

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology [Internet]. 3rd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015-2017. doi: 10.1101/glycobiology.3e.053


Essentials of Glycobiology [Internet]. 3rd edition.
Show details
Pure glycans of defined structure are essential research tools in glycobiology. Unlike proteins and nucleic acids, which can be obtained in homogeneous forms by recombinant expression and polymerase chain reaction (PCR), respectively, glycans produced in biological systems are heterogeneous. Furthermore, the quantities that can be obtained from biological systems are often small. Chemical synthesis can be used to obtain homogeneous glycans in larger quantities than are available from most cellular production systems. Chemical synthesis can be further used to incorporate glycans into homogeneous glycoproteins. This chapter summarizes the current state of the art in chemical methods to produce glycans. Enzymes can be used together with chemical methods to further simplify the process of glycan synthesis (Chapter 54).
CONTROLLING REGIOCHEMISTRY
The inherent chemical properties of oligosaccharides render their synthesis more complicated than the synthesis of the other major classes of biomolecules: oligonucleotides and oligopeptides. Fundamental challenges of glycan synthesis are required for modifying one specific hydroxyl group in the presence of many others and for control over the stereochemical outcome in the creation of an interglycosidic linkage. Commonly, glycan synthesis is characterized by the manipulation of various protecting groups, chemical moieties that mask the hydroxyl groups and prevent them from reacting with other chemical reagents. Hydroxyl-protecting groups are selectively added and removed from glycan structures, allowing for chemical alteration of the exposed hydroxyl groups. Subsequently, in a typical glycan synthesis scheme, the exposed hydroxyl group serves as a point for further elaboration. The selective exposure of one hydroxyl group allows for regioselective addition of another (mono)saccharide unit. Synthetic schemes of this type are commonly applied to the generation of O- and N-linked glycans (Chapter 9 and 10) as well as proteoglycans (Chapter 17) and glycosphingolipids (Chapter 11).
The choice of protecting groups and the order of protecting group installation are essential for a synthesis route to be successful. The most common protecting groups include persistent protecting groups, such as benzyl ethers that stay in place through many synthetic steps, and temporary protecting groups, such as carbonates or esters that are removed during intermediate steps in a synthesis. A wealth of information exists on the chemical generation of glycans and various protecting group manipulations used in the context of glycan synthesis.
CONTROLLING STEREOCHEMISTRY
A glycosidic linkage is generally formed through the activation of a glycosylating agent (glycosyl acceptor) to create an electrophilic species that reacts with the nucleophile—for example, a hydroxyl group present on the glycosyl acceptor or another class of biomolecule (serine/threonine in the case of glycopeptides or a sphingoid in the case of glycosphingolipids). The glycosylation reaction results in either the formation of an α- or β-glycosidic linkage (Chapter 2). A major challenge in glycan synthesis is the stereoselective formation of glycosidic bonds (Figure 53.1A; Chapter 2). A variety of methods are available to stereoselectively generate glycosidic linkages. The yield and the stereochemical outcome of these reactions depend on the steric and electronic nature of the glycosylating agent (the glycosyl donor), the nature of the nucleophile, and the reaction conditions chosen. One common method to control the stereochemistry at the anomeric center involves the use of certain protecting groups, such as esters or amides/carbamates, on the C-2-hydroxyl or C-2-amino group (Figure 53.1B). These “participating neighboring protecting groups” can form a cyclic oxonium ion intermediate during the glycosylation reaction, that shields one face of the molecule, leading exclusively to the formation of a “trans” glycosidic linkage (i.e., where the anomeric substituent and the C-2 group are on opposite sides of the ring, as in β-glucosides). The opposite anomeric stereochemistry, termed a “cis” glycosidic bond, is more difficult to construct with high selectivity, and neighboring group participation—with exceptions—is not possible. Many different synthetic procedures have been developed for the reliable stereoselective construction of cis-glycosidic linkages and linkages involving C-2-deoxy sugars. Although not as general as protecting group participation in the synthesis of trans-glycosidic linkages, insight into mechanisms of chemical glycosylation reactions continues to grow allowing for the stereocontrolled introduction of an increasing number of cis- and 2-deoxy glycosidic linkages.
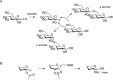
FIGURE 53.1.
(A) Stereospecific formation of glycosidic bonds as either an α- or β-linkage. LG, leaving group; R, protecting group. (B) Formation of a cyclic oxonium ion intermediate leading to the formation of a β-glycosidic linkage.
PROTECTIVE GROUP MANIPULATIONS
Carbohydrates are endowed with a wealth of functional groups. This complicates glycan synthesis, and considerable time and efforts are devoted to the development of suitable protective group schemes that allow the protection and deprotection of individual functional groups at will. Carbohydrate functional groups include hydroxyls, amines, and carboxylates, and distinguishing between these is relatively easy. Selective modification of hydroxyls—the most abundant functional groups in carbohydrates—is somewhat more complicated. Primary hydroxyls and anomeric hydroxyls are relatively easy to manipulate specifically, the former for steric reasons and the latter because they are part of a hemiacetal or hemiketal. Specific access to secondary hydroxyls requires chemistry tailored to capitalize on, for instance, their configuration (cis-hydroxyls versus trans-hydroxyls, equatorial versus axial) and their relative reactivity (i.e., exploiting steric and/or electronic differences). A representative protective group scheme starting from S-tolylglucopyranose (the S-tolyl group serves as both a masking group of the anomeric hydroxyl and as leaving group in glycosylation schemes) is depicted in Figure 53.2. In the first step all hydroxyls are transformed into the corresponding trimethylsilyl ethers—this to render the polar carbohydrate soluble in organic solvents. Next and in a series of complementary functional group manipulations, either O-2 (A), O-4 (B), or O-6 (C) is selectively liberated. In all instances, the first step comprises creation of the 4,6-O-benzilydene species. Reductive benzylation of O-3 followed by benzoylation of O-2 and reductive opening of the benzylidene toward the O-4-benzyl delivers glucose building block C. Alternative reductive opening of the benzylidene toward the O-6-benzyl delivers B, whereas leaving the benzylidene intact and removal of any remaining silyl protective groups delivers glucoside A.

FIGURE 53.2.
Protective group manipulations—which can be performed in one-pot procedures—toward a series of glucopyranose-derived building blocks for glycan assembly.
A REPRESENTATIVE SOLUTION PHASE CHEMICAL GLYCAN SYNTHESIS
In the synthesis of a fragment of a putative repeating unit of the exopolysaccharide of Pseudomonas aeruginosa (Figure 53.3), the interplay between protective group pattern and glycosylation strategies becomes apparent. Glycosylation of 4,6-O-benzylidene protected thiomannoside 1 with orthogonally protected fucoside 2 under the agency of benzenesulfinylpiperidine (BSP) and triflic anhydride (Tf2O) affords disaccharide 2. The stereoselective creation of the β-mannosidic linkage is explained by suppression of oxocarbenium ion formation due to torsional strain induced by the benzylidene moiety. Instead, SN2 displacement of an intermediate anomeric α-triflate provides the 1,2-cis β-mannoside species.

FIGURE 53.3.
Solution phase synthesis of Pseudomonas aeruginosa–derived decasaccharide 10.
Oxidative removal of the naphthyl (Nap) protective group and treatment with mannoside 4 gives trisaccharide 5. The anomeric allyl (All) group on the reducing fucoside is removed and transformed into the glycosyl trichloroacetimidate, which is condensed with the 3-hydroxyl group of orthogonally protected glucoside 6 to give tetrasaccharide 7. A 2-O-benzoyl (Bz) participating group in fucose building block 2 ensures the 1,2-trans-configuration. Elaboration of tetrasaccharide 7 led to fully protected decasaccharide 10 and illustrates the need for the two differently protected building blocks 1 and 4. The benzyl (Bn) protective group in 1 serves as a permanent protective group destined for removal at the final stages of the synthesis. The tertbutyldimethylsilyl (TBS) group in 4 in contrast can be removed selectively during the synthesis. The 2-hydroxyl groups in mannose residues are liberated and 1,2-trans (α) mannosylated with building block 8. Global deprotection of 9 is effected in two steps: first, base treatment to remove acyl protective groups, which is followed by catalytic hydrogenation to simultaneously remove benzyl, naphthyl, and benzylidene groups. The azide at the reducing end spacer is transformed into the amine for neoglycoprotein synthesis for immunization studies, or to produce glycan arrays.
REPRESENTATIVE SOLID-PHASE CHEMICAL GLYCAN SYNTHESES
Automated solid-phase synthesis has made a great impact on peptide/protein and nucleic acid chemistry and biology. This development in synthetic chemistry made peptides and oligonucleotides available to biologists and facilitated studies of their structures and functions. Likewise, the availability of a general automated (solid-phase) method for glycan synthesis would vastly accelerate access to homogeneous material for biological studies. Because oligosaccharide synthesis is much more complex than the assembly of oligopeptides and nucleotides, automated procedures for glycan assembly have been slower to evolve, but automated synthesizers are now commercially available. A prerequisite in automated glycan assembly is full control over the stereochemistry of a newly formed glycosidic linkage. Once suitable orthogonal protective groups and linker systems are defined, a solid-phase synthesis campaign is feasible. An important added value of solid-phase glycan assembly is the ability to rapidly generate libraries of related but distinct glycans, and the ability to elaborate fragments of a size not normally (because of aggregation) feasible in solution. Solid-phase synthesis allows for a large excess of the building blocks and activating agents to be used. Unreacted intermediates can be capped to avoid the accumulation of deletion sequences.
The synthesis of a library of plant cell wall arabinoxylans (Figure 53.4A) commences with charging tailor-made photocleavable linker 11 with orthogonally protected xylose 12 equipped with an anomeric phosphotriester as the leaving group. After triethylamine (Et3N)-induced removal of the fluorenylmethyloxycarbonyl (Fmoc) group the immobilized monoxylose is elongated with another copy of 12 and next (after Fmoc removal) with xylose derivative 13, featuring a 3-O-naphthyl (Nap) protective group. The Nap can be selectively removed under oxidative (DDQ) conditions after which L-arabinofuranose derivative 14 can be used in the next glycosylation cycle.
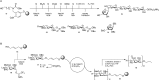
FIGURE 53.4.
Automated glycan assembly: (A) a tetrasaccharide and (B) an alginate dodecasaccharide.
The sequence of events outlined in Figure 53.4A can be readily adapted to produce larger xylan oligomer structures, or to install L-arabinofuranose moieties at different positions. In case coupling steps proceed inefficiently and truncated oligomers threaten to emerge, a capping step (with acetic anhydride and pyridine) can be programmed into the automated solid-phase glycan synthesizer to ensure facile separation of the full-length products from truncations. Once assembly is complete the oligomers are liberated from the solid support by exposure to 305 nm photons after which acyl protective groups are methanolized and benzyl/Nap groups are hydrogenolyzed. In this final step also the N-carboxybenzyl group that emerges upon photolytic cleavage is removed to yield an amine group at the reducing end sugar for bioconjugation purposes.
The synthesis of an alginate dodecamer composed of the β1-4-mannuronate moieties is of note because it required the repetitive installation of cis-glycosidic linkages. The synthesis makes use of immobilized allyl alcohol 15. Reaction with donor mannuronic ester 16 under catalytic amounts of TfOH (trifluoromethanesulfonic acid) yields immobilized monosaccharide 17. Removal of the levulinoyl (Lev) group at O-4 is accomplished by treatment with hydrazine (H2N-NH2) monoacetate, after which the coupling-deprotection sequence is repeated for the desired number of times. Upon completion of the glycan assembly the fully protected oligosaccharide is removed from the solid support by cross metathesis and subsequent removal of all protective groups by treatment with strong base and hydrogenolysis.
COMPUTATIONAL CHEMISTRY–ASSISTED GLYCAN ASSEMBLY
Significant progress in understanding the glycosylation mechanism (required to develop more effective and selective glycosylation procedures) has been made over the years by the application of computational methods. For example, density functional theory (DFT) calculations have been used to determine plausible transition states to explain high 1,2-cis-selectivity observed in reactions using 4,6-benzylidene mannosyl donors (as exemplified in Figure 53.3). These studies have revealed that the formation of the β-linked mannosides originate from an SN2-type displacement of the anomeric mannosyl α-triflate, where (the nondesired) α-linked products are formed. Computational methods guided the design of a 3,5-silylidene ketal to conformationally lock the arabinofuranose ring in an E3 envelope structure for selective cis-glycosylation reactions Figure 53.5. Selective attack of the envelope by the incoming nucleophile from the bottom (B)-face generates the 1,2-cis arabinofuranosyl linkage of a plant arabinogalactan fragment (Figure 53.5).
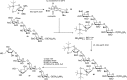
FIGURE 53.5.
cis-Arabinofuranosylation in the context of the synthesis of a plant cell wall arabinogalactan fragment 27.
A database has been used to design effective one-pot glycosylation sequences. Wong and coworkers determined the relative reactivity values (RRVs, a number indicating how reactive a glycosyl donor is) of hundreds of thioglycoside donors. This reactivity scale (spanning more than eight orders of magnitude) can be used to design one-pot oligosaccharide assembly strategies, in which several thioglycosides are combined and selective activation of one of the building blocks over the others can be achieved. A computer program, Optimer, aids the selection of the most effective combination of building blocks, equipped with the proper set of protecting groups that tune the reactivity. Figure 53.6 shows the one-pot assembly of the tumor-associated N3 antigen octasaccharide using the Optimer approach. Three building blocks with optimal RRVs were designed and synthesized: fucosyl donor 28 (RRV = 7.2 × 104), lactosamine donor 29 (RRV = 41) and lactose building block 30 (RRV = 0). Assembly of the latter disaccharides was also performed using chemoselective glycosylations by exploiting RRVs. Combination of the first two building blocks in a BSP/Tf2O-mediated condensation gave the trisaccharide thioglycoside. The addition of lactose 30 to the reaction vessel and NIS/TfOH to promote the ensuing double glycosylation provided the octasaccharide 31. After deprotection the spacer functionalized octasaccharide 32 was obtained in 11% yield.
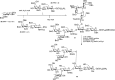
FIGURE 53.6.
One-pot synthesis of octasaccharide antigen 32.
PERSPECTIVES
Over the past two decades, the chemical synthesis of glycans has advanced from an art practiced by synthetic chemists who engaged in heroic total synthesis efforts, to a more and more routine task for glycans that contain common monomeric constituents and trans-glycosidic linkages. With the commercialization of automated glycan assembly in which a set of common building blocks is combined on a solid-phase resin, the synthesis process can be greatly accelerated. Polysaccharides as long as 50-mers, as well as glycosaminoglycans, are now rapidly accessible in multimilligram quantities. The production of longer sequences and highly branched structures will become routine in the coming years. Automated synthesis is expected to become an important part of carbohydrate synthesis in the years to come. Still, for challenging targets, containing rare monosaccharides, sialic acids, and 2-deoxy and 1,2-cis-linkages, improved methods for solution phase synthesis will have to be developed. These methods will greatly benefit from the advances in fundamental knowledge on the glycosylation reaction and will be aided by knowledge gathered in databases. In addition, enzymatic synthesis and combinations of chemical and enzymatic synthesis will become more and more important assets to procure larger glycans with more challenging sequences.
ACKNOWLEDGMENTS
The authors acknowledge contributions to previous versions of this chapter by Carolyn Bertozzi, Nathaniel Finney, and David Rabuka and appreciate helpful comments and suggestions from Yuta Maki, Keisuke Mizote, and Ryan Porell.
FURTHER READING
- Paulsen H. 1982. Advances in selective chemical syntheses of complex oligosaccharides. Angew Chem Int Ed 21: 155–173.
- Plante OJ, Palmacci ER, Seeberger PH. 2001. Automated solid-phase synthesis of oligosaccharides. Science 291: 1523–1527. [PubMed: 11222853]
- Zhu X, Kawatkar S, Rao Y, Boons GJ. 2006. Practical approach for the stereoselective introduction of β-arabinofuranosides. J Am Chem Soc 128: 11948–11957. [PubMed: 16953636]
- Wang CC, Lee JC, Luo SY, Kulkarni SS, Huang YW, Lee YW, Chang KL, Hung SC. 2007. Regioselective one-pot protection of carbohydrates. Nature 446: 896–899. [PubMed: 17443183]
- Zhu X, Schmidt RR. 2009. New principles for glycoside-bond formation. Angew Chem Int Ed 48: 1900–1934. [PubMed: 19173361]
- Crich D. 2010. Mechanism of a chemical glycosylation reaction. Acc Chem Res 43: 1144–1153. [PubMed: 20496888]
- Codée JDC, Ali A, Overkleeft HS, van der Marel GA. 2011. Novel protecting groups in carbohydrate chemistry. C R Chimie 14: 178–193.
- Hsu CH, Hung SC, Wu CY, Wong CH. 2011. Toward automated oligosaccharide synthesis. Angew Chem Int Ed 50: 11872–11923. [PubMed: 22127846]
- Walvoort MTC, van den Elst H, Plante OJ, Kröck L, Seeberger PH, Overkleeft HS, van der Marel GA, Codée JDC. 2012. Automated solid-phase synthesis of β-mannuronic acid alginates. Angew Chem Int Ed 51: 4393–4396. [PubMed: 22334421]
- Eller S, Collot M, Yin J, Hahm H.-S, Seeberger PH. 2013. Automated solid-phase synthesis of chondroitin sulfate glycosaminoglycans. Angew Chem Int Ed 52: 5858–5861. [PubMed: 23589381]
- Li H, Mo KF, Wang Q, Stover CK, DiGiandomenico A, Boons GJ. 2013. Epitope mapping of monoclonal antibodies using synthetic oligosaccharides uncovers novel aspects of immune recognition of the Psl exopolysaccharide of Pseudomonas aeruginosa. Chem Eur J 19: 17425–17431. [PubMed: 24248772]
- Seeberger PH. 2015. The logic of automated oligosaccharide assembly. Acc Chem Res 48: 1450–1463. [PubMed: 25871824]
- Review Chemical and Enzymatic Synthesis of Glycans and Glycoconjugates.[Essentials of Glycobiology. 2009]Review Chemical and Enzymatic Synthesis of Glycans and Glycoconjugates.Seeberger PH, Finney N, Rabuka D, Bertozzi CR. Essentials of Glycobiology. 2009
- Review Chemical Synthesis of Glycans and Glycoconjugates.[Essentials of Glycobiology. 2022]Review Chemical Synthesis of Glycans and Glycoconjugates.Seeberger PH, Overkleeft HS. Essentials of Glycobiology. 2022
- Review Advances in the Chemical Synthesis of Carbohydrates and Glycoconjugates.[Adv Biochem Eng Biotechnol. 2021]Review Advances in the Chemical Synthesis of Carbohydrates and Glycoconjugates.Malik A, Seeberger PH, Varón Silva D. Adv Biochem Eng Biotechnol. 2021; 175:201-230.
- Review Chemoenzymatic Synthesis of Glycans and Glycoconjugates.[Essentials of Glycobiology. 2022]Review Chemoenzymatic Synthesis of Glycans and Glycoconjugates.Overkleeft HS, Seeberger PH. Essentials of Glycobiology. 2022
- Review Understanding the Chemistry and Biology of Glycosylation with Glycan Synthesis.[Annu Rev Biochem. 2016]Review Understanding the Chemistry and Biology of Glycosylation with Glycan Synthesis.Krasnova L, Wong CH. Annu Rev Biochem. 2016 Jun 2; 85:599-630. Epub 2016 Apr 25.
- Chemical Synthesis of Glycans and Glycoconjugates - Essentials of GlycobiologyChemical Synthesis of Glycans and Glycoconjugates - Essentials of Glycobiology
Your browsing activity is empty.
Activity recording is turned off.
See more...