

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.
This publication is provided for historical reference only and the information may be out of date.

Scope
The process of annotating prokaryotic genomes includes prediction of protein-coding genes, as well as other functional genome units such as structural RNAs, tRNAs, small RNAs, pseudogenes, control regions, direct and inverted repeats, insertion sequences, transposons, and other mobile elements. Bacterial and archaeal genomes have the considerable advantage of usually lacking introns, which substantially facilitates the process of gene boundary identification. A protein coding gene in a prokaryotic genome can be defined as a single interval open reading frame (ORF)—a region starting with a valid start codon and ending with a stop codon bounding a region of in-frame translation covering three nucleotides per codon. In the absence of introns, it might seem that ORFs can be designated as any substring of DNA that begins with a start codon and ends with a stop codon. However, applying this straightforward and simple rule to any bacterial or archaeal genome will result in many overlapping and short ORFs. Determining which one of the overlapping ORFs represents a true gene is a particularly difficult task. In addition, designating the cutoff for filtering short ORFs that might encode small polypeptides presents a special challenge.
Additional complications arise from the fact that Bacteria and Archaea often use alternative start codons—codons other than the traditional ATG. Gene prediction tools must distinguish between six potential candidate start codons (ATG, GTG, TTG, and sometimes ATT, CTG and ATC). Several approaches have been developed for accurate prediction of translation initiation site in prokaryotes (1, 2). Stop codons can also have a dual function, as stop codons TGA or TAG may encode selenocysteine and pyrrolysine. With the rapid and continuous growth of prokaryotic genome sequencing, automated annotation techniques will remain the main approach in the future. Given advances in population studies and analysis of outbreaks, automated annotation processes will shift toward comparative analysis and away from individual genome annotation. On the other hand, diversity studies generate genome sequences from extreme environments and deep taxonomic lineages. These genomes may encode novel genes with no similarity to those available in public archive databases, and must be handled differently. NCBI has developed an automated pipeline that takes advantage of both statistical and similarity-based methods, using similarity when sufficient quantities of comparative data are available and relying more on statistical predictions in the absence of supporting material. NCBI provides a prokaryotic genome annotation service to GenBank submitters using this pipeline, which is also used to annotate RefSeq prokaryotic genomes.
History
Gene prediction or gene finding is one of the fundamental challenges in computational biology.
The main goal of gene prediction is to identify the regions of DNA that are biologically functional. The history of gene prediction dates to the works of Fickett, Gribskov, and Staden (3-5) that started in the early 1980s. The first generation of gene prediction algorithms used a local Bayesian approach analyzing one ORF at a time.
Generation of the first complete bacterial genome sequence of Haemophilus influenza in 1995 heralded a new era in genome sciences. The second-generation of gene prediction algorithms analyzed the global properties of the genomic sequence of a given organism and gave rise to several successful programs such as GeneMark (6) and Glimmer (7). These programs employ an inhomogeneous Markov model for short DNA segments (i.e., k-tuples), from which an estimate of the likelihood for the segment belonging to a protein coding sequence can be derived after training against existing validated gene data.
Similarity searches gave rise to another broad category of gene prediction methods (8). Experimentally derived or known protein sequences were used to determine putative placements and base gene models on those placements, using programs such as BLASTx and FASTA. The third generation of automated annotation programs combined execution of multiple gene-calling programs with similarity-based methods. These third generation approaches attempt to balance evidence-based gene model selection with computationally derived predictions. In 2013, NCBI released the current incarnation of its third-generation pipeline, based on an infrastructure that permits efficient parallel computation and high-throughput annotation.
The first version of NCBI’s Prokaryotic Genome Automatic Annotation Pipeline (PGAAP) using second-generation gene prediction approaches was developed in 2001-2002. The approach combined hidden Markov model (HMM)-based gene prediction algorithms with protein homology methods. Gene predictions were done using a combination of GeneMark (6) and Glimmer (7). Conserved proteins from curated clusters, Clusters of Orthologous Groups (9) and NCBI Prokaryotic Clusters (10), were used to search for genes that may have been missed by pure ab initio annotations. Ribosomal RNAs were predicted by sequence similarity searching using BLAST against an RNA sequence database and/or using Infernal and Rfam models. Transfer RNAs were predicted using tRNAscan-SE (11). This standard operating procedure was previously published (12).
With recent advances in genome sequencing technology, the paradigm has shifted from individual genomes to population studies represented by a so-called pan-genome. Newly submitted genomes can be annotated using data already available for closely related genomes. In order to incorporate information from closely related isolates, NCBI’s annotation pipeline was extensively redesigned. Our new approach is based on the assumption that proteins conserved in a genome clade (core proteins) should be found in a new genome of the same clade. The major difference compared to the previous pipeline is that the alignment-base information is calculated upfront and passed to a customized version of GeneMarkS (13), termed GeneMarkS+, which can incorporate external data in the analysis of the statistical evidence of coding potential and transcriptional start site.
Annotation Standards
Certain metrics can be used to assess the quality of the annotation of the prokaryotic genomes. NCBI has established a relationship with other major archive databases and major sequencing centers in an effort to develop standards for prokaryotic genome annotation. This collaboration has resulted in a set of annotation standards approved and accepted by all major annotation pipelines (14). Many groups still use a simplified set of rules for annotation, and as such may miss critical annotations. Some simplifications include eliminating alternative starts and applying hard-coded length cutoffs for acceptance of short proteins. In addition to these standards, many groups also apply “soft” validation checks.
Minimum standards for annotating complete genomes
- 1.
ANNOTATION SHOULD FOLLOW INSDC SUBMISSION GUIDELINES (GenBank/ENA/DDBJ)
- a.
Prior to genome submission a submitted Bioproject record with a registered locus_tag prefix is required according to accepted guidelines
http://www.ncbi.nlm.nih .gov/genomes/locustag/Proposal.pdf - b.
The genome submission should be valid according to feature table documentation
http://insdc.org/documents /feature_table.html
- 2.
MINIMAL GENOME ANNOTATION SHOULD HAVE
- a.
At least one copy of rRNAs (5S, 16S, 23S) of appropriate length and corresponding genes with locus_tags
- b.
At least one copy of tRNAs for each amino acid and corresponding genes with locus_tags
- c.
Protein-coding genes with locus_tags (see below) and corresponding CDS
- 3.
VALIDATION CHECKS AND ANNOTATION MEASURES
Validation checks should be done prior to the submission. NCBI has already provided numerous tools to validate and ensure correctness of annotation. Additional checks will be put in place to ensure the minimal standards are met.
Statistical measures that are used for annotation quality assessment include:- 4.
EXCEPTIONS
Exceptions (unusual annotations, annotations not within expected ranges) should be documented and strong supporting (experimental) evidence should be provided.
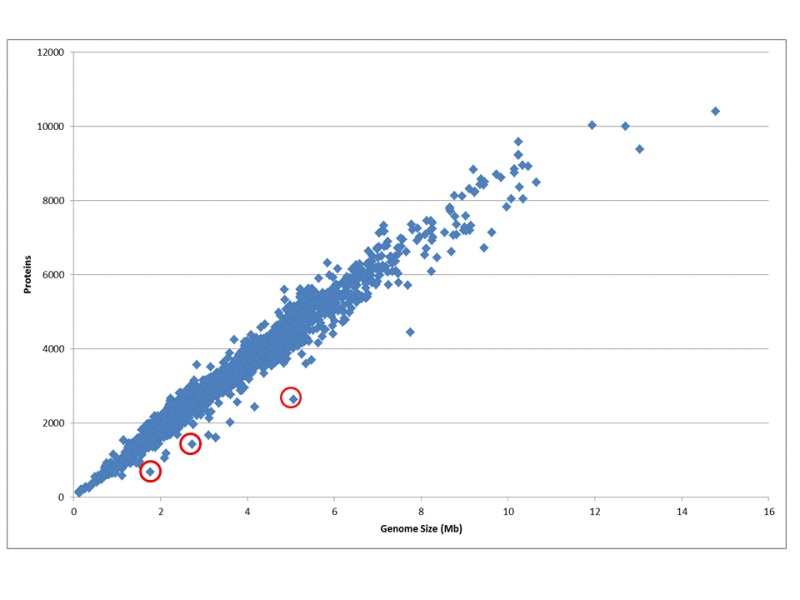
Figure 1.
The ratio of genome size to the number of protein coding genes for all genomes. The number of protein coding genes is directly proportional to the genome size; on average the density is one gene per 1000 kb. Small obligate parasites and symbionts that undergo rapid gene reduction have less protein coding genes than average. Some examples are indicated in red: Serratia symbiotica str. 'Cinara cedri', Synergistetes bacterium SGP1, and Yersinia pestis CO92.Methods
Non-coding RNA (structural RNA, small ncRNA, tRNA)
Structural ribosomal RNAs in prokaryotes (5S, 16S, and 23S) are highly conserved in closely related species. The NCBI Refseq collection contains a curated set of rRNA reference sequences for each of these three types of rRNA. The pipeline uses a nucleotide (BLASTn) search against the reference set. We further pass 5S rRNA hits through cmsearch for refinement against known structural motifs (15, 16). Partial alignments that fall below 50% of the average length are dropped. Prediction of small ncRNAs involves a two-step process similar to the identification of 5S rRNAs: first, we use a BLASTn search against sequences of selected Rfam families; second, we use cmsearch with default parameters to produce the final annotation.
TRNAscan-SE
The NCBI annotation pipeline uses tRNAscan-SE to identify tRNA placements. The tRNAscan-SE program identifies 99–100% of transfer RNA genes in DNA sequence with less than one false positive per 15 gigabases and is currently one of the most powerful and widely used tRNA identification tools. To identify tRNA genes, the input genome sequence is split into 200 nucleotide (nt) windows with overlap of 100 nt and run through tRNAscan-SE program (11). We automatically provide separate parameterization for Archaea and Bacteria. All tRNA calls with a score below 20 are discarded.
Protein Alignments—ProSplign
The current incarnation of NCBI’s automated annotation pipeline uses data derived from prokaryotic population studies. Our approach uses a pan-genome approach to identify the core set of proteins that we expect to find in all genomes belonging to a given group. We define several groups of proteins that can be used: a “target set,” comprising proteins that are expected to be found in all members of a group, such as universally conserved ribosomal proteins, clade-specific core proteins, and curated bacteriophage protein clusters; and a separate “search set,” comprising all automatic clusters, including curated and non-curated protein clusters, curated bacteriophage protein clusters, and all bacterial UniProtKB/Swiss-Prot proteins.
Proteins from the target set are aligned to genomic sequence using ProSplign, an application developed at NCBI for handling partial-frame and spliced protein alignments. ProSplign offers the advantage of being frameshift-aware and can align proteins correctly in the face of genome sequence errors.
Complete gapless alignments with 100% identity to a target protein are accepted for final annotation. Frameshifted alignments and partial alignments of good quality are passed to GeneMarkS+ for further refinement.
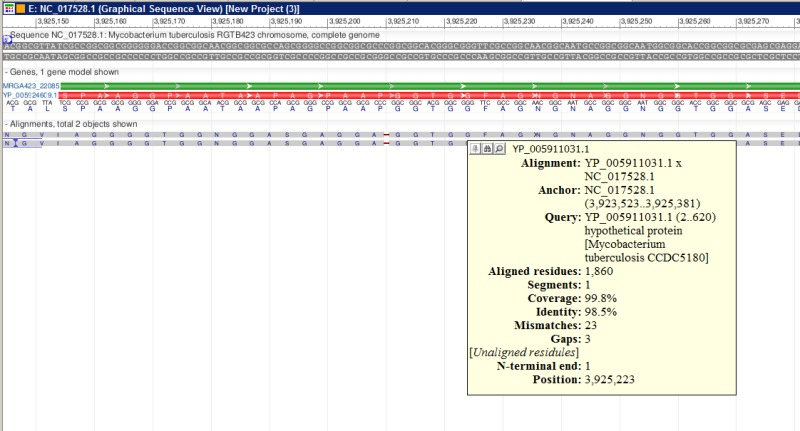
Figure 2.
A fragment of ProSplign alignment against an annotated peptide. The similarity is too low for BLAST to find a significant hit, but ProSplign was able to locate the corresponding protein and identify a frameshift as well.
Frameshift detection
Detecting frameshifted genes is a critical component of resolving ambiguities in automated annotation and provides important feedback in assessing the quality of an assembly. A shift of a reading frame in a coding region is caused by indels (insertions or deletions) of a number of nucleotides in a genomic sequence that is not divisible by three. These events may represent artifacts resulting from technical errors, or they may have biological causes. Sequencing errors are common with the Next Generation Sequencing (NGS) technologies leading to the potential for a high rate of frameshifted genes in assemblies generated using NGS techniques. In addition, gene inactivation during evolution allows selective mutation across ancient ORFs, representing a true biological event that can be marked as a pseudogene with a disrupted ORF. Further, programmed frameshift mutations that are tolerated during translation are known to play an important role in the evolution of novel gene function.
Two-pass improvement
The introduction of a two-pass method improved the original gene-calling procedure. An initial gene call is made by alignment or ab initio prediction, followed by extraction of the protein and BLAST comparison to a set of known conserved proteins. We scan these BLAST hits to identify candidates that are partial or incomplete matches to single or adjacent models. Candidate proteins are realigned to an expanded region using ProSplign. The candidate frameshift alignments are then combined with the original evidence and fed back to GeneMarkS+ in a two-step iterative process. This method is similar to a one-pass annotation with an extended protein reference set but is both more accurate and efficient.

Figure 3.
Two-pass protein alignment process produces improved gene model. The first pass, shown in the upper panel, does not get enough alignments; the second iteration, seen in the bottom panel, produced expanded evidence supporting a more conserved start.
GeneMarkS+
In collaboration with NCBI, The GeneMark team has developed GeneMarkS+, a special version of GeneMarkS that can integrate information about protein alignments and non-coding RNA features. NCBI’s pipeline first collects evidence based on placement of known proteins and structural elements as described above. These placements are then passed to GeneMarkS+, which combines information about statistical ribosomal binding sites and likelihood estimations for the start of transcription with provided evidence about high-quality placements to determine a final set of predictions.
Mobile or fast evolving genes (phage, CRISPR)
The annotation of phage related proteins is based on homology to a reference set of curated phage proteins. The bacteriophage protein reference data set comes from an independent effort to calculate and curate protein clusters from all complete bacteriophage genomes.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) are a family of DNA direct repeats of 20 to 40 nucleotides separated by unique sequences of similar length and are commonly found in prokaryotic genomes.
The CRISPR database (17) allows users to search and identify repeats of interest. These defense systems are encoded by operons that have an extraordinarily diverse architecture and a high rate of evolution for both the cas genes and the unique spacer content. For classification and nomenclature of CRISPR-associated genes see (18). For CRISPR prediction the pipeline uses a wrapper around CRISPR Recognition Tool (CRT) (19) and PILER-CR (20).
Protein naming
The final component of the pipeline is identifying protein function and naming the protein product of the coding region. Assignment of a predicted model to a cluster for purposes of naming is based on protein homology to members of the cluster: we require high coverage, high-scoring alignments to at least three members of the same cluster in order to assign a protein to a cluster.
Dataflow
NCBI’s Prokaryotic Genome Annotation Pipeline combines a computational gene prediction algorithm with a similarity-based gene detection approach. The pipeline currently predicts protein-coding genes, structural RNAs (5S, 16S, 23S), tRNAs, and small non-coding RNAs. The flowchart below describes the major components of the pipeline.

Figure 4.
NCBI Prokaryotic Genome Annotation Pipeline diagram. * Target and search sets are described in the Protein Alignments section. ** Described in the protein naming section.
GenBank Submission Service
The NCBI prokaryotic annotation pipeline is a genome annotation service that is intended to help GenBank submitters with prokaryotic genome annotation. The pipeline can be used with complete genomes as well as whole genome sequences (WGS) consisting of multiple contigs. NCBI’s submission standards require that genomic sequences deposited in GenBank meet a minimum level of quality, including passing contamination screening to eliminate foreign sequence elements and having all sequence contigs be at least 200 bases in length. The annotation pipeline is integrated into the submission system for those who choose this option: sequence data must pass initial validation within GenBank to ensure proper formatting and the presence of required information needed for annotation (organism information, genetic code, and locus-tag prefix). More details on the requirements for submission are available here: http://www.ncbi.nlm.nih.gov/genome/annotation_prok/
RefSeq Genome Annotations
In addition, the prokaryotic genome annotation pipeline is used to annotate NCBI reference sequence (RefSeq) genomes, with the exception of a small number that are manually curated by collaborating groups (for example, Escherichia coli K12 which is provided by EcoCyc). The RefSeq collection provides a comprehensive, integrated, well-annotated set of sequences, including genomic DNA, transcripts, and proteins. RefSeq sequences form the foundation of medical, functional, and diversity studies. They provide a stable reference for genome annotation, gene identification and characterization, mutation and polymorphism analysis, expression studies, and comparative analyses.
Autonomous Protein Records
In order to manage the flood of identical proteins and decrease representational redundancy, particularly from bacterial genomes, NCBI has introduced a new protein data type in the RefSeq collection signified by a ‘WP’ accession prefix. WP accessions provide non-redundant identifiers for protein sequences. This new data type is provided through NCBI’s prokaryotic genome annotation pipeline and is managed independently of the genome sequence data to ensure that the dataset remains non-redundant. There are two main reasons for this paradigm shift:
- 1.
Autonomous WP protein records represent a non-redundant protein collection that provides independent information about the protein sequence and name with linked information to genomic context and taxonomic sample.
- 2.
A WP accession may be annotated on numerous genomes (when the genome encoded proteins are identical) thereby providing a mechanism to avoid creating millions of redundant protein records in the RefSeq collection.
When the NCBI genome annotation pipeline annotates a bacterial protein that is 100% identical and the same length as an existing WP accessioned protein, NCBI is no longer creating a new protein record, with one exception (noted below). NCBI is instead annotating such proteins on the genome by referencing the existing WP accession in the annotated coding sequence (CDS) feature, indicating that the genome represents an exact example of that known protein sequence. Any annotation of protein function on the genome record (such as the product name and functional characteristics) reflects the independent WP record. WP records, therefore, always represent one exact sequence that may be observed one or many times in different strains or species. Also, WP records will always have a version of “1” and the sequence not be updated like taxon-specific RefSeq records.
Data Access
Genomes annotated by NCBI’s annotation pipeline include a relevant comment on the nucleotide record, and each feature specifies which gene prediction method was used. Within nucleotide records, users will find a generated comment and a structured comment block indicating the version of the annotation software used and the date on which a given genome was annotated:
LOCUS CP005492 1692823 bp DNA circular BCT 30-JUL-2013
DEFINITION Helicobacter pylori UM037, complete genome.
…
COMMENT Annotation was added by the NCBI Prokaryotic Genome Annotation
Pipeline (released 2013). Information about the Pipeline can be
found here: http://www.ncbi.nlm.nih.gov/genome/annotation_prok/
##Genome-Annotation-Data-START##
Annotation Provider :: NCBI Annotation Date :: 07/22/2013 11:47:17
Annotation Pipeline :: NCBI Prokaryotic Genome Annotation Pipeline
Annotation Method :: Best-placed reference protein set;GeneMarkS+
Annotation Software revision :: 2.1 (rev. 406590)
Features Annotated :: Gene; CDS; rRNA; tRNA; repeat_region
Genes :: 1,692
CDS :: 1,615
Pseudo Genes :: 35
rRNAs :: 6 ( 5S, 16S, 23S )
tRNAs :: 36
Frameshifted Genes :: 31
##Genome-Annotation-Data- END##
Within the Flat File report, users will find CDS regions marked up with information about evidence used and methods applied to generate such annotations:
CDS 17236..18585
/locus_tag="K750_07885"
/inference="EXISTENCE: similar to AA
sequence:RefSeq:YP_005789361.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="phospho-2-dehydro-3-deoxyheptonate aldolase"
/protein_id="AGL70499.1"
/db_xref="GI:499061585"
CDS complement(808991..809746)
/locus_tag="K750_04005"
/inference="COORDINATES: ab initio prediction:GeneMarkS+"
/note="Derived by automated computational analysis using
gene prediction method: GeneMarkS+."
/codon_start=1
/transl_table=11
/product="restriction endonuclease R.HpyAXII"
/protein_id="AGL69752.1"
/db_xref="GI:499060838"
Re-annotation Consortium
Historically, RefSeq prokaryotic genomes relied on author-supplied annotation. Curation focused primarily on the correction of protein names using protein clusters. Attempts to correct start sites based on manual review were not comprehensive. Because of the rapid increase in the number of genomes (many thousands), manual review became impractical. Moreover, the issue of genome submissions with unannotated genes (missing genes) was not resolved. As a result, the original RefSeq prokaryotic annotation dataset contained inconsistent annotation even in closely related genomes that had high-quality annotated references, such as E. coli. NCBI’s updated annotation pipeline can produce a consistent, high quality, automatic annotation that in many cases surpasses the original author-provided annotation. Therefore, NCBI is re-annotating prokaryotic RefSeq genomes to improve the overall consistency and quality of this dataset.
Related Tools and Resources
Protein Clusters—A collection of related protein sequences (clusters) consists of proteins derived from the annotations of whole genomes, organelles, and plasmids. It is currently limited to Archaea, Bacteria, Plants, Fungi, Protozoans, and Viruses. Protein clusters can be searched and viewed at http://www.ncbi.nlm.nih.gov/proteinclusters/
ProSplign—This tool produces accurate spliced alignments and locates alignments of distantly related proteins with low similarity and is an integral component of the NCBI's Genome Annotation Pipeline (Gnomon). The integration of ProSplign with the genome annotation pipeline significantly improved the quality of genome annotation over existing available methods. ProPSlign is available as an online tool at http://www.ncbi.nlm.nih.gov/sutils/static/prosplign/prosplign.html
Frameshift detection—This tool is available as a standalone tool or Web application. Adjacent genes on the same strand are analyzed for hits against the same subject (common BLAST hit) by comparing BLAST results. Since gene fusions and splits occur in prokaryotic genes, the BLAST hits are analyzed for any subject (not the common BLAST hit) that covers 90% of the query protein, in which case the frameshift is not reported under the assumption that this gene is “real.” Any pair of genes failing to meet these criteria is reported as potential frameshifted genes and should be manually inspected. The Web version is available at:http://www.ncbi.nlm.nih.gov/genomes/frameshifts/frameshifts.cgi
NCBI is hosting two ab initio prokaryotic gene prediction programs: GeneMark and Glimmer.
The programs can be used for a rapid draft annotation of prokaryotic genomes.
GeneMark—The GeneMark family of gene finding programs has been used for prokaryotic genome annotation since 1995 when GeneMark contributed to launching the genomic era by providing automatic gene annotation of complete genomes of Haemophilus influenza, Methanoccus jannaschii, as well as Escherichia coli and Bacillus subtilis. http://www.ncbi.nlm.nih.gov/genomes/MICROBES/genemark.cgi
Glimmer—GLIMMER (20) is a system for finding genes in microbial DNA, especially the genomes of bacteria and archaea. GLIMMER (Gene Locator and Interpolated Markov ModelER) uses interpolated Markov models to identify coding regions. Glimmer version 3.02b is the current version of the system. http://www.ncbi.nlm.nih.gov/genomes/MICROBES/glimmer_3.cgi
References
- 1.
- Yada T, Totoki Y, Takagi T, Nakai K. A novel bacterial gene-finding system with improved accuracy in locating start codons. DNA Res. 2001 Jun 30;8(3):97–106. [PubMed: 11475327]
- 2.
- Hu GQ, Zheng X, Zhu HQ, She ZS. Prediction of translation initiation site for microbial genomes with TriTISA. Bioinformatics. 2009 Jul 15;25(14):184–5. [PubMed: 19015130]
- 3.
- Staden R, McLachlan AD. Codon preference and its use in identifying protein coding regions in long DNA sequences. Nucleic Acids Res. 1982 Jan 11;10(1):141–56. [PMC free article: PMC326122] [PubMed: 7063399]
- 4.
- Gribskov M, Devereux J, Burgess RR. The codon preference plot: graphic analysis of protein coding sequences and prediction of gene expression. Nucleic Acids Res. 1984 Jan 11;12(1 Pt 2):539–49. [PMC free article: PMC321069] [PubMed: 6694906]
- 5.
- Fickett JW. Finding genes by computer: the state of the art. Trends Genet. 1996 Aug;12(8):316–20. [PubMed: 8783942]
- 6.
- Lukashin AV, Borodovsky M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 1998 Feb 15;26(4):1107–15. [PMC free article: PMC147337] [PubMed: 9461475]
- 7.
- Salzberg SL, Delcher AL, Kasif S, White O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998 Jan 15;26(2):544–8. [PMC free article: PMC147303] [PubMed: 9421513]
- 8.
- Guigó R, Burset M, Agarwal P, Abril JF, Smith RF, Fickett JW. Sequence similarity based gene prediction. In: Suhai S, editor. Genomics and proteomics: Functional and computational aspects. New York, NY: Kluwer Academic / Plenum Publishing; 2000. pp. 95–105.
- 9.
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001 Jan 1;29(1):22–8. [PMC free article: PMC29819] [PubMed: 11125040]
- 10.
- Klimke W, Agarwala R, Badretdin A, Chetvernin S, Ciufo S, Fedorov B, Kiryutin B, O'Neill K, Resch W, Resenchuk S, Schafer S, Tolstoy I, Tatusova T. The National Center for Biotechnology Information's Protein Clusters Database. Nucleic Acids Res. 2009 Jan;37(Database issue):D216–23. [PMC free article: PMC2686591] [PubMed: 18940865]
- 11.
- Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucl. Acids Res. 1997;25:955–964. [PMC free article: PMC146525] [PubMed: 9023104]
- 12.
- Angiuoli S, et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. OMICS. 2008;2008;12:137–41. [PMC free article: PMC3196215] [PubMed: 18416670]
- 13.
- Besemer J., Lomsadze A., Borodovsky M. 2001GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 26No. 4pp1107–1115. [PMC free article: PMC55746] [PubMed: 11410670]
- 14.
- Klimke W, O'Donovan C, White O, Brister JR, Clark K, Fedorov B, Mizrachi I, Pruitt KD, Tatusova T. Solving the Problem: Genome Annotation Standards before the Data Deluge. Stand Genomic Sci. 2011 Oct 15;5(1):168–93. [PMC free article: PMC3236044] [PubMed: 22180819]
- 15.
- Eddy S.R. A memory-efficient dynamic programming algorithm for optimal alignment of a sequence to an RNA secondary structure. BMC Bioinformatics. 2002;3:18. [PMC free article: PMC119854] [PubMed: 12095421]
- 16.
- Nawrocki EP, Eddy SR. Query-dependent banding (QDB) for faster RNA similarity searches. PLoS Comput Biol. 2007;3(3) [PMC free article: PMC1847999] [PubMed: 17397253]
- 17.
- Grissa I., Vergnaud G., Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. [PMC free article: PMC1892036] [PubMed: 17521438]
- 18.
- Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011 Jun;9(6):467–7. [PMC free article: PMC3380444] [PubMed: 21552286]
- 19.
- Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007 Jun 18;8:209. [PMC free article: PMC1924867] [PubMed: 17577412]
- 20.
- Biswas A., Gagnon J.N., Brouns S.J., Fineran P.C., Brown C.M. CRISPRTarget: Bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013;10(5):817–827. [PMC free article: PMC3737339] [PubMed: 23492433]
- 21.
- Delcher A.L., Harmon D., Kasif S., White O., Salzberg S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Research. 1999;27(23):4636–4641. [PMC free article: PMC148753] [PubMed: 10556321]
- Prokaryotic Genome Annotation Pipeline - The NCBI HandbookProkaryotic Genome Annotation Pipeline - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...