

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.
This publication is provided for historical reference only and the information may be out of date.

Scope
The Protein Clusters dataset consists of organized groups (clusters) of proteins encoded by complete and draft genomes from the NCBI Reference Sequence (RefSeq) collection of microorganisms: prokaryotes, viruses, fungi, protozoans; it also includes protein clusters from RefSeq genomes of plants, chloroplasts, and mitochondria. Clusters for each group are created and curated separately and given a different accession prefix. The primary goal of Protein Clusters is to provide the support to functional annotation of RefSeq genomes. Functional annotation of novel proteins is based on the assumption that proteins with high levels of sequence similarity are likely to share the same function. This oversimplified model of a linear evolution where similar proteins evolve from a single ancestor is further complicated by the events of gene duplication. Clusters of related (homologous) proteins include both orthologs and paralogs. Orthologs are genes in different organisms (species) that evolved from a common ancestral gene by speciation; paralogs are genes related by duplication within a genome. The definition was first introduced by Fitch in 1970 (7). The analysis of protein families from various organisms has shown that this definition does not embrace all the complexity of relationships of genes from different organisms. For more details see Koonin et al. 2005 (16). The NCBI Protein Clusters dataset contains automatically generated clusters that do not distinguish orthologs and paralogs. During manual evaluation some clusters containing paralogs can be split by curators, especially if the paralogs are known to have different functions.
History
The first complete bacterial genome of Haemophilus influenzae Rd KW20 sequenced and released in 1995 opened a new era in genome analysis (8). In the following year four more genomes were completed producing an unprecedented variety of protein sequences from all three major kingdoms (Archaea, Eubacteria, and Eukaryota). Comparative analysis of homologous genes has been used in evolutionary studies and functional classification since the first sequence became available, but for the first time the whole proteome of several organisms became available for comparison. New genome-scale methods were needed to provide an understanding of the true orthologous relationships of protein sequences. The protein database of Clusters of Orthologous Groups (COGs), a pioneering work of NCBI scientists, was the first attempt at creating a phylogenetic classification of the complete complement of proteins encoded by complete genomes (23). The COG approach is based on the simple notion that any group of at least three proteins from distant genomes that are more similar to each other than they are to any other proteins from the same genome are most likely to form an orthologous set. The COG project has proved to be an excellent approach for understanding microbial evolution and the diversity of protein functions encoded by their genomes. However, the major difficulty of any genomic data resource in the modern era of rapid genomic sequencing is keeping the genomic data and the annotations up-to-date.
The RefSeq project, which contains non-redundant sets of curated transcript, gene, and protein information in eukaryotes, and gene and protein information in prokaryotes, has been a very successful way to maintain and update annotated data. Given the increasing number of prokaryotic genomes being deposited, it became apparent that annotating protein families as a group was a convenient and efficient way to functionally annotate these data. The Protein Clusters database was constructed with two goals in mind: first, to routinely update RefSeq genomes with curated gene and protein information from such clusters; and second, to provide a central aggregation source for information collected from a wide variety of sources that would be useful for scientists studying protein-level or genomic-level molecular functions. In addition, curators routinely parse the scientific literature for reports of experimentally verified functions as the basis for existing or potential connections to genes/proteins, and such connections are added as annotations on each cluster. The first release of NCBI Protein Clusters in 2007 contained ~1 million proteins encoded by complete chromosomes and plasmids from three major groups: prokaryotes, bacteriophages, and the organelles (15). Since then the scope has been expanded to other taxonomic groups and proteins from draft genomes.
As of April 2013 the dataset represents more than 20 million proteins.
Data Model
Clustering is a well-known method in statistics and computer science. For a given set of entities, clusters are defined as subsets that are homogeneous and well separated. The cluster analysis should start from a definition of homogeneity and separation. Most clustering methods rely upon similarities (or distance) between entities. Protein clusters are aimed to be groups of homologous proteins. The similarity between two protein sequences is measured by maximum alignment between the sequences calculated by BLAST. There are multiple ways of defining various types of clusters that are based on criteria used to express separation or homogeneity of a cluster and separation from other clusters. NCBI Protein Clusters uses two methods for clustering, both resulting in building cliques, one based on partitioning and the other based on hierarchical aggregation.
Once clustered, each protein cluster is assigned a cluster ID and accession (letter prefix followed by digits) that is stable from version to version as long as the majority of its proteins don’t change. A protein cluster also includes certain attributes aggregated from proteins: “Gene names” (locus), “COG functional categories,” “EC numbers,” and “Conserved Domains.” An attribute “Conserved in” defines the common taxonomical name of genomes included in the cluster. The Protein Clusters database also includes a set of “Related Clusters”. Besides these attributes, each protein record in the database has “Organism name,” “Protein name,” “Protein accession,” “Locus tag,” “Length,” and UniProtKB / SwissProt accession. These attributes are easily searchable within a cluster and also through the whole database.
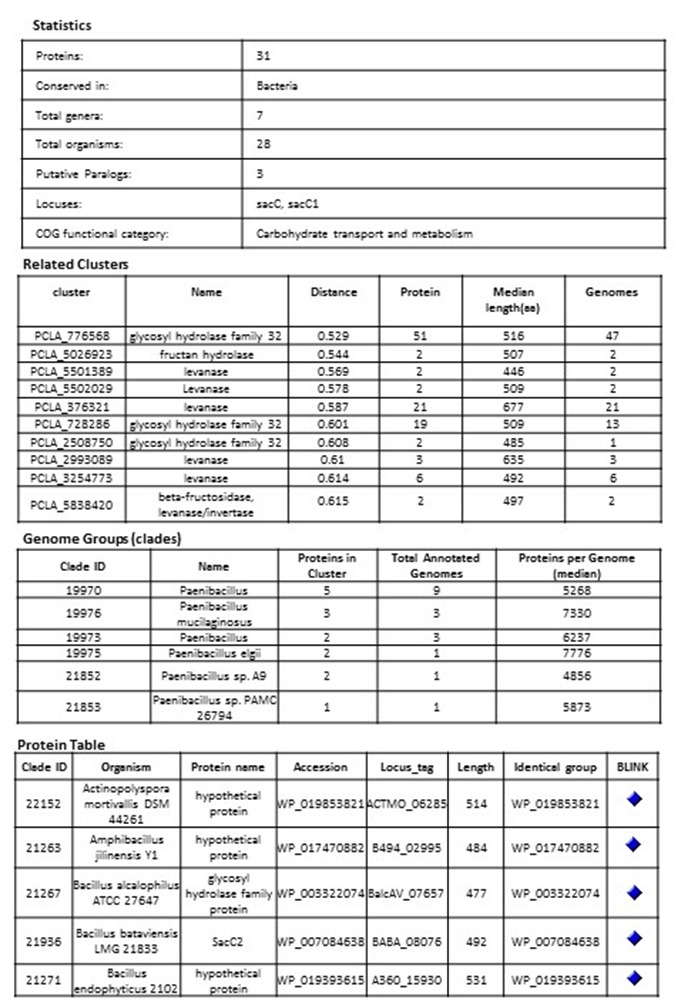
Example of a bacterial cluster PCLA_5029913 glycoside hydrolase
Clustering Methods
Partitioning in Cliques
Proteins are compared by sequence similarity using BLAST all against all (E-value cutoff 10E-05; effective length of the search space is set to 5 × 10E8). Each BLAST score is then modified by protein length × alignment length of the BLAST hit and the modified scores are sorted. Clusters (also known as cliques) consist of protein sets such that every member of the cluster hits every other protein member (reciprocal best hits by modified score). Cluster membership is such that for any given protein in the cluster (protein A), all the other members of the cluster will have a greater modified score to protein A than any protein outside of that cluster. During clustering, there are no cutoffs used nor strict requirements for clusters of orthologous groups, nor any check on phylogenetic distance. The initial set of uncurated clusters created in 2005 was used as a starting point for curation and has been updated periodically since then. During updates, new proteins are added to curated clusters. In the uncurated cluster set, proteins are allowed to repartition into different cluster sets, although this happens rarely and usually only in the case of smaller clusters.
Hierarchical Aggregation in Cliques
A new approach implemented for prokaryotic genomes is based on hierarchical clustering. While a hierarchical structure is conventionally represented by a dendrogram and clusters are selected as a sub-tree corresponding to a certain threshold (14, 17, 18), the hierarchical structure goes beyond simple clustering (1, 3). First, all the proteins are organized in global clusters, then links between clusters are calculated reflecting the similarity between the clusters based on several criteria.
Protein Clustering Procedure
The similarity of proteins is determined from the aggregated BLAST hits obtained by blastp with e-value . Two proteins are considered connected if there is an aggregated BLAST hit between them satisfying criteria on hit length and score. More specifically, we require the aggregated hit lengths on each protein, and , satisfy the inequalities and , where and are lengths of proteins, and , and the aggregated BLAST score satisfy the inequality , where and are self-scores.
The modified BLAST distance is defined as
,
where the score modifications are , 1- and , 1-, and Using allows some flexibility at the end of the sequences (the distance is reduced to when ). Clusters are aggregated in a hierarchical manner using the complete linkage distance, with an additional requirement that the minimum distance between clusters Ω) should not exceed threshold where for clusters and Ω to be merged. Note that we calculate and use both Ω) and Ω) in our clustering procedure (see Figure 1). Because of the sparse nature of connections and applied thresholds, we build a family of trees that we consider clusters. Currently, we use the values , , and .
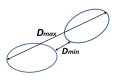
Figure 1.
Minimal and maximum distance between clusters
Establishing Links between Related Clusters
Each protein within a cluster should be similar to all other proteins in the same cluster, satisfying coverage and similarity criteria. Still, a pair of proteins in different clusters could be similar. Such clusters are designated as related clusters (1, 3, 12, 24). Links between related clusters are stored in link indexes, which are used to show neighborhoods of clusters in Entrez search.
Organization of computations. First, we eliminate redundancy and near-redundancy in the protein dataset (2, 12). Representative proteins from groups of redundant and nearly-redundant proteins are selected by the program USEARCH (5).
In order to perform clustering in parallel, the dataset is partitioned in disjoint sets (Figure 2) using a parallel implementation based on a disjoint-set forest with union-by-rank heuristics (4, 22), and then clustering is performed concurrently in partitions. When looking for disjoint sets, we only consider connections with
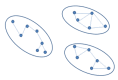
Figure 2.
Disjoint sets.
After the clustering is performed, link indexes are also calculated in parallel from the aggregated BLAST hit and protein assignment to clusters.
Dataflow
Input data are proteins from complete and draft (WGS) genomes that pass certain quality filters.
Proteins marked as incomplete in metadata (“incomplete,” “no start,” “no end,” “fragment,” etc.) are removed and only proteins that are presumed complete are analyzed. Bacterial genome clustering has a different dataflow compared to other genomes as indicated in Figure 3.
Figure 3.
Dataflow for prokaryotic and eukaryotic genomes
Manual Curation
One of the most important aspects of the curation process of Protein Clusters is the assignment of function that is obtained from the literature. Curated functional annotation can be propagated to all proteins within the cluster. That process improves the functional annotation of RefSeq genomes and unifies and standardizes the naming rules across various organisms and different annotation pipelines. In addition to providing functional annotation that is required for each cluster, other data are also added, such as the gene name, a detailed description about the protein, the E.C. number, and relevant publications.
Cluster Display
A protein cluster is represented by a list of protein identifiers (accessions) and the genomes that code for the proteins. Each cluster has a stable unique identifier and a functional cluster name. The cluster name is automatically calculated and followed by manual review. Each cluster provides statistics to indicate the number of proteins within that cluster and other important features including the Protein Table.
Cluster Examples
Viruses
The ease and efficiency of nucleic acid sequencing has led to an abundance of sequence data. Because of the relatively small genome size of viruses, the influx of sequence data has been particularly large. Likewise, the ever increasing advancements and publications in virology research make it difficult for researchers to keep up with new discoveries in protein structure and function. Rapid viral evolution, combined with the relatively large number of strains and closely related species in most viral families, makes the Protein Clusters resource an ideal channel through which viral RefSeq genomes can be curated.
The Poxviridae is an example of a virus family with a large set of proteins having varying degrees of similarity in function, homology, and structure (13). The poxvirus RNA helicase NPH-II belongs to a family of ubiquitous ATP-dependent helicases that are required for RNA metabolism in bacteria, eukaryotes, and many viruses (6). The NPH-II family of helicases found in hepatitis C and various poxviruses have similar sequence, structure, and mechanisms of action that are essential for viral replication. The protein cluster PHA2653 includes 27 NPH-II helicase proteins from various members of the Poxviridae. While they share a high level of homology, evolutionary pressures have resulted in changes to both sequence and activity. Of particular interest is the fact that the poxvirus NPH-II belongs to a superfamily, SF2, of which several eukaryotic helicases that play a major role in cellular responses to viral infection also belong (19). Furthermore, the helicase core domain is a component of the dicer complex which mediates RNAi in higher eukaryotes (9). Therefore, it stands to reason that study of the NPH-II helicases of the Poxviridae can serve as a model for understanding several distinct biological processes.
Frequently, several alternative names are used for viral proteins; this variation can lead to confusion for researchers and slow scientific progress. To standardize protein names, NCBI staff (viral genome curators) work closely with viral protein experts from UniProt. Such collaborations have resulted in functional naming for viral protein clusters from the family Adenoviridae. One of their representatives is cluster PHA3614. It combines related and highly conserved proteins from the genus Mastadenovirus, which presumably play an important role in host modulation (11). The old, commonly used name of proteins from the PHA3614 cluster was the E3 12.5 kDa protein. Because the size of the protein could vary in different viruses without its biological role changing, the molecular mass, as a component of protein name, was not informative and could be misleading. Therefore we proposed a new, functional name for this cluster: “putative host modulation protein E3.” All existing synonyms were included in the cluster’s functional description. Since the existence of the putative host modulation protein E3 was experimentally supported only for human adenovirus 2 and human adenovirus 5, this information was also included in the description of the cluster. These changes will be visible with the next cluster update.
Protozoa
The following is an example of the significance of publication links in a cluster of proteins as a tool to identify orthologs and paralogs.
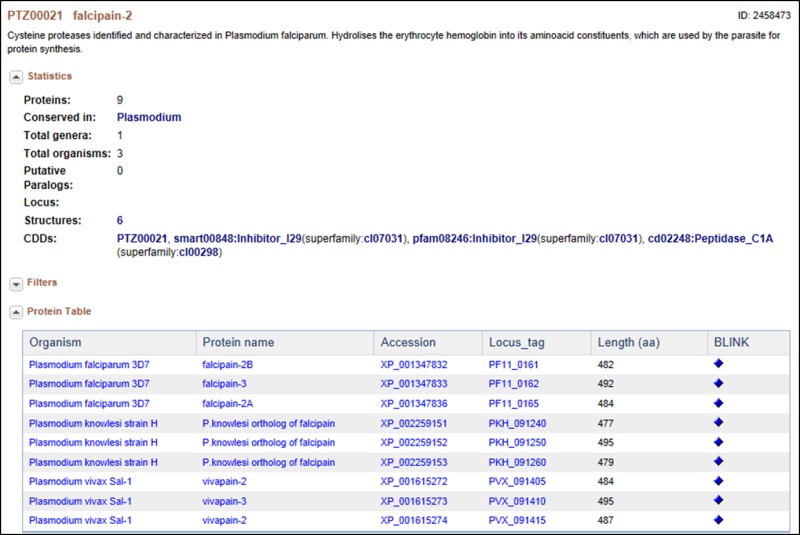
This is a cysteine protease, originally identified and characterized in Plasmodium falciparum; it hydrolyses the erythrocyte hemoglobin into its amino acid constituents, which are used by the parasite for protein synthesis. In this cluster of proteins, “facipain” is present in 4 different Plasmodium species. Falcipain-2 differs from the falcipains in other species (i.e., vivapain -2 and -3, berghepain, etc.,) as well as within the P. falciparum (i.e., falcipain -3) in sequence, in the timing of expression and in the acidic environment needed for enzymatic activation, but they all appear to have the same function (20). Interestingly, the two P. falciparum falcipain-2 proteins in this cluster are each located in a different part of chromosome 11, although they share high amino acid sequence homology and a seemingly identical function. The differences here also appear to be in expression timing and in the level of expression. Falcipain-2A (PF11_0165) appears to be expressed earlier in the trophozoite stage and in higher amounts than falcipain-2B (PF-11_0161) (10, 20).
Also of interest is the fact that cysteine protease inhibitors have been shown to have potent anti-malarial effects. Indeed, because this family of proteases shares low sequence identity with their human counterparts, they have been given serious consideration as potential drug targets.
Plants
Although protein clustering is not specifically geared towards clustering for orthologs or paralogs, clustering does provide a view into how different proteins are related as seen in the cluster PLN03595 shown below.
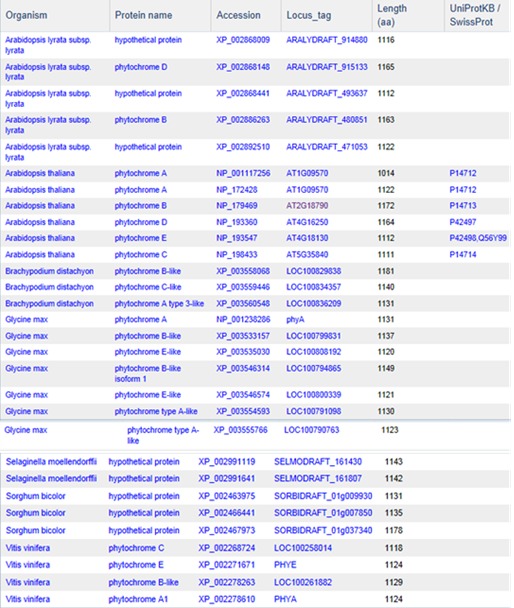
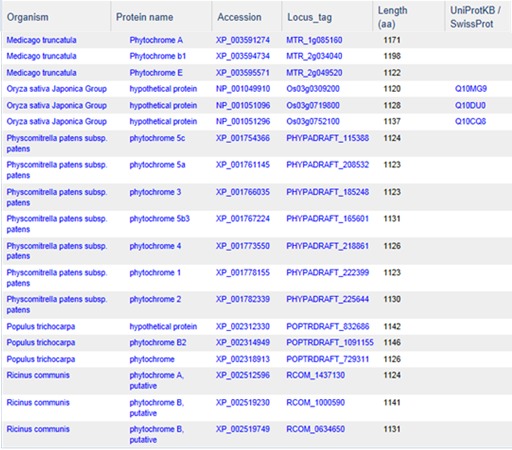
PLN03595 represents a family of photoreceptors involved in the photoperiodic control of plant growth and development. This family includes diverse but structurally conserved proteins. They are expressed in different plant organs under varying light conditions. Phylogenetic analyses suggest that the phytochrome gene family is composed of four subfamilies, PHYA, PHYB, PHYC/F, and PHYE. Arabidopsis thaliana has an additional PHYD gene that originated from the PHYB gene after a more recent gene duplication, and there is some functional redundancy between these two. PHYA and its paralog PHYC are found in monocots as well as in dicots, but PHYC is missing in some dicot lineages. Rice only has three phytochrome genes: PHYA, PHYB, and PHYC. Monocotyledonous plants are also known to lack several members of PHYB subfamily. Phytochromes exhibit distinct and cooperative functions. Mutant analysis has shown that, in rice, phyA and phyB act in a highly redundant manner to control de-etiolation under continuous red light. Under continuous far-red light, phyA and phyC are involved in photoperception, but the photoperception mode of phyC differs between rice and Arabidopsis (21).
We also used proteins of the photosynthesis system as a model for clustering validation. The photosynthesis system has been chosen as it is well conserved and characterized throughout the plant kingdom. As of now, 116 clusters were identified in plants using the “photos” keyword that were annotated and curated. The number of proteins per cluster ranged from 2 to 100. These photosynthesis proteins belong to 6 or more organisms out of 23 distinct genomes. One cluster, PLN00033, contains 22 proteins belonging to 19 organisms and corresponds to the photosystem II stability/assembly factor, which is coherent with the central role this protein plays in chloroplast biogenesis and photosystem stability (25, 26, and 27). Interestingly, 10 out of these 22 proteins from 8 different organisms are annotated as hypothetical proteins.
The second most conserved cluster, PLN00037, contains 34 proteins belonging to 18 organisms and corresponds to photosystem II oxygen-evolving enhancer protein 1 (Psb O). This situation is coherent with its crucial role in photosynthesis. Here again 11 proteins are annotated as hypothetical. Generally, the most conserved proteins in the plant kingdom are known for their central role in plant growth and development. The clustering can be used to hypothesize about the most important proteins whose function is worth analyzing further. For example, PLN03089, a cluster of 65 hypothetical proteins present in both monocots and dicots, should attract more interest. Although the proteins have homology with the Glutamate-gated kainate-type ion channel receptor subunit GluR5 in Medicago truncatula, there is no convincing evidence of such function.
The clusters containing protein specific to a group or subgroup of plants are also very interesting to study. Examples of such clusters are the ones with proteins present in all viridiplantae (PLN00046: photosystem I reaction center subunit O; PLN00054: photosystem I reaction center subunit N; PLN00049: carboxyl-terminal processing protease). The corresponding proteins would be among the most important in plant photosynthesis. Some other clusters contain proteins from a specific subgroup such as algae (PLN00100).
Access
Protein Clusters are presented in NCBI’s Entrez system (http://www.ncbi.nlm.nih.gov/sites/entrez?db=proteinclusters)
The first public release of the Protein Clusters in NCBI’s Entrez interface was in April 2007, and initially consisted of only prokaryotic clusters (15). The Entrez system provides a mechanism for the search, retrieval, and linkage between Protein Clusters and other NCBI databases, as well as external resources. Clusters can be searched by general text terms, and also by specific protein or gene names.
Limits and Advanced search allow clusters to be browsed by function and filtered by size and organism group. A table browser allows users to sort by the content of each column by clicking on the column header.
Protein clusters are available for download from the FTP directory (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/CLUSTERS/) by date and by major taxonomic groups.
Related Tools
Concise Protein BLAST
The Concise Protein database contains proteins from all clusters, as well as all singletons (not clustered proteins). From the clustered set, a representative at the genus level is chosen in order to reduce the data set. Results are therefore available rapidly and the results that are returned provide a broader taxonomic range due to this data reduction.
Concise BLAST provides an option for both protein and nucleotide searches using BLASTP and BLASTX, respectively.
RPS-BLAST
RPS-BLAST searches against pre-calculated position-specific scoring matrices (PSSMs) created during conserved domain processing for the CD-search tool. Therefore, only protein sequences are used for this type of search. PSSMs from the curated cluster set have been added to CDD and are also used in pre-calculated conserved domain hits available from the link menu on protein sequences and reported on each GenPept record. The curated set of PSSMs can be searched using RPS-BLAST and a protein sequence at
http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi or the full set of PSSMs for all curated clusters is available from FTP.
ProtMap
ProtMap is a graphical gene neighborhood tool that displays clickable, linked genes upstream and downstream of the target. The tool provides useful graphical representations of the members of a particular cluster in their genome environments. All members of the cluster of interest are mapped to their genome position, and the tool displays genomic segments coding for each member of the cluster. If the genome sequence is larger than 20KB, only the relevant 10KB portion of it is shown. Users can search for the cluster of interest by using cluster access or the COG/VOG attribute of the cluster. The display is centered on protein members of the cluster. Users can select additional sets of related proteins by clicking on the corresponding colored arrows depicting a protein, or find a cluster of interest by name, protein accession, or gene locus_tag. This resource is useful in identifying paralogs as well as missing or incorrectly annotated genes.
References
- 1.
- Ahn YY, Bagrow J, Lehmann S. Link communities reveal multiscale complexity in networks. Nature. 2010 Aug 5;466(5):761–765. [PubMed: 20562860]
- 2.
- Cameron M, Bernstein Y, Williams HE. Clustered Sequence Representation for Fast Homology Search. J Comput Biol. 2007 June;14(5):594–614. [PubMed: 17683263]
- 3.
- Clauset A, Moore C, Newman ME. Hierarchical structure and the prediction of missing links in networks. Nature. 2008 May 1;453:98–100. [PubMed: 18451861]
- 4.
- Cormen TH, Leiserson CE, Rivest RL, Stein C. Introduction to algorithms. 3rd Edition, The MIT Press; 2009.
- 5.
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010 Oct 1;26(19):2460–2461. [PubMed: 20709691]
- 6.
- Fairman-Williams ME, Jankowsky E. Unwinding initiation by the viral RNA helicase NPH-II. J Mol Biol. 2012 Feb 3;415(5):819–832. [PMC free article: PMC3267895] [PubMed: 22155080]
- 7.
- Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970 Jun;19:99–106. [PubMed: 5449325]
- 8.
- Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995 Jul 28;269(5223):496–512. [PubMed: 7542800]
- 9.
- Gargantini PR, Serradell MC, Torri A, Lujan HD. Putative SF2 helicases of the early-branching eukaryote Giardia lamblia are involved in antigenic variation and parasite differentiation into cysts. BMC Microbiol. 2012 Nov 28;12:284. [PMC free article: PMC3566956] [PubMed: 23190735]
- 10.
- Goh LL, Sim TS. Homology modeling and mutagenesis analyses of Plasmodium falciparum falcipain 2A: implications for rational drug design. Biochem Biophys Res Commun. 2004 Oct 15;323(2):565–572. [PubMed: 15369788]
- 11.
- Hawkins LK, Wold WS. A. 12,500 MW protein is coded by region E3 of adenovirus. Virology. 1992 Jun;188(2):486–494. [PubMed: 1585632]
- 12.
- Holm L, Sander C. Removing near-neighbor redundancy from large protein sequence collections. Bioinformatics. 1998 Jun;14(5):423–429. [PubMed: 9682055]
- 13.
- Hughes AL, Irausquin S, Friedman R. The evolutionary biology of poxviruses. Infect Genet Evol. 2010 Jan;10(1):50–59. [PMC free article: PMC2818276] [PubMed: 19833230]
- 14.
- Kaplan N, Sasson O, Inbar U, Friedlich M, Fromer M, Fleischer H, Portugaly E, Linial N, Linial M. ProtoNet 4.0: a hierarchical classification of one million protein sequences. Nucleic Acids Res. 2005 Jan 1;33(Database issue):D216–D218. [PMC free article: PMC539961] [PubMed: 15608180]
- 15.
- Klimke W, Agarwala R, Badretdin A, Chetvernin S, Ciufo S, Fedorov B, Kiryutin B, O'Neill K, Resch W, Resenchuk S, Schafer S, Tolstoy I, Tatusova T. The National Center for Biotechnology Information's Protein Clusters Database. Nucleic Acids Res. 2009 Jan;37(Database issue):D216–23. [PMC free article: PMC2686591] [PubMed: 18940865]
- 16.
- Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005;39:309–38. [PubMed: 16285863]
- 17.
- Krause A, Stoye J, Vingron M. Large scale hierarchical clustering of protein sequences. BMC Bioinformatics. 2005 Jan 22;6:6–15. [PMC free article: PMC547898] [PubMed: 15663796]
- 18.
- Loewenstein Y, Portugaly E, Fromer M, Linial M. Efficient algorithms for accurate hierarchical clustering of huge datasets: tackling the entire protein space. Bioinformatics. 2008 Jul 1;24(13):i41–49. [PMC free article: PMC2718652] [PubMed: 18586742]
- 19.
- Ranji A, Boris-Lawrie K. RNA helicases: emerging roles in viral replication and the host innate response. RNA Biol. 2010 Nov-Dec;7(6):775–87. [PMC free article: PMC3073335] [PubMed: 21173576]
- 20.
- Shenai BR, Sijwali PS, Singh A, Rosenthal PJ. Characterization of native and recombinant falcipain-2, a principal trophozoite cysteine protease and essential hemoglobinase of Plasmodium falciparum. J Biol Chem. 2000 Sep 15;275(37):29000–29010. [PubMed: 10887194]
- 21.
- Takano M, Inagaki N, Xie X, Yuzurihara N, Hihara F, Ishizuka T, Yano M, Nishimura M, Miyao A, Hirochika H, Shinomura T. Distinct and cooperative functions of phytochromes A, B, and C in the control of deetiolation and flowering in rice. Plant Cell. 2005 Dec;17(12):3311–3325. [PMC free article: PMC1315371] [PubMed: 16278346]
- 22.
- Tarjan RE, Data structures and network algorithms, CBMS 44, Society for Industrial and Applied Mathematics, Philadelphia, PA; 1983.
- 23.
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997 Oct 24;278(5338):631–637. [PubMed: 9381173]
- 24.
- Zaslavsky L, Tatusova T. Mining the NCBI influenza sequence database: adaptive grouping of BLAST results using precalculated neighbor indexing. PLoS Curr. 2009;1:RRN1124. [PMC free article: PMC2771650] [PubMed: 20029662]
- 25.
- Plücken H1. Müller B, Grohmann D, Westhoff P, Eichacker LA. The HCF136 protein is essential for assembly of the photosystem II reaction center in Arabidopsis thaliana. FEBS Lett. 2002 Dec 4;532(1-2):85–90. [PubMed: 12459468]
- 26.
- Meurer J, Plücken H, Kowallik KV, Westhoff P. A nuclear-encoded protein of prokaryotic origin is essential for the stability of photosystem II in Arabidopsis thaliana. EMBO J. 1998 Sep 15;17(18):5286-5297. [PMC free article: PMC1170856] [PubMed: 9736608]
- 27.
- Peltier JB, Emanuelsson O, Kalume DE, Ytterberg J, Friso G, Rudella A, Liberles DA, Söderberg L, Roepstorff P, von Heijne G, van Wijk KJ. Central functions of the lumenal and peripheral thylakoid proteome of Arabidopsis determined by experimentation and genome-wide prediction. Plant Cell. 2002 Jan;14(1):211–236. [PMC free article: PMC150561] [PubMed: 11826309]
- Protein Clusters - The NCBI HandbookProtein Clusters - The NCBI Handbook
- Bookshelf - The NCBI HandbookBookshelf - The NCBI Handbook
- "628-35-3"[CompleteSynonym] (1)PubChem Compound
Your browsing activity is empty.
Activity recording is turned off.
See more...