., Veenendaal R. A., Vandenbroucke J. P. Peptic ulcer bleeding: interaction between non-steroidal anti-inflammatory drugs, Helicobacter pylori infection, and the ABO blood group system. Scand. J. Gastroenterol. 1999;34:1082–1086. [PubMed: 10582757]
- 43.
- Labenz J., Peltz U., Kohl H., Kaiser J., Malfertheiner P., Hackelsberger A., Borsch G. Helicobacter pylori increases the risk of peptic ulcer bleeding: a case-control study. Ital. J. Gastroenterol. Hepatol. 1999;31:110–115. [PubMed: 10363194]
- 44.
- Lanza F. L., Evans D. G., Graham D. Y. Effect of Helicobacter pylori infection on the severity of gastroduodenal mucosa injury after the acute administration of naproxen or aspirin to normal volunteers. Am. J. Gastroenterol. 1991;86:735–737. [PubMed: 2038996]
- 45.
- Maaroos H. I., Vorobjova T., Sipponen P., Tammur R., Uibo R., Wadstrom T., Keevallik R., Villako K. An 18-year follow-up study of chronic gastritis and Helicobacter pylori association of CagA positivity with development of atrophy and activity of gastritis. Scand. J. Gastroenterol. 1999;34:864–869. [PubMed: 10522603]
- 46.
- Marshall B., Armstrong J., McGechie D., Glancy R. Attempt to fulfill Koch's postulates for pyloric Campylobacter. Med. J. Austr. 1985;142:436–439. [PubMed: 3982345]
- 47.
- Marshall B. J., McGeehie D. B., Rogers P. A., Glancy R. J. Pyloric Campylobacter infection and gastroduodenal disease. Med. J. Austr. 1985;142:439–444. [PubMed: 3982346]
- 48.
- Meyer-Roseberg K., Scott D. R., Melchers K., Sachs G. The effect of environmental pH on the proton motive force of H. pylori. Gastroenterology. 1996;111:886–900. [PubMed: 8831583]
- 49.
- Moayyedi, P., C. Wason, R. Peacock, K. Gillon, K. Bardhan, A. T. R. Axon, and M. F. Dixon. Changing patterns of gastritis in long-standing acid suppression: effect of Helicobacter pylori eradication. Helicobacter, in press. [PubMed: 11179985]
- 50.
- Mohammadi M., Nedrud J., Redline R., Lycke N., Czinn S. J. Murine CD4 T-cell response to Helicobacter infection: TH1 cells enhance gastritis and TH2 cells reduce bacterial load. Gastroenterology. 1997;113:1848–1857. [PubMed: 9394724]
- 51.
- Morris A., Nicholson G. Ingestion of Campylobacter pyloridis causes gastritis and raised fasting gastric pH. Am. J. Gastroenterol. 1987;82:192–199. [PubMed: 3826027]
- 52.
- Mukawa K., Nakamura T., Nakano G., Nagamachi Y. Histopathogenesis of intestinal metaplasia: minute lesions of intestinal metaplasia in ulcerated stomachs. J. Clin. Pathol. 1987;40:13–18. [PMC free article: PMC1140822] [PubMed: 3818971]
- 53.
- Negrini R., Savio A., Poiesi C., Appelmelk B. J., Buffoli F., Paterlini A., Cesari P., Graffeo M., Vaira D., Franzin G. Antigenic mimicry between Helicobacter pylori and gastric mucosa in the pathogenesis of body atrophic gastritis. Gastroenterology. 1996;111:655–665. [PubMed: 8780570]
- 54.
- Ogata T. Electron microscopic study on the regenerating epithelium of the chronic gastric ulcer. J. Submicroscop. Cytol. Pathol. 1995;27:171–182. [PubMed: 7757944]
- 55.
- Oi M., Oshida K., Sugimura S. The location of gastric ulcer. Gastroenterology. 1959;36:45–59. [PubMed: 13620016]
- 56.
- Oi M., Sakurai Y. The location of duodenal ulcer. Gastroenterology. 1959;36:60–64. [PubMed: 13620018]
- 57.
- Olso E. R. Influence of pH on bacterial gene expression. Mol. Microbiol. 1993;8:5–14. [PubMed: 8388532]
- 58.
- Oohara T., Tohma H., Aono G., Ukawa S., Kondo Y. Intestinal metaplasia of the regenerative epithelia in 549 gastric ulcers. Hum. Pathol. 1983;14:1066–1071. [PubMed: 6642497]
- 59.
- Parrish J. A., Rawlins D. C. Intestinal mucosa in the Zollinger-Ellison syndrome. Gut. 1965;6:286–289. [PMC free article: PMC1552290] [PubMed: 18668786]
- 60.
- Rektorschek M., Weeks D., Sachs G., Melchers K. Influence of pH on metabolism and urease activity of Helicobacter pylori. Gastroenterology. 1998;115:628–641. [PubMed: 9721160]
- 61.
- Roland M., Berstad A., Liavag I. A histological study of gastric mucosa before and after proximal gastric vagotomy in duodenal ulcer patients. Scand. J. Gastroenterol. 1975;10:181–186. [PubMed: 1124352]
- 62.
- Sakagami T., Dixon M. F., O'Rourke J., Howlett R., Alderuccio F., Vella J., Shimoyama T., Lee A. Atrophic gastric changes in both Helicobacter felis and Helicobacter pylori infected mice are host dependent and separate from antral gastritis. Gut. 1996;39:639–648. [PMC free article: PMC1383385] [PubMed: 9026476]
- 63.
- Scott D. R., Marcus E. A., Weeks D. L., Lee A., Melchers K., Sachs G. Expression of the Helicobacter pylori urel gene is required for acidic pH activation of cytoplasmic urease. Infect. Immun. 2000;68:470–477. [PMC free article: PMC97165] [PubMed: 10639406]
- 64.
- Scott D. R., Weeks D., Hong C., Postius S., Melchers K., Sachs G. The role of internal urease in acid resistance of Helicobacter pylori. Gastroenterology. 1998;114:58–70. [PubMed: 9428219]
- 65.
- Shaoul R., Marcon P., Okada Y., Cutz E., Forstner G. The pathogenesis of duodenal gastric metaplasia: the role of local goblet cell transformation. Gut. 2000;46:632–638. [PMC free article: PMC1727926] [PubMed: 10764705]
- 66.
- Silberg D. A., Furth E. E., Taylor J. K., Schuck T., Chiou T., Traber P. G. CDX1 protein expression in normal, metaplastic, and neoplastic human alimentary tract epithelium. Gastroenterology. 1997;113:478–486. [PubMed: 9247467]
- 67.
- Slomiany B. L., Piotrowski J., Slomiany A. Induction of caspase-3 and nitric oxide synthase-2 during gastric mucosal inflammatory reaction to Helicobacter pylori lipopolysaccharide. Biochem. Mol. Biol. Int. 1998;46:1063–1070. [PubMed: 9861460]
- 68.
- Sobala G. M., Axon A. T. R., Dixon M. F. Morphology of chronic antral gastritis: relationship to age, Helicobacter pylori status and peptic ulceration. Eur. J. Gastroenterol. Hepatol. 1992;4:825–829.
- 69.
- Sobala G. M., Crabtree J., Dixon M. F., Schorah C. J., Taylor J. D., Rathbone B. J., Heatley R. V., Axon A. T. R. Acute Helicobacter pylori infection: clinical features, local and systemic immune response, gastric mucosal histology and gastric juice ascorbic acid concentrations. Gut. 1991;32:1415–1418. [PMC free article: PMC1379180] [PubMed: 1752479]
- 70.
- Sobala G. M., O'Connor H. J., Dewar E. P., King R. F. J., Axon A. T. R., Dixon M. F. Bile reflux and intestinal metaplasia in gastric mucosa. J. Clin. Pathol. 1993;46:235–240. [PMC free article: PMC501177] [PubMed: 8463417]
- 71.
- Sobala G. M., Schorah C. J., Shires S., Lynch D. A. F., Gallacher B., Dixon M. F., Axon A. T. R. Effect of eradication of Helicobacter pylori on gastric juice ascorbic acid concentrations. Gut. 1993;34:1038–1041. [PMC free article: PMC1374349] [PubMed: 8174949]
- 72.
- Stadelmann K., Elster K., Stolte M., Miederer S. E., Deyhle P., Demling L., Siegenthaler W. The peptic gastric ulcer—histotopographic and functional investigations. Scand. J. Gastroenterol. 1971;6:613–623. [PubMed: 5128070]
- 73.
- Steer H. W. Ultrastructure of cell migration through the gastric epithelium and its relationship to bacteria. J. Clin. Pathol. 1975;28:639–646. [PMC free article: PMC475793] [PubMed: 1184762]
- 74.
- Steer H. W. Surface morphology of the gastro-duodenal mucosa in duodenal ulceration. Gut. 1984;25:1203–1210. [PMC free article: PMC1432316] [PubMed: 6500361]
- 75.
- Steer H. W. The gastroduodenal epithelium in peptic ulceration. J. Pathol. 1985;146:355–359. [PubMed: 4032128]
- 76.
- Stolte M., Baumann K., Bethke B., Ritter M., Lauer E., Eidt H. Active autoimmune gastritis without total atrophy of the glands. Zentralbl. Gastroenterol. 1992;30:729–735. [PubMed: 1441676]
- 77.
- Stolte M., Meining A. A., Schmitz J. M., Alexandris T., Seifert E. Changes in Helicobacter pylori-induced gastritis in the antrum and corpus during 12 months of treatment with omeprazole and lansoprazole in patients with gastrooesophageal reflux disease. Aliment. Pharmacol. Ther. 1998;12:247–253. [PubMed: 9570259]
- 78.
- Tatsuta M., Ishii H., Yamamura H., Yamamoto R., Taniguchi H. Enhancement by tetragastrin of experimental induction of gastric epithelium in the duodenum. Gut. 1989;30:311–315. [PMC free article: PMC1378451] [PubMed: 2707631]
- 79.
- Terres A. M., Pajares J. M., O'Toole D., Ahern S., Kelleher D. H. pylori infection is associated with down-regulation of E-cadherin, a molecule involved in epithelial cell adhesion and proliferation control. J. Clin. Pathol. 1998;51:410–412. [PMC free article: PMC500709] [PubMed: 9708215]
- 80.
- Veldhuyzen van Zanten, S. J. O. 1994. H. pylori and NSAIDs: a meta-analysis on interactions of acute gastroduodenal injury, gastric and duodenal ulcers and upper gastrointestinal symptoms, p. 449–457. In R. H. Hunt and G. N. J. Tytgat (ed.), H. pylori—Basic Mechanisms to Clinical Cure. Kluwer Academic Publishers, London, United Kingdom.
- 81.
- Walker M. M., Dixon M. F. Gastric metaplasia: its role in duodenal ulceration. Aliment. Pharmacol. Ther. 1996;10(Suppl. 1):119–128. [PubMed: 8730266]
- 82.
- Walker M. M., Logan R. P. H., Gummett P. A., Baron J. H., Misiewicz J. J. The influence of Helicobacter pylori eradication on the structure of duodenal ulcer scars. Eur. J. Gastroenterol. Hepatol. 1993;5(Suppl. 3):S97–S98.
- 83.
- Warburton V. J., Everett S., Mapstone N. P., Axon A. T. R., Hawkey P., Dixon M. F. Clinical and histological associations of cagA and vacA genotypes in Helicobacter pylori gastritis. J. Clin. Pathol. 1998;51:55–61. [PMC free article: PMC500433] [PubMed: 9577374]
- 84.
- Warren J. R., Marshall B. J. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet. 1983;i:1273–1275. [PubMed: 6134060]
- 85.
- Wu C. Y., Poon S. K., Chen G. H., Chang C. S., Yeh H. Z. Interaction between Helicobacter pylori and non-steroidal anti-inflammatory drugs in peptic ulcer bleeding. Scand. J. Gastroenterol. 1999;34:234–237. [PubMed: 10232865]
- 86.
- Wyatt J. I., Rathbone B. J., Dixon M. F., Heatley R. V. Campylobacter pyloridis and acid-induced gastric metaplasia in the pathogenesis of duodenitis. J. Clin. Pathol. 1987;40:841–848. [PMC free article: PMC1141122] [PubMed: 3654985]
- 87.
- Yang H. T., Dixon M. F., Zuo J. S., Xu Z., Zhou D., Blum A. L. Helicobacter pylori infection and gastric metaplasia in China. J. Clin. Gastroenterol. 1995;20:110–112. [PubMed: 7769188]
- 88.
- Yoshimura T., Shimoyama T., Fukuda S., Tanaka A., Axon A. T. R., Munakata A. Most gastric cancer occurs on the distal side of the endoscopic atrophic border. Scand. J. Gastroenterol. 1999;34:1077–1081. [PubMed: 10582756]
- Helicobacter pylori infection, glandular atrophy, intestinal metaplasia and topography of chronic active gastritis in the Nepalese and Japanese population: the age, gender and endoscopic diagnosis matched study.[Kathmandu Univ Med J (KUMJ). 2...]Helicobacter pylori infection, glandular atrophy, intestinal metaplasia and topography of chronic active gastritis in the Nepalese and Japanese population: the age, gender and endoscopic diagnosis matched study.Matsuhisa T, Miki M, Yamada N, Sharma SK, Shrestha BM. Kathmandu Univ Med J (KUMJ). 2007 Jul-Sep; 5(3):295-301.
- Helicobacter pylori eradication in the healing of atrophic gastritis: a one-year prospective study.[Scand J Gastroenterol. 2006]Helicobacter pylori eradication in the healing of atrophic gastritis: a one-year prospective study.Arkkila PE, Seppälä K, Färkkilä MA, Veijola L, Sipponen P. Scand J Gastroenterol. 2006 Jul; 41(7):782-90.
- Increase of Helicobacter pylori-associated corpus gastritis during acid suppressive therapy: implications for long-term safety.[Am J Gastroenterol. 1995]Increase of Helicobacter pylori-associated corpus gastritis during acid suppressive therapy: implications for long-term safety.Kuipers EJ, Uyterlinde AM, Peña AS, Hazenberg HJ, Bloemena E, Lindeman J, Klinkenberg-Knol EC, Meuwissen SG. Am J Gastroenterol. 1995 Sep; 90(9):1401-6.
- Review Pathophysiology of Helicobacter pylori infection.[Scand J Gastroenterol Suppl. 1...]Review Pathophysiology of Helicobacter pylori infection.Dixon MF. Scand J Gastroenterol Suppl. 1994; 201:7-10.
- Review Helicobacter pylori, chronic gastritis and peptic ulcer.[Mater Med Pol. 1992]Review Helicobacter pylori, chronic gastritis and peptic ulcer.Sipponen P. Mater Med Pol. 1992 Jul-Sep; 24(3):166-8.
- Pathology of Gastritis and Peptic Ulceration - Helicobacter pyloriPathology of Gastritis and Peptic Ulceration - Helicobacter pylori
Your browsing activity is empty.
Activity recording is turned off.
See more...