This chapter discusses inherited human diseases that are caused by defects in glycan biosynthesis and metabolism (congenital disorders of glycosylation, CDGs). Representative examples are described of genetic defects in the major glycan families and what lessons we can learn from them about glycobiology. Among genetic disorders of glycosylation, those caused by somatic mutations are described in Chapter 46. Disorders affecting the lysosomal degradation of glycans are described in Chapter 44. Although the term “congenital disorders” by definition include those caused by nongenetic, unfavorable in utero conditions, the term “congenital disorders of glycosylation (CDG)” is now widely used as an equivalent of inherited disorders of glycosylation.
BACKGROUND AND DISCOVERY
Following the discovery of the genetic defect in human I-cell disease in 1980 (Chapter 33) it was anticipated that many more human defects in glycan biosynthesis would be found, but more than a decade went by before the next example was found. In retrospect, the difficulty arose from the pleiotropic multisystem nature of the clinical presentations. Meanwhile Belgian pediatrician Jaak Jaeken noted unusual profiles of serum proteins in some children with multisystem disorders and decided to apply a previously established test to separate transferrin isoforms. The test was positive in these children, thereby, for the first time, indicating a generic defect in protein N-glycosylation. Lacking further details about glycan structures and the underlying genetic variants, Jaeken decided to call these cases “carbohydrate-deficient glycoprotein syndromes” (CDGS). Several years later, many people defined primary inherited genetic defects in glycosylation pathways in many such cases. To keep the acronym “CDG,” those involved also decided to call them “congenital disorders of glycosylation,” rather than inherited genetic defects in glycosylation.
INHERITED PATHOLOGICAL MUTATIONS OCCUR IN ALL MAJOR GLYCAN FAMILIES
Nearly all inherited disorders in glycan biosynthesis were discovered in the last 20 years. They are rare, biochemically and clinically heterogeneous, and usually affect multiple organ systems. CDGs are rare primarily because embryos with complete defects in a step of glycosylation do not usually survive to be born, documenting the critical biological roles of glycans in humans. CDG patients that survive are usually hypomorphic, retaining at least some activity of the pathways involved. Although rare, research on this group of genetically defined glycosylation disorders reveals important novel insights into the biology of the glycosylation process.
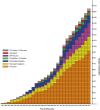
Some defects strike only a single glycosylation pathway, whereas others impact several. Defects occur in (1) the activation, presentation, and transport of sugar precursors; (2) glycosidases and glycosyltransferases; and (3) proteins that traffic glycosylation machinery or maintain Golgi homeostasis. A few disorders can be treated by the consumption of monosaccharides. The rapid growth in the number of discovered disorders, shown in Figure , has resulted in an evolution of disease nomenclature. Since 1999, CDGs were defined as genetic defects in N-glycosylation, but now the term is applied to any glycosylation defect. Nowadays, CDGs are categorized in four groups, comprising defects in N-glycosylation, O-glycosylation, and lipid and GPI-anchor glycosylation and defects that impact multiple glycosylation pathways. CDGs are named by the mutated gene followed by “-CDG” suffix (e.g., PMM2-CDG). Selected disorders are listed in Table and all known disorders in Online Appendix 45A.
Glycosylation-related disorders. The graph shows the cumulative number of human glycosylation disorders in various biosynthetic pathways and the year of their identification. (For simplicity, disorders affecting multiple pathways are included in disorders (more...)
Selected congenital disorders of glycosylation in humans
DEFECTS IN N-GLYCAN BIOSYNTHESIS
Clinical and Laboratory Features and Diagnosis
The broad clinical features of disorders in which N-glycan biosynthesis is defective involve many organ systems, but are especially common in the central and peripheral nervous systems and hepatic, visual, and immune systems. The generality and variability of clinical features makes it difficult for physicians to recognize CDG patients with defective N-glycosylation. The first were identified in the early 1980s based primarily on deficiencies in multiple plasma glycoproteins. The patients were also delayed in reaching growth and developmental milestones and had low muscle tone, incomplete brain development, visual problems, coagulation defects, and endocrine abnormalities. However, many of these symptoms are also seen in patients with other multisystemic genetic syndromes. CDG patients with defective N-glycosylation can be distinguished because they often have abnormal glycosylation of common liver-derived serum proteins containing disialylated, biantennary N-glycans. Serum transferrin is especially convenient because it has two N-glycosylation sites, each normally containing disialylated, biantennary N-glycans. Different glycoforms can be resolved by isoelectric focusing (IEF) or ion-exchange chromatography, but better accuracy and sensitivity is achieved by mass spectrometry (MS) of purified transferrin. This simple test alerts physicians to likely CDG patients without knowing the genetic or molecular basis of the disease.
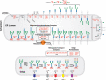
N-glycosylation defects may be divided into two types based on transferrin glycoforms. Type I (CDG-I) patients lack one or both N-glycans because of defects in the biosynthesis of the lipid-linked oligosaccharide (LLO) or its transfer to proteins. Type II (CDG-II) patients have incomplete protein-bound glycans because of abnormal processing. The biosynthetic pathways and locations of N-glycan defects are shown in Figure .
Congenital disorders of glycosylation in the N-glycosylation pathway. The figure shows individual steps in lipid-linked oligosaccharide (LLO) biosynthesis, glycan transfer to protein, and N-glycan processing similar to Figure 9.3 and Figure 9.4. The shuttling (more...)
Defects in ER N-Glycosylation
A complete absence of N-glycans is lethal. Therefore, known mutations mostly generate hypomorphic alleles encoding proteins with diminished activity. A deficiency in any of the steps required for the assembly of LLOs in the endoplasmic reticulum (ER) (e.g., nucleotide sugar synthesis or sugar addition catalyzed by a glycosyltransferase) (Chapter 9) produces a structurally incomplete LLO. Because the oligosaccharyltransferase (OST) prefers full-sized LLO glycans, this results in hypoglycosylation of multiple glycoproteins. This means that some N-glycan sites are not modified. In recent years, it has been shown that some intermediate LLO structures can also be transferred to proteins, although with much lower efficiency. For example, low amounts of an extended chitobiose glycan were identified in ALG1-CDG, which is deficient in the mannosyltransferase that adds mannose to the chitobiose core. Importantly, many deficiencies in LLO synthesis produce incomplete LLO intermediates. Most of the LLO assembly steps are not easy to assay, but LLO assembly is conserved from yeast to humans, and intermediates that accumulate in CDG patients often correspond to the intermediates seen in mutant Saccharomyces cerevisiae strains with known defects in LLO assembly. Some mutant mammalian cells (e.g., Chinese hamster ovary cells) have been shown to have similar defects (Chapter 49). The close homology between yeast and human genes for ER N-glycosylation enables the normal human orthologs to rescue defective glycosylation in mutant yeast strains, whereas mutant orthologs from patients do not. This provides substantial clues to the likely human defect, along with a system in which to perform functional assays.
PMM2-CDG in Table is the most common CDG with more than 1000 cases identified worldwide. The patients have moderate to severe developmental and motor deficits, hypotonia, dysmorphic features, failure to thrive, liver dysfunction, coagulopathy, and abnormal endocrine functions. More than 100 mutations found in phosphomannomutase 2 (PMM2) impair conversion of mannose-6-P (Man-6-P) to Man-1-P, which is a precursor required for the synthesis of GDP-mannose (GDP-Man) and dolichol-P-mannose (Dol-P-Man). Both donors are substrates for the mannosyltransferases involved in the synthesis of Glc3Man9GlcNAc2-P-P-Dol which is decreased in cells from PMM2-CDG patients. Patients have hypomorphic PMM2 alleles because complete loss of PMM2 function is lethal. Mouse embryos lacking Pmm2 die 2–4 days after fertilization, whereas some of those with hypomorphic alleles survive. There are currently no approved therapeutic options for PMM2-CDG patients.
MPI-CDG (CDG-Ib) in Table is caused by mutations in MPI (mannose-6-phosphate isomerase). This enzyme interconverts fructose-6-P and Man-6-P. In contrast to PMM2-CDG, these patients do not have intellectual disability or developmental abnormalities. Instead, they have impaired growth, hypoglycemia, coagulopathy, severe vomiting and diarrhea, protein-losing enteropathy, and hepatic fibrosis. Several patients died of severe bleeding before the basis of this CDG was known. Mannose dietary supplements effectively treat these patients. Man-6-P can be generated directly by hexokinase-catalyzed phosphorylation of mannose (Chapter 5). This pathway is intact in MPI-CDG patients and its high activity in brain is thought to be the reason for the absence of neurological symptoms. Human plasma contains ∼50 µm mannose because of export following glycan degradation and processing. Mannose supplements correct coagulopathy, hypoglycemia, protein-losing enteropathy, and intermittent gastrointestinal problems, as well as normalize the glycosylation of plasma transferrin and other serum glycoproteins. Because orally administered mannose is well-tolerated, this approach is clearly a satisfyingly effective therapy for this life-threatening condition, although not curative for long-term symptoms.
Patients with mutations in nearly all the remaining steps of LLO biogenesis have been found (Table ; Online Appendix 45A; Figure ) They have a broad range of clinical phenotypes including low low-density lipoprotein (LDL), low immunoglobulin G (IgG), kidney failure, genital hypoplasia, and cerebellar hypoplasia. The pathophysiology underlying these symptoms is still unknown. Defects in dolichol biosynthesis present with more specific symptoms as outlined below in the section Defects in Multiple Glycosylation Pathways. Mutations in six of the OST subunits also cause a CDG. The OST is a complex of multiple subunits of which STT3 is the catalytic subunit that contains OST activity. Two different genes encode STT3—namely, STT3A and STT3B. They are part of two different OST complexes, one with STT3A for cotranslational glycosylation and one with STT3B for glycosylation of proteins that escape glycosylation via the STT3A complex. Patients with defects in either subunit present with the typical multisystem phenotype. Interestingly, transferrin glycosylation is dependent on the STT3A-containing OST complex and thus abnormal in STT3A-CDG, but normal in STT3B-CDG. STT3B dependent protein substrates, such as sex hormone binding globulin, show reduced glycosylation in STT3B-CDG specifically. The phenotype of MAGT1-CDG is very different, presenting as a primary immunodeficiency characterized by chronic infections with the Epstein–Barr virus (EBV). Transferrin glycosylation is abnormal in MAGT1-CDG. MAGT1 is an oxidoreductase that is mostly associated with the STT3B-OST complex. It is required for glycosylation of protein sequences containing cysteine residues. Reduced expression on natural killer cells of the NKG2D protein that requires MAGT1 for glycosylation is proposed to contribute to the immunodeficiency against EBV infections.
Defects in Golgi N-Glycosylation
Golgi disorders (Figure ) affect N-glycan processing and include defects in glycosyltransferases, nucleotide sugar transporters, vacuolar pH regulators, and multiple cytoplasmic proteins that traffic glycosylation machinery within the cell and maintain Golgi homeostasis. Most of these processes are required for multiple glycosylation pathways and are discussed below (see the section Defects in Multiple Glycosylation Pathways).
In B4GALT1-CDG, glycans show a dramatic loss of both galactose (Gal) and sialic acid (Sia) from transferrin because of deficient β1-4 galactosyltransferase activity. Patients present with a relatively mild clinical course without neurological symptoms, which can be explained by the fact that related genes are expressed in different tissues, such as B4GALT2 in the brain. A similar glycan pattern occurs in the X-linked SLC35A2-CDG, because of loss of UDP-Gal transporter activity. Surprisingly, within a few years after birth, abnormal glycosylation becomes normal. This is probably caused by somatic mosaicism of cells carrying the mutated SLC35A2 gene along with unaffected cells and by selection against the affected cells. Patients suffer from a mainly neurological phenotype. Defects in the glycosyltransferase MGAT2 and glycosidase MAN1B1 also result in characteristic glycan abnormalities on transferrin and other proteins that are now used to diagnose patients, whereas FUT8-CDG shows normal glycosylation of transferrin but severe fucosylation defects on total serum proteins. These CDGs present with neurological symptoms and other multisystem features, whereas MAN1B1-CDG additionally gives rise to obesity.
Disorders of N-Glycoprotein Deglycosylation
Early on, it was assumed that glycosylation disorders would mainly result from defects in glycan biosynthetic enzymes, but that perspective has changed. Discovery of defects in Golgi organization and homeostasis, in ER chaperones such as COSMC or EDEM, and in ER quality control (such as EDEM-3-CDG) have broadened the perspective. A new defect in the ER-associated degradation (ERAD) continuum (Chapter 39) is caused by mutations in NGLY1, an enzyme that cleaves N-glycans from misfolded glycoproteins transported into the cytoplasm before their proteasomal degradation (Table ). NGLY1 defects do not appear to induce the ERAD pathway, accumulate undegraded glycoproteins in vesicles, or trigger autophagy. It is unclear how these defects cause symptoms such as developmental delay, movement disorder, seizures, and a curious lack of tear production, but their clinical similarity to other CDGs emphasizes that glycosylation defects cannot simply be divided into “synthesis” or “degradation.”
DEFECTS IN O-GLYCAN BIOSYNTHESIS
Defects in O-Man Synthesis (Congenital Muscular Dystrophies)
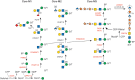
Mutations altering O-Man glycans (Chapter 13), primarily on α-dystroglycan (α-DG), cause a range of congenital muscular dystrophies (CMDs) termed dystroglycanopathies (Figure ). α-DG at the sarcolemma links skeletal muscle cells to laminin in the extracellular matrix. A disruption of this linkage results in muscular dystrophy. Similarly, defective interaction of α-DG with other protein ligands in eye and brain results in a broad clinical spectrum of dystroglycanopathies, ranging from very severe, and often lethal, musculo-oculo-encephalopathies, such as Walker–Warburg syndrome (WWS), muscle–eye–brain disease (MEB), and Fukuyama congenital muscular dystrophy (FCMD), to milder forms of isolated limb-girdle muscular dystrophy in adults. Genetic analysis of these disorders has been indispensable for discovering α-DG's functional glycans and their biosynthesis. The complex pathway is presented in Figure and Chapter 13. The pathway is initiated in the ER by the transfer of mannose to Ser/Thr of α-DG via the protein O-mannosyltransferase complex POMT1/POMT2 (Table ). In the Golgi, a polymer glycan (matriglycan) is generated, recognized by several diagnostic monoclonal antibodies and necessary for the binding of laminin and other molecules to α-DG. An unusual feature on one subset of O-Man glycans is the existence of a Man-6-P generated by the kinase POMK. The Man-6-P-containing Core M3 glycan consists of Man-6-P, GlcNAc (transferred by POMGNT2), and GalNAc (transferred by B3GALNT2). The trisaccharide core is extended by two units of ribitol-5-phosphate sequentially transferred by FKTN (fukutin) and FKRP (fukutin-related protein) from CDP-ribitol, which is generated by CRPPA. This “core” glycan is elongated by an alternating disaccharide (β1-3Xylα1-3GlcA) to form matriglycan.
O-Man glycan biosynthetic pathway. The biosynthetic pathways of representative O-Man glycans are shown. Enzymes whose defects cause a disorder are indicated in red. Three main groups are identified: cores M1–M3. All O-Man glycans are initiated (more...)
O-Man glycans can also play a traitorous role, because they are receptor molecules for Lassa virus entry into cells. This feature was cleverly exploited to screen libraries and identify genes required for virus entry. The method correctly identified all previously known WWS-causing genes and predicted new culprits. Dystroglycanopathies can be divided into primary (caused by mutations in the DAG1 gene, encoding the α-DG core protein), secondary (caused by mutations in one of 11 glycosyltransferases [POMT1, POMT2, POMGNT1, POMGNT2, FKTN, FKRP, LARGE1, RXYLT1, B3GALNT2, POMK, or B4GAT1]) or tertiary (caused by defects in the synthesis of sugar building blocks [CRPPA for CDP-ribitol, GMPPB, DOLK, DPM1, DPM2, and DPM3 for Dol-P-Man]).
WWS is the most severe CMD. Patients live about one year and have multiple brain abnormalities, and severe muscular dystrophy. About 20% of patients have mutations in POMT1 and a few have mutations in POMT2. Others have defects in FKTN and FKRP, but mutations in both also cause milder forms of muscular dystrophy. POMGNT1 is mutated in MEB, which is characterized by symptoms similar to, but milder than, WWS. The most severely affected MEB patients die during the first years of life, but the majority of mild cases survive to adulthood. FCMD is caused by a single 3-kb 3′-retrotransposon insertional event into the FKTN gene, which occurred 2000–2500 years ago. This partially reduces the stability of the mRNA, making it a relatively mild mutation. FCMD is one of the most common types of CMD in Japan with a carrier frequency of 1/188. Fktn-null mice die by E9.5 in embryogenesis and appear to have basement membrane defects. Congenital muscular dystrophy type 1C (MDC1C) is a relatively mild disorder that is caused by mutations in FKRP. Patients with MDC1D, a limb-girdle muscular dystrophy, contain mutations in LARGE1, originally described in myodystrophic mice (myd; now called Largemyd). The protein has two glycosyltransferase signatures (DXD) in different domains that account for xylosyl- and glucuronosyl-transferase activities, respectively (Chapter 13).
Defects in O-GalNAc Synthesis
Polypeptide GalNAc-transferases (ppGalNAcTs) are required for initiation of mucin type O-glycosylation (Chapter 10). Clinical phenotypes of defects in this family of enzymes depend on their tissue-specific expression and substrate specificity. A defect in O-GalNAc synthesis by a particular polypeptide GalNAc-transferase (GALNT3) causes familial tumoral calcinosis. This severe autosomal-recessive metabolic disorder causes phosphatemia and massive calcium deposits in the skin and subcutaneous tissues. Mutations in the O-glycosylated fibroblast growth factor 23 (FGF23) also cause phosphatemia, and further research has shown that ppGALNT3 glycosylates FGF23. Mutations in GALNT2, which encodes a ubiquitous ppGalNAcT, result in a multisystem neurological disease with remarkably low HDL cholesterol. Further research identified ANGPTL3 and APOCIII as substrates, the latter of which can be used for easy diagnostic screening by IEF. Tn syndrome, a rare autoimmune disease, is caused by somatic mutations in the X-linked gene C1GALT1C1, which encodes a highly specific chaperone COSMC required for the proper folding and normal activity of the β1-3galactosyltransferase C1GALT1, needed for synthesis of core 1 and 2 O-glycans (Chapters 10 and 46).
Defects in Other O-Glycosylation Families
The identification of patients with defects in other types of O-glycosylation (O-glucose, O-fucose, and O-GlcNAc) highlights the physiological importance of this class of glycans (Chapter 13). Each of these O-glycans is found on certain epidermal growth factor (EGF)-like repeats in the extracellular domain of Notch receptors, and mutations in the glycosyltransferases that modify NOTCH1 have been identified in different diseases, including Dowling–Degos disease (caused by haploinsufficiency of POFUT1 or POGLUT1), a form of limb-girdle muscular dystrophy (caused by autosomal-recessive mutations in POGLUT1), Adams–Oliver syndrome (autosomal-recessive mutations in EOGT), and some cancers (amplification, gain or loss-of-function of POFUT1, Fringe GlcNAc-transferases, POGLUT1, MGAT3). Mutations in OGT, encoding the GlcNAc-transferase that acts on cytoplasmic and nuclear proteins (Chapter 18), cause intellectual disability. The mutations cluster in the region encoding TRP-repeats of the OGT enzyme, implicating altered substrate recognition and O-GlcNAc modification of specific cytosolic and nuclear proteins as a pathogenic mediator.
Defects in Glycosaminoglycan (GAG) Synthesis
Proteoglycans and their GAG chains are critical components in extracellular matrices. For a discussion of their biosynthesis, core proteins, and function, see Chapter 17. Genetic defects in GAG synthesis can be divided into so-called linkeropathies, because of defects in the synthesis of the tetrasaccharide linker, defects in the synthesis of the polysaccharides, and defects in GAG modification, mainly sulfation.
Genetic defects have been reported for all enzymes involved in biosynthesis of the linker tetrasaccharide (XYLT1, XYLT2, B4GALT7, B3GALT6, B3GAT3), linking the core proteins to the glycosaminoglycan. Skeletal and bone defects, joint laxity, and short stature are characteristic symptoms for this group of diseases. Bikunin has been used as a plasma proteoglycan marker for the biochemical confirmation of defects in this group of GAG biosynthesis defects. Ehlers–Danlos syndrome (progeroid type) is a connective tissue disorder characterized by failure to thrive, loose skin, skeletal abnormalities, hypotonia, and hypermobile joints, together with delayed motor development and delayed speech. The molecular basis of the disorder is reduced synthesis of the core region of GAGs initiated by Xyl. Galactosyltransferase I (B4GALT7) the enzyme that adds Gal to Xyl-Ser is mutated in this disease. A defect in the phosphorylation of the core xylose sugar by mutations in FAM20B results in Desbuquois dysplasia, a disease characterized by dwarfism, joint laxity, and skeletal abnormalities. This also shows the importance of Xyl phosphorylation in the regulation of GAG biosynthesis.
Linker elongation produces different polysaccharides, such as heparan sulfate, dermatan sulfate, chondroitin sulfate, and keratan sulfate. Defects in the formation of heparan sulfate (HS) cause hereditary multiple exocytosis (HME), an autosomal-dominant disease with a prevalence of about 1:50,000 births (Table ). It is caused by mutations in one of two genes, EXT1 or EXT2, that are both involved in HS synthesis. HME patients have bony outgrowths, usually at the growth plates of the long bones. Normally, the growth plate contains chondrocytes in various stages of development, which are enmeshed in an ordered matrix composed of collagen and chondroitin sulfate (CS). In HME, however, the outgrowths are often capped by disorganized, cartilaginous masses with chondrocytes in different stages of development. About 1%–2% of patients also develop osteosarcoma. HME mutations occur in EXT1 (60%–70%) or EXT2 (30%–40%). The encoded proteins probably form a complex in the Golgi as both are required for polymerizing N-acetylglucosamine (GlcNAc)α1-4 and glucuronic acid (GlcA)β1-3 into HS. However, the partial loss of one allele of either gene appears sufficient to cause HME. This means that haploinsufficiency decreases the amount of HS, and that EXT activity is rate-limiting for HS biosynthesis. This is unusual because most glycan biosynthetic enzymes are in substantial excess. Defects in GAG biosynthesis may also be caused by a lack of available nucleotide sugars. Defects in the UDP-GlcA/UDP-GalNAc Golgi transporter (SLC35D1) cause Schneckenbecken dysplasia. Patients have bone abnormalities similar to those seen in other chrondrodysplasias, and a mouse model of the disease shows similar features.
Modification of GAGs by sulfation is important for several functions. Three autosomal-recessive disorders—diastrophic dystrophy (DTD), atelosteogenesis type II (AOII), and achondrogenesis type IB (ACG-IB)—result from defective cartilage proteoglycan sulfation. These forms of osteochondrodysplasia have various outcomes. AOII and ACG-IB are perinatal lethal because of respiratory insufficiency, whereas DTD patients develop symptoms only in cartilage and bone, including cleft palate, club feet, and other skeletal abnormalities. Those DTD patients surviving infancy often live a nearly normal life span. All of these disorders result from different mutations in the DTD gene (SLC26A2), which encodes a plasma membrane sulfate transporter. Unlike monosaccharides, sulfate released from degraded macromolecules in the lysosome is not salvaged well. The heavy demand for sulfate in bone and cartilage proteoglycan synthesis probably explains why the symptoms are most evident in these locations.
DEFECTS IN LIPID AND GPI-ANCHOR BIOSYNTHESIS
Defects in GPI-Anchored Proteins
Complete deletion of the GPI pathway in mice causes embryonic lethality. This is not surprising because more than 150 membrane proteins require a GPI anchor for cell surface expression (Chapter 12). Hypomorphic mutations in multiple genes in the pathway lead to a partial reduction in GPI-anchored proteins. These include PIGA, PIGH, PIGQ, PIGY, PIGC, PIGP, PIGL, PIGW, PIGM, PIGV, PIGB, PIGF, and PIGO in GPI-anchor assembly (Table ), and PIGK, GPAA1, PIGS, PIGT, and PIGU in the transfer of the GPI anchor to proteins. Defects in side-chain modifications (PIGN and PIGG) and maturation of the GPI glycan following attachment to proteins (PGAP1, PGAP2, and PGAP3) also cause inherited GPI deficiency, but not embryonic death. GPI deficiency has immense and variable consequences including neurologic symptoms, particularly developmental delay/intellectual disability and seizures, epileptic encephalopathy, progressive cerebral and/or cerebellar atrophy, hypotonia, cortical visual impairment, sensorineural deafness, and Hirschsprung disease. Nonneurologic phenotypes include brachytelephalangy, anorectal anomaly, renal abnormality, cleft palate, heart defect, and characteristic facial features such as hypertelorism, broad nasal bridge, and tented mouth. Other symptoms such as ichthyosis, iron deposition, hepatosplenomegaly, diaphragmatic hernia, and hepatic and/or portal vein thrombosis are reported in small fractions of the affected individuals. It is not easy to causally relate specific symptoms to deficiency of particular GPI-anchored proteins except in a few instances. Deficiency of tissue-nonspecific alkaline phosphatase (TNALP) accounts for seizures in some patients. Death within a year after birth due to aspiration or status epilepticus is not rare among severely affected individuals with GPI deficiency, whereas mildly affected individuals can live with a GPI deficiency.
Defects in Glycosphingolipid (GSL) Synthesis
Only three disorders in GSL synthesis (Chapter 11) are known in humans. Mutations in ST3GAL5 cause autosomal-recessive, Amish infantile epilepsy syndrome and also “salt-and-pepper syndrome.” This gene encodes a sialyltransferase required for the synthesis of the ganglioside GM3 (Siaα2-3Galβ1-4Glc-ceramide) from lactosylceramide (Galβ1-4Glc-ceramide). GM3 is also a precursor for some more complex gangliosides. The patients’ plasma GSLs are nonsialylated. In contrast to the human form of the disease, mice that lack GM3 do not have seizures or a shortened life span. However, mouse strains that are null for the sialyltransferase and an N-acetylgalactosaminyltransferase that is required for making other complex gangliosides do develop seizures, suggesting that it is the absence of these more complex gangliosides that may be the underlying problem (Chapter 11). Mutations in B4GALNT1 (also known as GM2/GD2 synthase) cause hereditary spastic paraplegia subtype 26 (SPG26). These patients have developmental delays and varying cognitive impairments with early-onset progressive spasticity owing to axonal degeneration. Cerebellar ataxia, peripheral neuropathy, cortical atrophy, and white-matter hyperintensities are also consistent across the disorder. A B4galnt1−/− mouse recapitulates several of the neurological characteristics of SPG26, most prominently the progressive gait disorder.
ST3GAL3 makes more complex gangliosides as well as N- and O-glycans. It is required for the development of high cognitive functions and is mutated in some individuals with West syndrome. An St3gal3−/− mouse model also exists, but these mice appear to have no overt neurological phenotype. GSL disorders are difficult to identify biochemically because no convenient biomarker tests have been developed. Therefore, novel genetic defects are likely to be found via next-generation sequencing.
DEFECTS IN MULTIPLE GLYCOSYLATION PATHWAYS
Defects in the Synthesis of Sugar Precursors
Sugar metabolism is essential to produce nucleotide sugars and dolichol-linked sugars for glycosylation reactions in the ER and Golgi. Because activated sugars are required to synthesize multiple glycan classes, genetic disorders affect multiple pathways. Below, some defects in the mannose, galactose, Sia, and fucose sugar activating pathways are discussed.
Galactose Pathway
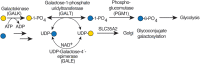
Galactosemia refers to mutations in three genes involved in Gal metabolism. In “classical galactosemia,” Gal-1-P uridyltransferase (GALT; Figure ) is deficient. This results in excess Gal-1-P and decreased synthesis and availability of UDP-Gal. Defects in UDP-Gal-4′-epimerase (GALE; Figure ) or galactokinase (GALK; Figure ) also cause the disease, but they are rarer.
CDGs related with UDP-Gal metabolism. The most common form of galactosemia is caused by a deficiency of galactose-1-phosphate uridyltransferase (GALT). This enzyme uses Gal-1-P derived from dietary Gal. In the absence of GALT, Gal-1-P accumulates along (more...)
GALT-deficient infants fail to thrive and have an enlarged liver, jaundice, and cataracts. A lactose-free diet ameliorates most of the acute symptoms by reducing the amount of Gal entering the pathway and the accumulation of Gal and Gal-1-P. Reducing Gal decreases galactitol and galactonate, which are produced via reductive or oxidative metabolism of Gal, respectively. Galactitol is not metabolized further and has osmotic properties that contribute to cataract formation. Unfortunately, a Gal-free diet apparently does not prevent the appearance of cognitive disability, ataxia, growth retardation, and ovarian dysfunction that are characteristic of this disease. Abnormal glycosylation of glycoproteins and glycolipids has been observed in some GALT-deficient individuals; however, this has not been directly related to a lack of galactose. For example, some patients who mistakenly are given Gal or those before start of a Gal-free diet synthesize transferrin that is missing both N-glycans. In GALT deficiency, a Gal-free diet normalizes glycosylation. However, dietary supplementation with Gal was found to be an effective treatment for some of the symptoms in another CDG, PGM1-CDG. In PGM1-CDG, phosphoglucomutase activity results in a reduction of UDP-Gal and UDP-Glc, leading to hypogalactosylation and missing N-glycans. These biochemical abnormalities are restored by Gal feeding. Clinically, the hypoglycemia, liver and coagulation abnormalities, and gonadal hormones improved on treatment. Still, the exercise intolerance and dilated cardiomyopathy, linked to other functions of PGM1, did not improve.
Sialic Acid Pathway
Disorders in four of the Sia biosynthesis enzymes (Chapter 15) are known, resulting in rather different phenotypes. Recessive mutations in GNE cause adult-onset GNE myopathy (previously named hereditary inclusion body myopathy type 2 [HIBM2] or Nonaka myopathy) (Table ). It occurs worldwide, but one mutation (p.Met745Thr) is especially common among Persian Jews (1:1500) and occurs in the kinase domain (Chapter 5). GNE mutations occur in various combinations in both GNE enzymatic domains and variably affect enzyme activity. The mutations moderately reduce enzymatic activity and reduce sialylation in mouse models. Oral ManNAc, as precursor for Sia synthesis, is being tested as a therapy for GNE myopathy patients, as well as for patients with primary glomerular diseases (focal segmental glomerulosclerosis, minimal change disease, and membranous nephropathy). Recessive mutations in other genes—NANS, CMAS, and SLC35A1—result in a neurological phenotype with intellectual disability and epilepsy. Additionally, varying degrees of thrombocytopenia may be seen. Interestingly, specific mutations in the GNE gene also give rise to a thrombocytopenia syndrome without the presence of neurological symptoms. The biological mechanism behind these differences is not yet understood. The catabolism of Sia is executed by neuraminate pyruvate lyase (NPL), the role of which was unknown in humans until the identification of a human genetic disease. Recessive mutations in NPL result in a phenotype of myopathy and cardiomyopathy, possibly indicating a role of Sia in (cardiac) muscle besides glycosylation.
Fucose Pathway
GDP-Fucose (Fuc) can be synthesized from GDP-Man or obtained via fucose salvage. In the salvage pathway, Fuc is converted into Fuc-1-P by fucose kinase and then to GDP-Fuc by GDP-Fuc pyrophosphorylase (Chapter 5). The contribution of the endogenous versus the salvage pathway in different cells and tissues is not yet known. A disease in the salvage pathway has been reported due to mutations in FCSK (fucose kinase), resulting in a severe neurological syndrome with encephalopathy, intractable seizures, and intellectual disability. Patients with leukocyte adhesion deficiency type II (LAD-II or SLC35C1-CDG) have mutations in SLC35C1, encoding the GDP-Fuc transporter in the Golgi. Here, transferrin sialylation was normal, so this defect was not detected by the usual test, but some serum proteins and O-linked glycans on leukocyte surface proteins were deficient in Fuc. One leukocyte protein carries a selectin ligand glycan, sialyl-Lewis x, that mediates leukocyte rolling before extravasation of leukocytes from capillaries into tissues (Chapter 34). This defect greatly elevates circulating leukocytes and decreases leukocyte extravasation, so patients have frequent infections. A few patients responded to dietary fucose therapy. Sialyl-Lewis x reappeared on their leukocytes, and circulating neutrophils promptly returned to normal levels. Fucose supplements must increase the amount of GDP-Fuc enough to correct the defect. A mouse model of Fuc deficiency lacks de novo biosynthesis of GDP-Fuc from GDP-Man (Chapter 5). The mice die without Fuc supplements, but providing Fuc in the drinking water rapidly normalizes their elevated neutrophils. The treatment also corrects abnormal hematopoiesis resulting from disrupted O-Fuc-dependent Notch signaling.
Defects in the Biosynthesis of Dolichol-Monosaccharides
Numerous defects in the biosynthesis of dolichol, the primary lipid carrier for monosaccharides and oligosaccharides in glycosylation, have been identified. These disorders share many of the features of other CDGs, although some of the mutations are associated with more specific phenotypes. As an example, variants in DHDDS have been identified in patients with retinitis pigmentosa, and heterozygous variants in NUS1 have been found in patients with various neurological phenotypes. The NUS1 and DHDDS gene products are involved in the initial steps of dolichol synthesis. The two proteins form a complex with cis-prenyltransferase activity that catalyzes polyprenolpyrophosphate formation. SRD5A3 acts as a polyprenol reductase to form dolichol (Chapter 9). This dolichol is then phosphorylated by DOLK to make the activated lipid, dolichol-phosphate, on which both monosaccharide and oligosaccharide precursors are built. Although the primary functions of dolichol-linked sugars lie within the N-glycosylation pathway, Dol-P-Glc and Dol-P-Man are both integral for different O-glycosylation pathways, and Dol-P-Man is required for biosynthesis of GPI. Clinical symptoms of some of these defects, including muscular dystrophy and dilated cardiomyopathy in patients with DPM1-3 and DOLK defects, have been associated with abnormal O-mannosylation of dystroglycan.
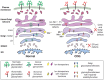
Defects in Golgi Homeostasis
The discovery of CDG defects associated with trafficking proteins showed that the abnormal glycosylation linked to these disorders could arise from alterations in Golgi homeostasis and not only from impairment of the enzymes and transporters needed for glycosylation (Figure ). Among the first of these trafficking proteins identified are subunits of the conserved oligomeric Golgi (COG) complex. This complex has multiple roles in trafficking within the Golgi, including the tethering of COPI-coated vesicles and recycling of Golgi-localized glycosyltransferases. COG7-CDG (Table ) was discovered first. Trafficking of multiple glycosyltransferases and nucleotide sugar transporters were disrupted in COG7-CDG. The mutation affects the synthesis of both N- and O-glycans and glycosaminoglycan (GAG) chains. Mutations have now been found in all COG subunits except COG3. Mammalian cells deficient in the different COG subunits also show various degrees of altered glycosylation.
Golgi homeostasis defects. Congenital disorders of glycosylation (CDGs) are also caused by defects in proteins and transporters that control Golgi homeostasis. These CDGs include members of the vesicle tethering conserved oligomeric Golgi (COG) complex, (more...)
An unusual disorder called HEMPAS (hereditary erythroblastic multinuclearity with positive acidified-serum test) leads to abnormal red cell shape and instability (hemolysis) caused by mutations in SEC23B, another intracellular trafficking protein that produces abnormal red blood cell glycans in several pathways.
In addition to trafficking proteins, a number of mutations in Golgi-associated ion transporters and vacuolar H+-ATPase subunits have also been shown to disrupt multiple glycosylation pathways, presumably because of an increase in Golgi pH, decrease in metal ion-dependent glycosyltransferase activities, or a more general disruption in Golgi recycling. Mutations in two Golgi manganese transporters, TMEM165 and ZIP8 (SLC39A8), cause CDGs highlighting the role of manganese homeostasis in glycosylation.
FUTURE OUTLOOK—PATHOPHYSIOLOGY AND TREATMENT
Expression of the same CDG mutation can have highly variable impacts, even among affected siblings. Explanations based on residual activity from hypomorphic alleles of these “simple Mendelian disorders” are neither simple nor generally satisfying. The variability is often attributed to “genetic background.” A knockout mutation may be lethal in one highly inbred mouse strain, but not in another because compensatory pathways exist in the latter. Dietary and environmental impacts are substantial as seen in MPI-CDG patients treated by oral mannose therapy. Multiple simultaneous or sequential environmental insults may impinge on borderline genetic insufficiencies to produce overt disease. A major question for future investigations is how CDG defects cause pathology. Addressing this challenge will require the implementation of analytical approaches like glycomics and glycoproteomics and of model systems that allow identification of sensitive pathways and glycoproteins in affected tissues.
In an interesting twist, it is possible to have diseases caused by “excessive” glycosylation. For example, Marfan syndrome results from mutations in fibrillin1 (FBN1) and one of these creates an N-glycosylation site that disrupts multimeric assembly of FBN1. This may not be an isolated case. A survey of nearly 600 known pathological mutations in proteins traveling the ER–Golgi pathway showed that 13% of them create novel glycosylation sites. This is far greater than predicted by random missense mutations and may mean that hyperglycosylation leads to a new class of CDGs.