

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology [Internet]. 3rd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015-2017. doi: 10.1101/glycobiology.3e.050


Essentials of Glycobiology [Internet]. 3rd edition.
Show details
This chapter surveys methods for the structural characterization of glycans, including residue composition, linkage types, and attachment to aglycones. It covers methods for detection of specific glycan sequences on glycoproteins and cellular surfaces, and it covers methods for characterizing structures in three dimensions. The methods vary widely, ranging from classical chemical methods for detection and characterization of glycans on isolated products, to sensitive fluorescence methods used in combination with glycan-binding proteins and microscopy on cells and tissues. Methods also include nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS) approaches that allow more detailed structural characterization.
BACKGROUND
The primary structure of a glycan is defined by the nature and order of constituent monosaccharide residues, by the configuration and position of glycosidic linkages, and by the nature and location of nonglycan entities to which they are attached (aglycones; Chapters 2 and 3). Beyond this, different glycans can be attached to different sites on a glycoprotein, and these glycans can vary when that glycoprotein is made in different cell types or at different stages of development. Moreover, it is often three-dimensional (3D) features, or particular surface distributions of glycans, that are recognized by glycan-binding proteins. Characterization of these diverse structural features requires an array of different methods, with the choice of methodology depending very much on the problem.
For a typical mammalian glycoprotein, the aim is often to identify the correct structure from a range of known or predictable candidate structures, and a limited amount of structural data may suffice (Chapter 51). For glycans from bacteria or less well-characterized organisms, it is hard to make predictions, and a more complete data set may be required. Choice of methodology also depends on the amount and purity of material available, as well as the context in which data must be collected (e.g., tissue surface vs. isolated glycoprotein). If quantities are not limiting, the complete primary structure and even the tertiary (3D) structure may be determined. The need to respond to these diverse circumstances and to understand this more complex picture of glycan structure has driven the development of many of the methods described in the following sections.
DETECTION OF GLYCANS
Methods for glycan detection in glycoconjugates include direct chemical reactions with the constituent monosaccharides, metabolic labeling with either radioactive or chemically reactive monosaccharides, and detection with specific glycan-recognizing proteins (including lectins and antibodies) (Chapter 48). A general method for detecting the presence of glycans on proteins involves periodate oxidation of their hydroxyl groups followed by Schiff base formation with amine- or hydrazide-based probes (Chapter 2). This chemical modification, also known as the periodic acid–Schiff (PAS) reaction, can identify glycoproteins in gels. Commercially available kits allow detection of 5–10 ng of glycoprotein, using the periodate reaction with subsequent amplification by means of biotin-hydrazide/streptavidin-alkaline phosphatase. Lectin overlay of a blot of a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel can detect the presence of specific glycans with comparable sensitivity and greater specificity. For example, the agglutinins from Sambucus nigra (SNA) bind to glycans that terminate with α2-6 sialic acid (Sia). Lectins (Chapters 31–36) recognizing terminal fucose (Fuc), galactose (Gal), N-acetylgalactosamine (GalNAc), and N-acetylglucosamine (GlcNAc) are also commercially available.
Metabolic labeling of glycoconjugates with radioactive sugars is another powerful tool for determining the composition of attached glycans. Cells incubated in media containing 3H- or 14C-labeled monosaccharides will incorporate the label into the glycans of glycoconjugates. A particularly powerful method has been the use of [2-3H]mannose labeling, as all of the label remains in mannose and fucose, or becomes vastly diluted in 56 m water. Radiolabeled glycans can be detected following gel electrophoresis (SDS-PAGE) or thin-layer chromatography (TLC) by autoradiography or fluorography. They can also be released and studied in significant detail by various methods. Proteins with a glycosylphosphatidylinositol (GPI) anchor may also be selectively labeled with radioactive precursors such as myo-inositol or ethanolamine (Chapter 12). Glycosaminoglycan (GAG) chains on proteoglycans (Chapter 17) can be metabolically labeled with 35SO4 or 3H-glucosamine and separated from other glycoproteins by ion-exchange chromatography or cetylpyridinium chloride/ethanol precipitation. In recent years, the use of fluorescent probes and labels has enabled a reduction in the use of radioisotopes in applications in which the detection and quantitative determination of glycans, as opposed to glycosylation pathway information, is the primary objective. Fluorescent labels for sensitive detection of glycans after liquid chromatography (LC) include the readily available 2-amino benzoic acid (2-AA) and 2-aminobenzamide (2-AB), that may be attached to reducing sugars by reductive amination following release from an aglycone.
Metabolic labeling can also be performed with synthetic monosaccharides that are modified with chemically reactive groups. For example, the azido monosaccharide N-azidoacetylmannosamine (ManNAz) is converted by cells to N-azidoacetyl Sia, which is incorporated into sialylated glycans in place of a natural Sia. The azide group can then be selectively reacted with phosphine or alkyne reagents (Chapter 53) that introduce a fluorescent dye or an affinity probe such as biotin, thereby enabling detection of the sialic acid. Azido analogs of GalNAc and GlcNAc can be used to label O-GalNAc glycans (Chapter 10) and O-GlcNAc (Chapters 13 and 19), respectively. Use of fluorescent labels can also be coupled with confocal microscopy to give important insight into the location of glycans in cells and tissues. However, such modifications may change the biosynthesis and/or biology of glycans they are on, creating some uncertainity about the observed results. In the final analysis no method has completely supplanted radioactive metabolic labeling for pulse-chase studies of some naturally occurring glycans.
Glycoproteins
A glycosylated protein typically presents one or more diffuse bands during gel electrophoresis, resulting from heterogeneity of the attached glycans. Even when visualized by protein staining reagents, this phenomenon is often the first indication of the presence of glycans. Some glycoconjugates of very high molecular weight, such as mucins and proteoglycans, do not enter ordinary gels or, if they do, they migrate as heterogeneous smears. Agarose gels or combination polyacrylamide-agarose gels may be useful in this situation. Several analytical options are available to investigate the presence of glycans further (e.g., the PAS stain described above). Treatment of glycoproteins with endoglycosidases (e.g., peptide-N-glycosidase F [PNGase F], endoglycosidase F2 [Endo F2], endoglycosidase H [Endo H]; see Table 50.1 and Figure 50.1) is another option. If this results in a mobility change of one or more of the bands on the gel, the presence of N-glycans is indicated. O-glycanase (endo-α-N-acetylgalactosaminidase; Table 50.1) can be used for the specific identification of O-glycans, sometimes requiring pretreatment with other enzymes to expose the disaccharide core. Removal of individual sugars by exoglycosidases such as sialidase or β-galactosidase (Figure 50.1) may also result in a mobility change depending on the number of residues removed. However, some glycans cannot be detected by these treatments as they are resistant to the enzymes used. Resistance can result from modifications to glycan hydroxyl groups (e.g., sulfation, acetylation, or phosphorylation; Chapter 2), glycosidic linkages that are not recognized by the enzymes, or steric inaccessibility of the glycan. Complete removal of N- and O-glycans can be achieved by chemical treatments (e.g., hydrazinolysis or β-elimination) but peptide damage usually precludes further analysis by gel electrophoresis. The glycan may also be partially degraded (e.g., by loss of O-acetylation).

FIGURE 50.1.
Glycosidases used for structural analysis. (Left) A biantennary N-glycan is shown with exoglycosidases that remove each monosaccharide sequentially. Exoglycosidases act only on terminal sugars. Also shown are endoglycosidases that remove an intact N-glycan. (more...)
TABLE 50.1.
Table of enzymes for glycan analysis
Proteoglycans
Proteoglycans (Chapter 17) may be separated by agarose gel electrophoresis and by ion-exchange chromatography, which separates on the basis of charge conferred by sulfate groups. Treatment of proteoglycans with GAG lyases (Table 50.1) will produce a shift in mobility on a gel, condensing the proteoglycan smear into discrete bands. After removal of much of the glycan portion, antibodies that recognize the remaining “stub” glycans may be used in western analysis.
Glycolipids
Typically, the analysis of glycolipid glycans by NMR or MS is preceded by their purification using chromatographic methods. Mixtures of glycolipids can be fractionated by TLC, and staining of TLC plates with glycan-reactive reagents may allow detection of individual glycolipids. Using different reagents, it is possible to recognize gangliosides (e.g., resorcinol-HCl detects Sia) or neutral monosaccharides (e.g., orcinol-sulfuric acid detects all monosaccharides) in a TLC band. Reagents are also available for detection of sulfate and phosphate groups on glycolipids. Some prepurification of the crude extract is usually preferred (e.g., Folch partitioning and ion-exchange chromatography). These procedures separate nonpolar or nonionic lipids from polar lipids (e.g., glycosphingolipids; Chapter 11) and those that contain charged groups (i.e., gangliosides, phospholipids, and sulfatides). It is also common practice to deduce the presence of specific sugars by evaluating the shifts produced in the migration position of a band following a chemical or enzymatic treatment. Glycolipids on TLC plates can also be detected by monoclonal antibodies, lectins, or even intact microorganisms expressing glycan-specific receptors that recognize specific glycan features (Chapter 48). Detailed structural features may be identified by running the TLC in a second dimension following a specific treatment. On a larger scale, glycolipids are separated using column chromatography or by high-performance TLC on silica plates.
GPI Anchors
GPI-anchored proteins (Chapter 12), with their lipid, protein, and glycan moieties, have unique physicochemical properties that can be exploited for their detection. The nonionic detergent Triton X-114 at low temperature (4°C) extracts soluble and integral membrane proteins as well as GPI-anchored proteins. When the solution is warmed, two phases separate, and GPI-anchored and other amphiphilic proteins remain associated with the detergent-enriched phase. GPI-specific phospholipases can be used to cleave GPI anchors for further characterization. Successful cleavage by GPI-specific phospholipases can be assessed by subsequently analyzing samples by SDS-PAGE, because removal of the GPI anchor causes a shift in molecular mass. This is a common diagnostic method for identifying the presence of a GPI anchor on a protein of interest. Another method is to treat the GPI-anchored protein with nitrous acid, which cleaves the unsubstituted glucosamine residue that links the glycan to the phosphatidylinositol.
Plant and Bacterial Polysaccharides
This class of glycans contains many structures, including homo- and heteropolysaccharides, neutral and ionic polysaccharides, and linear and branched structures, with widespread molecular sizes ranging from a few monosaccharide units to thousands (Chapters 3, 21, and 22). These polysaccharides are typically extracted with water, salts, chaotropic agents, or detergents and isolated by precipitation with alcohols. Detection is based on refractive index (RI) or colorimetric reactions, because sample quantity is not usually a limitation.
RELEASE AND SEPARATION OF GLYCANS
Once the presence and general type of glycan has been established, the next challenge is to release specific types of glycans and separate different classes in sufficient quantities for structural characterization.
Release of Glycans from Glycoconjugates
When glycans are released before structural analysis, it is best to use a quantitative release procedure that neither destroys nor alters the glycan. Ideally, information regarding the type of linkage between the glycan and its liberated protein or lipid should be retained, although this is not always possible.
Glycolipids can often be isolated and characterized by MS and/or NMR without the need for release of glycans, but if necessary enzymatic methods can be used, or for glycosphingolipids, ozonolysis will separate lipid from glycan.
Complex, hybrid and oligomannose N-glycans can be released from glycoproteins with N-glycosidases such as PNGase F or PNGase A. Endo H may be used for the selective release of high-mannose and hybrid N-glycans, but complex N-glycans are resistant (Figure 50.1).
Chemical approaches suitable for release of glycans from a protein include hydrazinolysis, which releases N-glycans and/or O-glycans, depending on the experimental conditions. Strong base treatment can, under carefully controlled conditions, release only O-glycans in a process called β-elimination; it is generally accompanied by reduction with borohydride to give an alditol. More recently methods have been developed in which base treatment is accompanied by derivatization with pyrazolone, which acts as a UV-absorbing label during chromatographic separation. However, all of the chemical methods can result in partial or complete loss of labile modifications, such as O-acetylation or sulfation.
Separation of Glycans
Glycans released from a glycoconjugate usually form a complex mixture. Even when only one glycosylation site in the protein is occupied, individual molecules can bear different glycan species generating multiple glycoforms. The high-throughput analysis of these glycan mixtures using glycomics technologies is described in Chapter 51.
Preparation of individual glycan samples for structural analysis, whatever their source, will rely on separation techniques. Chromatographic separations commonly used to isolate pure glycans, or at least glycan mixtures of reduced complexity, include size exclusion chromatography (SEC), strong or weak anion exchange chromatography (SAX or WAX), and some forms of reverse-phase high-pressure liquid chromatography (HPLC). Electrophoretic methods include capillary electrophoresis (CE) and fluorophore-assisted carbohydrate electrophoresis (FACE).
HPLC-based separation is feasible, even for large samples (>10 mg), if the mixture contains fewer than ∼50 glycan species. Individual fractions can be recovered for further structural analysis by MS or NMR. Once liberated from their glycoconjugates, glycans with free-reducing termini (Chapter 2) can be chemically labeled with fluorescent tags, such as 2-amino-pyridine (2-AP) or 2-AB, providing detection sensitivity that rivals the sensitivity achieved with radiolabels. Advantages of this method include more facile purification of the labeled glycans and a wider variety of options for chromatographic separations and structural analysis techniques. Nonlabeled glycans may be detected by (relatively insensitive) RI methods, or by more sensitive pulsed amperometric detection (PAD).
If a label is introduced at the reducing end, it is possible to use sequential exoglycosidase treatments (Figure 50.1) and look for shifts of the oligosaccharide in a chromatographic system (e.g., paper chromatography, HPLC, TLC) as an indication of susceptibility to the enzyme. Comparison with known standards treated in the same manner allows tentative glycan identification. However, well-characterized standards of known structure are difficult to obtain in a pure form and there are nearly always “contaminating” peaks in a chromatogram that cannot be readily identified. It is very important to note that separation profiling should not be confused with actual structural analysis, because coelution with a standard could occur with a variety of different structures.
MONOSACCHARIDE COMPOSITION ANALYSIS
Some qualitative information on monosaccharide composition of a glycan may be derived from the procedures described above. However, it is often convenient and informative to determine monosaccharide composition without prior release of glycans. After total hydrolysis of a glycan into its monosaccharide constituents, colorimetric reactions can be used to determine the total amount of hexose, hexuronic acid, or hexosamine in a sample. These approaches only require common reagents and a spectrophotometer, but determination of total glycan content may not always be accurate because of variations in the sensitivities of different linkages to hydrolysis, variations in the degradation of individual saccharides, or a lack of specificity and/or sensitivity in the assays.
Quantitative monosaccharide analysis provides estimated molar ratios of individual monosaccharide components of glycans and may suggest the presence of specific oligosaccharide classes (e.g., N-glycans vs. O-glycans). The analysis involves the following steps: cleavage of all glycosidic linkages (typically by acid hydrolysis), fractionation of the resulting monosaccharides, detection, and quantification. Since the early 1960s, a variety of gas chromatography (GC) methods have been developed to quantify monosaccharides. The most useful involve coupling of GC and MS for linkage and composition information. GC requires volatile samples so the monosaccharides are first chemically modified at their hydroxyl, amide, carboxyl, and aldehyde groups. Reduction of the aldehyde of free monosaccharides followed by acetylation of their hydroxyl groups provides derivatives termed “alditol acetates.” These modified monosaccharides can be readily analyzed by GC-MS and compared with authentic standards. The hydroxyl and amino groups of free monosaccharides generated by glycan hydrolysis can also be converted to trimethylsilyl ethers. These per-O-trimethylsilyl derivatives are widely used for monosaccharide compositional analysis by GC-MS. Incorporation of an optically pure chiral aglycone (e.g., [–]-2-butyl alcohol), in combination with trimethylsilylation, allows the GC separation of the D and L isomers and thus determination of the absolute configuration of each monosaccharide.
An alternative to GC-MS is high-pH anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD), a special type of ion exchange chromatography which does not require monosaccharide derivatization. This technique is especially useful for analyzing acidic sugars such as Sia, which are refractive to GC-MS. A convenient method for quantitating Sia, that does not require expensive equipment, involves tagging with 1,2-diamino-4,5-methylene-dioxybenzene and measuring fluorescence. This method has a detection sensitivity in the femtomole range, and can also pick up many of the common naturally occurring modifications of this diverse class of monosaccharides (Chapter 15). Other popular techniques for defining monosaccharide composition are HPLC and high-performance capillary electrophoresis (HPCE). The monosaccharides are usually tagged with fluorescent derivatives for high-sensitivity detection. Tagging with 8-amino-1,3,6-naphthalene trisulfonic acid yields anionic derivatives that can be conveniently analyzed by gel electrophoresis. This is referred to as fluorophore-assisted carbohydrate electrophoresis (FACE).
LINKAGE ANALYSIS
Determination of Linkage Positions
Linkage analysis is a well-established and ingenious approach for determining linkage positions. The principle of this method is to introduce a stable substituent (an ether-linked methyl group) onto each free hydroxyl group of the native glycan. Glycosidic linkages are then cleaved by acid hydrolysis, producing partially methylated monosaccharides with free hydroxyl groups at the positions that were previously involved in a linkage. The partially methylated monosaccharides are ring-opened with a reducing agent (normally borodeuteride) to introduce a new hydroxyl group and, more importantly, a deuterium atom at C-1, which helps identify the reducing end of each monosaccharide. All the free hydroxyl groups are then acetylated resulting in partially methylated alditol acetates (PMAAs) that can be identified by a combination of GC retention times and electron impact (EI)-MS (Figure 50.2). The masses of fragments produced by impact of high-energy electrons on PMAAs identify substitution sites in some cases, but fragmentation patterns of similarly substituted isomeric monosaccharides (e.g., Glc and Gal) can be nearly identical. Thus, definitive identification of monosaccharides requires, in addition to the analysis of the MS pattern, the comparison of GC retention times with those of known standards (e.g., all peracetylated 2,3,4-tri-O-methyl-hexoses produce the same EI-MS spectrum, but peracetylated 2,3,4-tri-O-methylgalactitol elutes later than peracetylated 2,3,4-tri-O-methylglucitol). This type of analysis identifies terminal residues (they are methylated at every position except the hydroxyl group at C-1 and the ring oxygen), indicates how each monosaccharide is substituted, including the occurrence of linkage and branching points, and allows the determination of the ring size (pyranose [p] or furanose [f]) for each monosaccharide. However, linkage analysis provides no sequence information and cannot determine the α- or β-anomeric configuration.
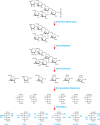
FIGURE 50.2.
An example of linkage analysis showing a bacterial O-linked branched hexasaccharide with a sequence of Rha1-3Glc1-(Glc1-3GlcNAc1-)2,6Glc1-6GlcNAc. The O-glycan is reductively eliminated from proteins before successive steps of permethylation, acid hydrolysis, (more...)
Mass Spectrometry
EI-MS in monosaccharide composition and linkage analyses is covered above. In this section, three other types of ionization used in MS—fast atom bombardment (FAB), matrix-assisted laser desorption/ionization (MALDI), and electrospray ionization (ESI)—are described. All three technologies permit the direct ionization of nonvolatile substances, and are applicable to intact glycoconjugates, as well as fragments thereof. FAB-MS was the first to be developed and was the premier MS method for sequencing glycans and glycoconjugates through the 1980s and 1990s. Although FAB-MS has now been largely replaced by ESI-MS and MALDI-MS (which offer greater sensitivity and applicability to high molecular weight glycans and glycoconjugates), the latter techniques continue to exploit sample handling strategies originally developed for FAB-MS. In addition, the knowledge of glycan fragmentation pathways that emerged from the FAB-MS research of the 1980s underpins today's ESI- and MALDI-MS analyses. Among the structural features that can be defined by MS methods are (1) degree of heterogeneity and type of glycosylation (e.g., N-glycan vs. O-glycan; oligomannose, hybrid, or complex N-glycans); (2) sites of glycosylation and identity of the protein/lipid carrier; (3) glycan branching; (4) the number and lengths of antennae, their composition, and substitution with Fuc, Sia, or other capping groups such as sulfate, phosphate, or acetyl esters; and (5) complete sequences of individual glycans.
In the FAB-MS experiment, samples are dissolved in a liquid matrix and ionization/desorption is effected by a high-energy beam of particles fired from an atom or ion gun. In MALDI-MS experiments, the sample is dried on a metal target in the presence of a light-absorbing matrix until matrix crystals containing trapped sample molecules are formed. Ionization of the sample is effected by energy transfer from matrix molecules that have absorbed energy from laser pulses. MALDI-MS is the preeminent technique for screening for molecular ions in complex mixtures and it is a powerful tool for glycomic profiling (Chapter 51). For ESI-MS, a stream of liquid containing the sample enters the source through a capillary interface, where the sample molecules are stripped of solvent, leaving them as multiply charged species. ESI-MS can be coupled to nano- or capillary-bore LC permitting online LC/ESI-MS analysis. This method is particularly useful when complex mixtures of peptides and glycopeptides are being examined (e.g., after proteolytic digestion of a glycoprotein).
In principle, MS provides two types of structural information—the masses of intact molecules (the molecular ions) and the masses of fragment ions. However, in both ESI- and MALDI-MS, the ionization process is usually not sufficiently energetic for fragment ion formation. To overcome this, ESI and MALDI instruments normally have two analyzers in tandem, which allows detection by the second analyzer, of fragment ions produced after molecular ions selected by the first analyzer undergo collisions with an inert gas in a chamber placed between the two analyzers. This is referred to as collision-activated dissociation (CAD) or collision-induced dissociation (CID). MALDI-MS/MS is commonly performed on instruments that have two time-of-flight (TOF) analyzers in tandem (TOF-TOF), whereas the most popular instruments for ESI-MS/MS have either a quadrupole (Q) as the first analyzer and an orthogonal TOF as the second analyzer (QTOF) or a linear ion trap as the first analyzer and an Orbitrap as the second analyzer (LTQ-Orbitrap). Some MS/MS instruments have a single “ion trap” analyzer that traps ions and functions as the collision chamber. Ion traps are relatively inexpensive but they lack the sensitivity, versatility, and mass range of most tandem instruments. The Fourier transform mass spectrometer (FTMS), a very expensive but very powerful instrument, has extremely high mass accuracy. An alternative to CAD for fragment ion production has been developed for FTMS. This is called electron capture dissociation (ECD) and involves the capture of an electron by the multiply charged molecular ion. A related technique, called electron transfer dissociation (ETD) was subsequently developed for tandem instruments. Both ECD and ETD yield radical cations generated by different fragmentation pathways than ions produced by CAD. ECD and ETD are especially useful for defining glycosylation sites in glycopeptides.
Although underivatized glycans can be analyzed by MALDI- or ESI-MS/MS, superior data can be obtained if the glycans are derivatized before MS analysis. Derivatization methods can be broadly divided into two categories: (1) “tagging” of reducing ends and (2) protection of all functional groups. Commonly used tagging reagents include p-aminobenzoic acid ethyl ester, 2-AP and 2-AB. This type of derivatization facilitates chromatographic purification and enhances useful fragmentation. Protection of hydroxyl groups by permethylation is by far the most important type of full derivatization used in glycan MS (although with accompanying destruction of acetyl esters and other base-labile functional groups during the derivatization process). In MS/MS experiments, permethylated derivatives form abundant fragment ions produced by cleavage at susceptible glycosidic linkages, notably on the reducing side of each HexNAc residue. In N- and O-glycans, this preferred fragmentation unambiguously establishes the antennae sequences.
Broadly speaking, the unique strengths of MS can be exploited in two general ways in glycobiology. The first is to obtain detailed characterization of purified individual glycans, or mixtures of glycans. In this type of study, it is essential to acquire sufficient rigorous data to define structure unambiguously; many different MS-based experiments will be required, often complemented by NMR, linkage analysis, and profiling of enzyme digests. The second is to pursue glycomics investigations in situations in which it may not be essential to define structures fully, and high-throughput glycomic profiling or mass-mapping procedures can be exploited (Chapter 51).
NMR Spectroscopy
NMR spectroscopy is a powerful tool capable of full de novo structural characterization of isolated glycans and simple glycan mixtures. Among its several advantages are its broad applicability (glycans contain an abundance of protons, the most easily detected magnetic nucleus), its nondestructive nature, the quantitative relationship between resonance intensity and residue concentration, the diverse set of experiments available, and the ability to return information on both primary and tertiary structure. Its principal limitation is sensitivity, typically requiring several nanomoles of glycan for even the simplest experiment directed at structure determination. However, there are emerging experiments using methods, such as dynamic nuclear polarization, that provide several orders of magnitude improvement in sensitivity for applications that can monitor rapid in vivo conversions of metabolites such as glucose and fructose. Other limitations for both structural and metabolic applications are the relatively high cost of spectrometers and the level of expertise required for acquiring and interpreting NMR spectra.
When small amounts of sample are available and the source of the sample is known, the acquisition of a 1D proton NMR spectrum, combined with simple 2D coupling correlated spectroscopy (COSY) or total correlation spectroscopy (TOCSY), can frequently identify a glycan. Figure 50.3 shows a 1D proton NMR spectrum collected on 30 µg of PNGase F-released N-glycans from the Fc fragment of a human IgG1 antibody engineered to have a near homogeneous glycan compostion. The collection required ∼20 min on a currently state-of-the-art spectrometer (900 MHz with a cryoprobe). The anomeric proton resonances appear in a well-resolved region of the spectrum and show characteristic doublets with a splitting that is small for most α anomers and is significantly larger for most β anomers. Thus, a first glance at the 1H-NMR spectrum gives a good estimate of how many residues there are (one anomeric proton per residue except for Sia), and how many of them belong to each anomeric type. Connections from the anomeric (H1) resonances to the H2 and subsequently other resonances can usually be determined from simple COSY or TOCSY data, which show chemical shifts (resonance positions) for these protons as cross peaks on columns or rows emanating from an anomeric resonance (an ∼24-h acquisition in the example given). The downfield (higher ppm) position and small couplings of the H2 resonances connected by cross peaks to three of the α-anomeric resonances identify these as belonging to core Man residues of an N-glycan. These, including other easily resolved resonances (historically called reporter group resonances), are sensitive to both monosaccharide type and linkage type, and often prove sufficient to identify a glycan, particularly when options are restricted by knowledge of biosynthetic pathways in the system under study.
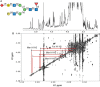
FIGURE 50.3.
Section of a 900-MHz proton nuclear magnetic resonance (NMR) spectrum of N-glycans released from the Fc of human IgG1. (A) 1D proton NMR spectrum, ∼30 µg in ∼300 µL of D2O. (B) COSY spectrum of the same sample showing connectivities (more...)
Accessible structures are more diverse in other organisms (e.g., bacteria and plants), and it is often advantageous to include 13C chemical shifts. These can be correlated with resonances of directly bonded protons using 2D heteronuclear single quantum coherence (HSQC) spectra. 13C is magnetically active, but 13C spectroscopy is less sensitive than proton spectroscopy, so chemical shifts are indirectly detected through protons. Even with the improved detection, 13C is just 1% naturally abundant, and unless the sample is isotopically enriched, approximately two orders of magnitude more material is required for collection of these data. Web-based tools that rely on empirical rules or databases that correlate chemical shifts with structural features now exist, and these allow facile prediction of possible structures from 1D and 2D NMR data sets (e.g., CASPER).
When unprecedented structures emerge, it is also possible to perform a de novo structure determination using NMR data alone. This usually begins with a full assignment of the 1H and 13C resonances for each residue of a glycan using a combination of COSY, TOCSY, and HSQC spectra. In addition, the two-dimensional heteronuclear multiple-bond correlation (HMBC) experiment is used to detect through-bond coupling between the anomeric proton and the carbon atom on the opposite side of the glycosidic linkage. This is, however, an even less sensitive and more time-consuming experiment. It is possible to avoid both 13C-based HSQC and HMBC experiments by substituting a 2D nuclear Overhauser effect (NOE) spectrum or its rotating frame analog, an ROE spectrum. These experiments correlate proton resonances based on interproton distances, as opposed to through-bond couplings; they typically show cross peaks for pairs of protons that are within ∼4 Å of one another. Most connections are between protons in the same residue, but typically an extra connection will be between an anomeric proton and a proton in another residue. This proton is frequently across the glycosidic bond at the linkage site, providing a way to assemble residues into a complete structure. However, caution must be exercised. The identification of linkage sites is not as definitive as with 13C-based experiments, because the distance for the trans-glycosidic pair is dependent on glycosidic torsion angles, and these NOE cross peaks can be weak. Also, other non-linkage site protons can come within NOE distance and give false identification.
THREE-DIMENSIONAL GLYCAN STRUCTURE
The same NOE cross peaks that assist in identifying linkages between residues can, in principle, be used to define the tertiaryl (3D) structure of a glycan. However, the interpretation is complicated by the presence of internal molecular motion, resulting in many different conformations in solution. This motion is nicely illustrated in ϕ, ψ energy plots much like the Ramachandran plots used to describe the energetics of torsional angles in the polypeptide backbone of a protein. For glycans, these plots are usually based on parameterized functions that represent a combination of quantum mechanical and experimental data. Such a plot is shown for a Glcβ1-4Glc linkage in Figure 50.4. The minima in these plots correlate well with angles found in structures of glycans deposited in databases such as the Cambridge Structural Database or the Protein Data Bank (PDB). Thermal energies are ∼0.6 kcal, so in solution, states within ∼1.4 kcal of the minimum have appreciable populations (>10%), representing a ±20° variation in torsion angles of the Glcβ1-4Glc linkage. In NMR experiments, intensities of NOE cross peaks are roughly proportional to the average of the inverse sixth power of the distance sampled over these states, and the steep distance dependence results in skewing distances toward smaller numbers. Nevertheless, NOEs, particularly those representing close approach of remote parts of larger glycans, can guide construction of 3D structural models. Convenient tools for building structures are available, such as the carbohydrate builder of the GLYCAM package. Initial structures can be further refined with molecular mechanics or molecular dynamics packages, or they can be manipulated in common molecular graphics packages to match solution data coming from NOEs and other NMR measurements, including residual dipolar couplings and paramagnetic effects, which provide angular and long range distance constraints respectively (Chapter 30).
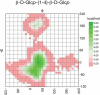
FIGURE 50.4.
ϕ–ψ energy plot for the glycosidic torsion angles between Glc residues in Glcβ1-4Glc-OMe. The plot was obtained with the Web-based tool CARP. ϕ, ψ angles are in the “NMR” convention (ϕ (more...)
ACKNOWLEDGMENTS
The authors acknowledge contributions to previous versions of this chapter by Carolyn R. Bertozzi and appreciate helpful comments and suggestions from Morten Thaysen-Andersen, Keisuke Mizote, Arun Everest-Dass, and Anne Q. Phan.
FURTHER READING
- Li E, Tabas I, Kornfeld S. 1978. The synthesis of complex-type oligosaccharides. I. Structure of the lipid-linked oligosaccharide precursor of the complex-type oligosaccharides of the vesicular stomatitis virus G protein. J Biol Chem 253: 7762–7770. [PubMed: 212434]
- Zitzmann N, Ferguson MA. 1999. Analysis of the carbohydrate components of glycosylphosphatidylinositol structures using fluorescent labeling. Methods Mol Biol 116: 73–89. [PubMed: 10399147]
- Duus J, Gotfredsen CH, Bock K. 2000. Carbohydrate structural determination by NMR spectroscopy: Modern methods and limitations. Chem Rev 100: 4589–4614. [PubMed: 11749359]
- Harvey DJ. 2001. Identification of protein-bound carbohydrates by mass spectrometry. Proteomics 1: 311–328. [PubMed: 11680878]
- Mechref Y, Novotny MV. 2002. Structural investigations of glycoconjugates at high sensitivity. Chem Rev 102: 321–369. [PubMed: 11841246]
- J Jiménez-Barbero, T Peters, editors. . 2003. NMR spectroscopy of glycoconjugates. Wiley-VCH, Weinheim, Germany.
- Lamari FN, Kuhn R, Karamanos NK. 2003. Derivatization of carbohydrates for chromatographic, electrophoretic and mass spectrometric structural analysis. J Chromatogr B Analyt Technol Biomed Life Sci 793: 15–36. [PubMed: 12880852]
- Haslam SM, North SJ, Dell A. 2006. Mass spectrometric analysis of N- and O-glycosylation of tissues and cells. Curr Opin Struct Biol 16: 584–591. [PubMed: 16938453]
- Vliegenthart JFG, Woods RJ. 2006. NMR spectroscopy and computer modeling of carbohydrates: Recent advances. ACS Symposium; Washington, DC.
- Tissot B, Gasiunas N, Powell AK, Ahmed Y, Zhi ZL, Haslam SM, Morris HR, Turnbull JE, Gallagher JT, Dell A. 2007. Towards GAG glycomics: Analysis of highly sulfated heparins by MALDI-TOF mass spectrometry. Glycobiology 17: 972–982. [PubMed: 17623722]
- Jiménez-Barbero J, Díaz MD, Nieto PM. 2008. NMR structural studies of oligosaccharides related to cancer processes. Anticancer Agents Med Chem 8: 52–63. [PubMed: 18220505]
- Battistel MD, Azurmendi HF, Yu B, Freedberg DI. 2014. NMR of glycans: Shedding new light on old problems. Prog Nucl Magn Reson Spectrosc 79: 48–68. [PubMed: 24815364]
- Balagurunathan K, Nakato H, Desai UR. eds. 2015. Glycosaminoglycans: Chemistry and biology. In Methods in molecular biology, Vol. 1229. Springer, New York. [PubMed: 25489643]
- Yan H, Yalagala RS, Yan F. 2015. Fluorescently labelled glycans and their applications. Glycoconj J 32: 559–574. [PubMed: 26239924]
- Review Structural Analysis of Glycans.[Essentials of Glycobiology. 2022]Review Structural Analysis of Glycans.Haslam SM, Freedberg DI, Mulloy B, Dell A, Stanley P, Prestegard JH. Essentials of Glycobiology. 2022
- Chromatographic Profiling of N-Glycans.[Methods Mol Biol. 2019]Chromatographic Profiling of N-Glycans.Gohlke M, Blanchard V. Methods Mol Biol. 2019; 1934:65-81.
- Chemical Structure and Composition of Major Glycans Covalently Linked to Therapeutic Monoclonal Antibodies by Middle-Down Nuclear Magnetic Resonance.[Anal Chem. 2018]Chemical Structure and Composition of Major Glycans Covalently Linked to Therapeutic Monoclonal Antibodies by Middle-Down Nuclear Magnetic Resonance.Peng J, Patil SM, Keire DA, Chen K. Anal Chem. 2018 Sep 18; 90(18):11016-11024. Epub 2018 Aug 27.
- Separation of N-glycans by HPLC.[Methods Mol Biol. 2008]Separation of N-glycans by HPLC.Gohlke M, Blanchard V. Methods Mol Biol. 2008; 446:239-54.
- Review Mass spectrometry of N-linked glycans.[Methods Mol Biol. 2009]Review Mass spectrometry of N-linked glycans.Azadi P, Heiss C. Methods Mol Biol. 2009; 534:37-51.
- Structural Analysis of Glycans - Essentials of GlycobiologyStructural Analysis of Glycans - Essentials of Glycobiology
Your browsing activity is empty.
Activity recording is turned off.
See more...