

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.
This publication is provided for historical reference only and the information may be out of date.

Scope
UniGene is a largely automated analytical system for producing an organized view of the transcriptome. By analyzing sequences known to be expressed, and the libraries or samples from which they were derived, it is possible to organize the data into gene-specific clusters, and, in some cases, evaluate the patterns of expression by tissue, health status, and age. In this chapter, we discuss the properties of the input sequences, the process by which they are analyzed in UniGene, and some pointers on how to use the resource.
History
The task of assembling an inventory of all genes of Homo sapiens and other organisms began more than two decades ago with large-scale survey sequencing of transcribed sequences. The resulting Expressed Sequence Tags (ESTs) continue to be an invaluable component of efforts to characterize the transcriptome of many organisms. These efforts which rely on ESTs include genome annotation (2-4), expression systems (5), and full-length cDNA cloning projects (6). In addition, targeted gene-hunting projects have benefited from the availability of these sequences and the physical clone reagents. However, the high level of redundancy found among transcribed sequences, not to mention a variety of common experimental artifacts, made it difficult for many people to make effective use of the data. This problem was the motivation for the development of UniGene.
Now that the genomes of many species have been sequenced completely, a fundamental resource expected by many researchers is a simple list of all of an organism's genes. However, many species of medical and agricultural importance do not yet have a complete annotated genome available. Furthermore, when the genomic sequence of an organism is made public, a collection of cDNA sequences provides the best tool for identifying genes within the DNA sequence. When the source material for cDNA sequences is drawn from diverse tissues, an approximate expression profile for the organism's transcriptome can be computed. This approximate expression profile can serve to at least identify transcripts of interest to researchers interested in a particular system, and at best to characterize the function of novel transcripts. Thus, we can anticipate that the sequencing of transcribed products will remain a significant area of interest well into the future.
Data Model
The data model for UniGene is straightforward. Identify sequences of RNA molecules, the source of those sequences (species, tissue, age, health status), compute when independent sequences are derived from the same gene based on sequence similarity, and report the results. Historically this computation was based on ESTs (Extended Sequence Tags), but now the vast majority of sequences are either full-length clones or RNAseq data.
ESTs
The basic strategy of generating ESTs involves selecting cDNA clones at random and performing a single, automated, sequencing read from one or both ends of their inserts. This class of sequence is characterized by being short (typically about 400-600 bases) and relatively inaccurate (around 2% error). In most cases, there is no initial attempt to identify or characterize the clones. Instead, they are identified using only the small bit of sequence data obtained, comparing it to the sequences of known genes and other ESTs. It is fully expected that many clones will be redundant with others already sampled and that a smaller number will represent various sorts of contaminants or cloning artifacts. There is little point in incurring the expense of high-quality sequencing until later in the process, when clones can be validated and a non-redundant set selected.
Despite their fragmentary and inaccurate nature, ESTs were found to be an invaluable resource for the discovery of new genes, particularly those involved in human disease processes. After the initial demonstration of the utility and cost effectiveness of the EST approach, many similar projects were initiated, resulting in an ever-increasing number of human ESTs. In addition, large-scale EST projects were launched for several other organisms of experimental interest. In 1992, a database called dbEST was established to serve as a collection point for ESTs, which are then distributed to the scientific community as the EST division of GenBank.
Dataflow
The number of transcribed sequences is large enough that interactive analysis of each sequence by a researcher is impossible. A major challenge is to make putative gene assignments for these sequences, recognizing that many of these genes will be anonymous, defined only by the sequences themselves. Computationally, this can be thought of as a clustering problem in which the sequences are vertices that may be coalesced into clusters by establishing connections among them.
Experience has shown that it is important to eliminate low-quality or apparently artifactual sequences before clustering because even a small level of noise can have a large corrupting effect on a result. Thus, procedures are in place to eliminate sequences of foreign origin (most commonly Escherichia coli) and identify regions that are derived from the cloning vector or artificial primers or linkers. At present, UniGene focuses on protein-coding genes of the nuclear genome; therefore, those identified as rRNA or mitochondrial sequence are eliminated. Through the NCBI Trace Archive, an increasing number of EST sequences now have base-level error probabilities that are used to identify the highest quality segment of each sequence. Repetitive sequences sometimes lead to false alignments and must be treated with caution. Simple repeats (low-complexity regions) are identified using a word-overrepresentation algorithm called DUST, and transposable repetitive elements are identified by comparison with a library of known repeats for each organism. Rather than eliminating them outright, subsequences classified as repetitive are soft-masked, which is to say that they are not allowed to initiate a sequence alignment, although they may participate in one that is triggered within a unique sequence. For a sequence to be included in UniGene, the clone insert must have at least 100 base pairs that are of high quality and not repetitive.
With a given a set of sequences, a variety of different sources of information may be used as evidence that any pair of them is or is not derived from the same gene. The most obvious type of relationship would be one in which the sequences overlap and can form a near-perfect sequence alignment. One dilemma is that some level of mismatching should be tolerated because of known levels of base substitution errors in ESTs, whereas allowing too much mismatching will cause highly similar paralogous genes to cluster together. One way to improve the results is to require that alignments show an approximate dovetail relationship, which is to say that they extend about as far to the ends of the sequences as possible. Values of specific parameters governing acceptable sequence alignments are chosen by examining ratios of true to false connections in curated test sets. It is important to note that the resulting clusters may contain more than one alternative-splice form.
Multiple incomplete but non-overlapping fragments of the same gene are frequently recognized in hindsight when the gene's complete sequence is submitted. To minimize the frequency of multiple clusters being identified for a single gene, UniGene clusters are required to contain at least one sequence carrying readily identifiable evidence of having reached the 3 terminus. In other words, UniGene clusters must be anchored at the 3 end of a transcription unit. This evidence can be either a canonical polyadenylation signal or the presence of a poly(A) tail on the transcript, or the presence of at least two ESTs labeled as having been generated using the 3 sequencing primer. Because some clusters do not contain such evidence (typically, they are single ESTs), not all uncontaminated sequences in dbEST appear in UniGene clusters. Of course, alternatively spliced terminal 3 exons will appear as distinct clusters until sequence that spans the distinct splice forms is submitted.
With the availability of genome sequence, a more stringent test of 3’ anchoring is possible, because internal priming can be recognized. Clusters that satisfy this more-stringent requirement can be identified by adding the term has_end to any query. Specific query possibilities such as this one are listed under the rubric Query Tips on the UniGene homepage.
Access
The UniGene website allows the user to search for particular genes of interest, or to browse UniGene entries related by expression or sequence similarity. Each UniGene Web page includes a header with a query bar and a sidebar providing links to related online resources. UniGene is also the basis for other NCBI resources:
- ProtEST, a facility for browsing protein similarities;
- a library browser and display, which support exploration of cDNA libraries from
tissues of interest.
Looking for sequences expressed under a particular circumstance (a body site or developmental stage, for example) is a common method by which users identify individual genes or sets of genes that are of interest. There are several interfaces into UniGene's data to help users do this. Most broadly, there is a straightforward way to browse all cDNA libraries prepared with RNA from a particular biological source. Properties of individual libraries are summarized as well; the library submitter's description of source material and protocol, and a summary of the UniGene clusters expressed by the library's sequences.
UniGene Cluster Browser
The UniGene Cluster page summarizes the sequences in the cluster and additional derived information that may be used to infer the identity and in some cases function of the gene. Figure 1 shows an example of such a view for the human SERPINF2 gene. When available, links are provided to a corresponding entry in other NCBI resources (e.g., Gene, HomoloGene, OMIM) or external databases (e.g., Mouse Genome Informatics (MGI) at the Jackson Laboratory and the Zebrafish Information Network (ZFIN) at the University of Oregon). Additional sections on the page provide protein similarities, mapping data, expression information, and lists of the clustered sequences.
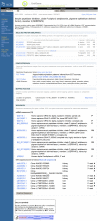
Figure 1.
Web view of a UniGene cluster.
Possible protein products for the gene are suggested by providing protein similarities between one representative sequence from the cluster and protein sequences from selected model organisms with an annotated geneome. For each model organism, the protein with the highest degree of sequence similarity to the nucleotide sequence is listed, with its title and GenBank accession. The sequence alignment is summarized using the percent identity and length of the aligned region. Also provided is a link (a popup menu attached to the protein accession) to other protein-oriented NCBI resources including ProtEST, which summarizes translating searches of the model organism protein against all organisms in UniGene.
The next section summarizes information on the aligned or inferred map position of the gene. For human and some other annotated genomes, the map position and link to the genomic neighborhood as represented in Map Viewer. Absent these aligned map positions, radiation hybrid (RH) maps have been constructed using Sequence Tagged Site (STS) markers derived from ESTs. In these cases, the UniGene cluster can be associated with a marker in the UniSTS database, and a map position can be assigned from the RH map. More recently, map positions have been derived by alignment of the cDNA sequences to the finished or draft genomic sequences present in the NCBI MapViewer. For example, the SERPINF2 gene in Figure 1 has a link to human chromosome 17 in the Map Viewer. The map is initially shown with a few selected tracks that are likely to be of interest, but others may be added by the user.
Although ESTs are not an optimal probe of gene expression, both the total number of ESTs and the tissues from which they originated are often useful. In the Gene Expression section of the cluster browser, a link to a summary of the gene's expression in cDNA libraries is available, with an example shown in Figure 2. Expression in each body site or developmental stage available in ESTs (excepting those from normalized or subtracted libraries, also excepting those which come from libraries of mixed source material) is reported, expresssed as counts of ESTs transcribed per miliion sequenced. Expression data is also available by FTP. UniGene clusters with similar expression profile are precomputed, and available under the link labelled "show more like this." This is most likely to be informative when expression differs markedly from uniform expression. Clusters that are predominantly expressed in a single body site or developmental stage are searchable in Entrez, by using the field "Restricted Expression." More specifically, these are clusters where 2/3 or more of the detected gene expression expresssed in normalized units of transcripts per million is from a single source. Links to NCBI's GEO computed from the GenBank accessions in the UniGene cluster are also present in this section of the cluster view.

Figure 2.
Expression profile view of a UniGene cluster.
The component sequences of the cluster are listed, with a brief description of each one and a link to its UniGene Sequence page. The Sequence page provides more detailed information about the individual sequence, and in the case of ESTs, includes a link to its corresponding UniGene Library page. On the cluster page, the EST clones that are considered by the Mammalian Gene Collection (MGC) project to be putatively full length are listed at the top, whereas others follow in order of their reported insert length. At the bottom of the UniGene Cluster page is a button for users to download the sequences of the cluster in FASTA format.
An FTP representation of UniGene is available as well. Sequence sets as FASTA (both aggregate and best representative per cluster), a summary of the mapping of sequences to UniGene clusters with library of origin for ESTs, and an expression summary. A common use of UniGene is to use a single representative sequence from each cluster for primer design or as a BLAST database. In this case, researchers are advised to retain both the sequence accession number as well as the cluster identifier for later reference. This is because the cluster identifier is not guaranteed to be indefinitely stable. While most UniGene builds differ only through incremental changes to existing clusters or the addition of newly represented transcripts, new sequences or new genome mapping can provide information that leads to substantial reorganization of previously identified clusters.
Related Tools
Protein Similarity Browser
The ProtEST section of UniGene allows the user to explore precomputed alignments for a selected protein to the cDNA sequences found in any cluster. Especially for cases where looking at alignments to the same protein of transcripts from multiple organisms, this interface provides a single concise overview. In the cluster viewer's protein similarity section, this overview is under the "Protein/EST matches” link in the popup menu that appears on mouseover of a protein accession; this popup is shown in Figure 3. BLASTX has been used to compare each sequence in UniGene to selected protein sequences drawn from model organisms with a fully annotated genome. By default, only alignments to the organisms in the same broad taxonomic group as the original organism are shown (primates, rodents, etc), though alignments to a broader set of organisms can be selected from the protEST pulldown menu. These alignments include alignments both to RefSeqs, which are based on a sequenced and annotated mRNA, as well as RefSeqs, which are gene predictions.
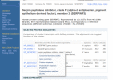
Figure 3.
Popup menu in UniGene cluster browser providing links to information about proteins similar to the transcript sequences in the cluster.
The sequence alignments in ProtEST are summarized in tabular form (Figure 4). The first column is a schematic representation of the nucleotideprotein alignment. The width of the column represents the entire length of the protein, whereas the unaligned nucleotide sequence is represented as a thin gray line and the aligned region is represented as a thick magenta bar. The alignment representation is a hyperlink to the full alignment regenerated on-the-fly using BLAST. Other information in the table includes the frame and strand of the alignment the UniGene cluster ID, the GenBank accession, and a summary of the aligned region and percent identity.
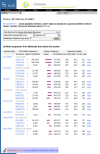
Figure 4.
Protein-transcript alignment summary.
Digital Differential Display (DDD)
DDD is a tool for comparing EST-based expression profiles among the various libraries, or pools of libraries, represented in UniGene. These comparisons allow the identification of those genes that differ among libraries of different tissues, making it possible to determine which genes may be contributing to a cell's unique characteristics, e.g., those that make a muscle cell different from a skin or liver cell. Along similar lines, DDD can be used to try to identify genes for which the expression levels differ between normal, premalignant, and cancerous tissues or different stages of embryonic development.
As is UniGene, the DDD resource is organism specific and is available from the UniGene website for that organism. For those libraries that have sequences in UniGene, DDD lists the title and tissue source and provides a link to the UniGene Library page, which gives additional information about the library. From the libraries listed, the user can select two for comparison. DDD then displays those genes for which the frequency of the transcript is significantly different between the two libraries. The output includes, for each gene, the frequency of its transcript in each library and the title of the gene's corresponding UniGene cluster. Results are sorted by significance, with the genes having the largest differences in frequencies displayed at the top. Libraries can be added sequentially to the analysis, and DDD will perform an analysis on each possible library gene pair combination. Similarly, groups of libraries can be pooled together and compared with other pools or single libraries. An example comparing two pools of libraries with similar sequence counts from human muscle and human brain is shown in Figure 5.
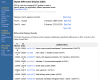
Figure 5.
Differential expression assessment comparing libraries from muscle and from brain.
DDD uses the Fisher Exact test to restrict the output to statistically significant differences (P 0.05). The analysis is also restricted to deeply sequenced libraries; only those with over 1000 sequences in UniGene are included in DDD. These requirements place limitations on the capabilities of the analysis. Unless there are a large number of sequences in each pool, the frequencies of genes are generally not found to be statistically significant. Furthermore, the wide variety of tissue types, cell types, histology, and methods of generating the libraries can make it difficult to attribute significant differences to any one aspect of the libraries. These issues underscore the need for more libraries to be made public and the need for the comparisons to be made using proper controls.
cDNA Library Browser and UniGene cDNA Library Display
Researchers frequentlly wish to identify particular cDNA libraries that interest them. In addition to the gene-oriented resources described above, UniGene offers an overview of all libraries from an organism of interest in the library browser. Libraries are grouped by their source material (body site or developmental stage where these are described by the library’s submitter), with the Web interface for browsing shown in Figure 6. For individual libraries, the library summary aggregates information provided by the submitter with the UniGene clusters that contain sequences from a library, with the Web interface for an individual library summary shown in Figure 7. Researchers may download all sequences from the library in FASTA format whether they are in any UniGene cluster or not from this page.
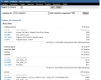
Figure 6.
UniGene library browser.
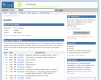
Figure 7.
UniGene library summary.
References
- 1.
- Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, Xiao H, Merril CR, Wu A, Olde RF, Moreno RF., et al. Complementary DNA sequencing: expressed sequence tags and human genome project. Science. 1991;252(5013):1651–1656. [PubMed: 2047873]
- 2.
- Lu F, Jiang H, Ding J, Mu J, Valenzuela JG, Ribeiro JM, Su XZ. cDNA sequences reveal considerable gene prediction inaccuracy in the Plasmodium falciparum genome. BMC Genomics. 2007;8:255. [PMC free article: PMC1978503] [PubMed: 17662120]
- 3.
- Shangguan L, Han J, Kayesh E, Sun X, Zhang C, Pervaiz T, Wen X, Fang J. Evaluation of genome sequencing quality in selected plant species using expressed sequence tags. PLoS One. 2013 Jul 29;8(7) [PMC free article: PMC3726750] [PubMed: 23922843]
- 4.
- Head and Neck Annotation Consortium. Large-scale transcriptome analyses reveal new genetic marker candidates of head, neck, and thyroid cancer. Cancer Res. 2005 Mar 1;65(5):1693–9. [PubMed: 15753364]
- 5.
- Zhang YE, Landback P, Vibranovski MD, Long M. Accelerated recruitment of new brain development genes into the human genome. PLoS Biol. 2011 Oct;9(10) [PMC free article: PMC3196496] [PubMed: 22028629]
- 6.
- MGC Project Team. The completion of the Mammalian Gene Collection (MGC). Genome Res. 2009 Dec;19(12):2324–33. [PMC free article: PMC2792178] [PubMed: 19767417]
- UniGene - The NCBI HandbookUniGene - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...