

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology [Internet]. 3rd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2015-2017. doi: 10.1101/glycobiology.3e.038


Essentials of Glycobiology [Internet]. 3rd edition.
Show details
Glycosaminoglycans bind to many different classes of proteins mostly through electrostatic interactions between negatively charged sulfate groups and uronic acids and positively charged amino acids in the protein. This chapter focuses on examples of glycosaminoglycan (GAG)-binding proteins, methods for measuring GAG–protein interaction, and information about three-dimensional structures of the complexes.
GAG-BINDING PROTEINS ARE COMMON
Several hundred GAG-binding proteins have been discovered, which make up the GAG-interactome and fall into the broad classes presented in Table 38.1. To a large extent, studies of the GAG-interactome have focused on protein interactions with heparin, a more highly sulfated, iduronic acid (IdoA)-rich form of heparan sulfate (HS; Chapter 17). This bias reflects, in part, the commercial availability of heparin and heparin-Sepharose, which are frequently used for fractionation studies, and the partially incorrect assumption that binding to heparin mimics binding to HS present on cell surfaces and in the extracellular matrix. In comparison, relatively few proteins are known to interact with chondroitin sulfate (CS) or keratan sulfate (KS) with comparable avidity and affinity. In some cases, CS and the related GAG, dermatan sulfate (DS), may be physiologically relevant binding partners because these GAGs predominate in many tissues. Determining the physiological relevance of these interactions is a major area of research.
TABLE 38.1.
Examples of glycosaminoglycan (GAG)-binding proteins and their biological activity
In contrast to lectins, which tend to fall into evolutionarily conserved families (Chapters 28–37), GAG-binding proteins do not have common folds and instead appear to have evolved by convergent evolution. Interactions between GAGs and proteins can have profound physiological effects on processes such as hemostasis, lipid transport and absorption, cell growth and migration, and development (Table 38.1). Binding to GAGs can result in immobilization of proteins at their sites of production or in the extracellular matrix for future mobilization, regulation of enzyme activity, binding of ligands to their receptors, protein oligomerization, and protection of proteins against degradation. In some cases, the interaction may reflect complementarity of charge (e.g., histone–heparin interactions) rather than any specific biologically relevant interaction. In other cases, the interaction has been shown to depend on rare but very specific sequences of modified sugars in the GAG chain (e.g., antithrombin binding).
METHODS FOR MEASURING GAG–PROTEIN BINDING
Numerous methods are available for analyzing GAG–protein interactions, and some provide a direct measurement of Kd values. A common method involves affinity fractionation of proteins on Sepharose columns containing covalently linked GAG chains, usually heparin. The bound proteins are eluted with different concentrations of sodium chloride, and the concentration required for elution is generally proportional to the Kd. High-affinity interactions require at least 1 m NaCl to displace bound ligand, which translates into Kd values of 10−7–10−9 m (determined under physiological salt concentrations by equilibrium binding). Proteins with low affinity (10−4–10−6 m) either do not bind under “normal” conditions (0.15 m NaCl) or require only 0.3–0.5 m NaCl to elute. This method is based on the assumption that GAG–protein interaction is entirely ionic, which is not entirely correct. Nevertheless, it can provide an assessment of relative affinity, when comparing different GAG-binding proteins.
A number of more sophisticated methods are now in use that provide detailed thermodynamic data (ΔH [change in enthalpy], ΔS [change in entropy], ΔCp [change in molar heat capacity], etc.), kinetic data (association and dissociation rates), and high-resolution data on atomic contacts in GAG–protein interactions (Table 38.2). Regardless of the technique one uses, it must be kept in mind that in vitro binding measurements are not likely to be the same as those when the protein binds to proteoglycans on the cell surface or in the extracellular matrix, where the density and variety of GAG-binding proteins, proteoglycans, and other interacting factors varies greatly. To determine the physiological relevance of the interaction, one should consider measuring binding under conditions that can lead to a biological response. For example, one can measure binding to cells with altered GAG composition (Chapter 49) or after treatment with specific lyases to remove GAG chains from the cell surface (Chapter 17) and then determine whether the same response occurs as observed in the presence of GAG chains. The interaction can then be studied more intensively using the in vitro assays described above.
TABLE 38.2.
Methods to measure glycosaminoglycan (GAG)–protein interaction
CONFORMATIONAL AND SEQUENCE CONSIDERATIONS
As mentioned above, most GAG-binding proteins interact with HS or heparin. The likely basis for this preference is greater sequence heterogeneity and more variable sulfation, compared with other GAGs. The unusual conformational flexibility of iduronic acid, which is found in heparin, HS, and DS, also has a role in their ability to bind proteins. GAGs are linear helical structures, consisting of alternating residues of N-acetylglucosamine (GlcNAc) or N-acetylgalactosamine (GalNAc) with glucuronic acid (GlcA) or IdoA (with the exception of KSs, which consist of alternating GlcNAc and galactose residues; Chapter 17). Inspection of heparin oligosaccharides containing highly modified domains ([GlcNS6S-IdoA2S]n) shows that the N-sulfo and 6-O-sulfo groups of each disaccharide repeat lie on opposite sides of the helix from the 2-O-sulfo and carboxyl groups (Figure 38.1). Analysis of the conformation of individual sugars shows that GlcNAc and GlcA residues assume a preferred conformation in solution, designated 4C1 (indicating that carbon 4 is above the plane defined by carbons 2, 3, and 5 and the ring oxygen, and that carbon 1 is below the plane; Chapter 2). In contrast, IdoA2S assumes the 1C4 or the 2SO conformation (Figure 38.1), which reorients the position of the sulfo substituents, thereby creating a different orientation of charged groups. In many cases when a protein binds to an HS chain, it induces a change in conformation of the IdoA2S residue resulting in a better fit and enhanced binding. IdoA2S residues have always been found in domains rich in N-sulfo and O-sulfo groups (for biosynthetic reasons; Chapter 17), which is also where proteins usually bind. Thus, the greater degree of conformational flexibility in these modified regions may explain why so many more proteins bind with high affinity to heparin, HS, and DS than to other GAGs. The presence of an N-acetyl group in a GlcNAc residue changes the preferred conformation of the neighboring IdoA2S residue, showing that even minor modifications can influence conformation and chain flexibility. Binding to GAGs that have a low degree of sulfation may require larger domains in the protein to interact with longer stretches of an oligosaccharide. Molecular dynamic simulations on large heparin oligosaccharides are possible with the availability of supercomputers (Online Appendix 38A). Such simulations can be used to predict the conformational flexibility of different domains within the chain and, when combined with recent advances in protein–GAG docking, can provide additional insights into GAG–protein interactions.
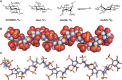
FIGURE 38.1.
Conformation of heparin oligosaccharides. (A) Glucosamine (GlcN) and glucuronic acid (GlcA) exist in the 4C1 conformation, whereas iduronic acid (IdoA) exists in equally energetic conformations designated 1C4 and 2S0. (B) Space-filling model of a heparin (more...)
HOW SPECIFIC ARE GAG–PROTEIN INTERACTIONS?
The discovery of multiple GAG-binding proteins led a number of investigators to examine whether there is a consensus amino acid sequence for GAG binding. In retrospect, this strategy was overly simplistic because it assumed that all GAG-binding proteins have a common evolutionary origin, and would recognize the same oligosaccharide sequence within heparin, or at least, sequences that would share many common features. It is now known that the convergently evolved GAG-binding proteins interact with different oligosaccharide sequences. The binding sites in the protein always contain basic amino acids (lysine and arginine), whose positive charges presumably interact with the negatively charged sulfates and carboxylates of the GAG chains. However, the arrangement of these basic amino acids can be quite variable, consistent with the variable positioning of sulfo groups in the GAG partner.
Most proteins are formed from α-helices, β-strands, and loops. Therefore, to engage a linear GAG chain, the positively charged amino acid residues must align along the same side of the protein segment. α-Helices have periodicities of 3.4 residues per turn, which would require the basic residues to occur every third or fourth position along the helix to align with an oligosaccharide. In β-strands, the side chains alternate sides every other residue. Thus, to bind a GAG chain, the positively charged residues in a β-strand would be located quite differently than in an α-helix.
On the basis of the structure of several heparin-binding proteins that were available in 1991, Alan Cardin and Herschel Weintraub proposed that typical heparin-binding sites had the sequence XBBXBX or XBBBXXBX, where B is lysine or arginine and X is any other amino acid. From the structural arguments provided above, it should be obvious that only some of the basic residues in these sequences could participate in GAG binding, the actual number being determined by whether the peptide sequence exists as an α-helix or a β-sheet. It is now known that the presence of these sequences in a protein merely suggests a possible interaction with heparin (or another GAG chain), but it does not prove that the interaction occurs under physiological conditions. In fact, the predicted binding sites for heparin in fibroblast growth factor 2 (FGF2) turned out to be incorrect once the crystal structure was determined. It is likely that binding involves multiple protein segments that juxtapose positively charged residues into a three-dimensional turn-rich recognition site. In many cases the binding involves loops which make the positioning more variable. An example of this phenomenon is observed in the chemokine CCL5, which contains a BBXB motif in a loop. The specific arrangement of residues should vary according to the type and fine structure of those oligosaccharides involved in binding.
In lectins, and in antibodies that recognize glycans, the glycan recognition domains are typically shallow pockets that engage the terminal sugars of the oligosaccharide chain (Chapters 29, 30, and 37). In GAG-binding proteins, the protein usually binds to sugar residues that lie within the chain or near the terminus. Therefore, the binding sites in GAG-binding proteins consist of clefts or sets of juxtaposed surface residues rather than pockets. These GAG-binding sites on the protein surface give rise to more rapid GAG–protein binding kinetics than are typically observed for protein–protein interactions. Given that GAG chains generally exist in a helical conformation, only those residues on the face toward the protein interact with amino acid residues; the ones on the other side of the helix are potentially free to interact with a second ligand (e.g., as observed in FGF dimers). Alternatively, residues in a binding cleft could interact with both sides of the helix (e.g., in dengue envelope protein). Finally, one should keep in mind that binding occurs to only a small segment of the GAG chain. Thus, a single GAG chain can potentially bind multiple protein ligands facilitating cooperative binding that can lead to protein oligomerization (e.g., some chemokines).
ANTITHROMBIN–HEPARIN: A PARADIGM FOR STUDYING GAG-BINDING PROTEINS
Perhaps the best-studied example of a protein–GAG interaction is the binding of antithrombin to heparin and HS (see cover image and Figure 38.2). This interaction is of great pharmacological importance because heparin is widely used clinically as an anticoagulant. Binding of antithrombin to heparin has a dual effect: first, it causes a conformational change in the protein and activation of the protease inhibiting action, resulting in a 1000-fold enhancement in the rate at which it inactivates thrombin and factor Xa. Second, the heparin chain acts as a template, enhancing the physical apposition of thrombin and antithrombin. Thus, both the protease (thrombin) and the inhibitor have GAG-binding sites. Heparin acts as a catalyst in these reactions by enhancing the rate of the reaction through apposition of substrates and conformational change. After the inactivation of thrombin by antithrombin occurs, the complex loses affinity for heparin and dissociates. The heparin is then available to participate in another activation/inactivation cycle. Antithrombin is a member of the serpin family of protease inhibitors, many of which bind to heparin.
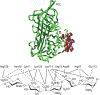
FIGURE 38.2.
(Top) Crystal structure of the antithrombin–pentasaccharide complex (from Protein Data Bank). A and D are α-helices that make contact with heparin; RCL is the reactive center loop that inactivates thrombin and factor X; and F is another (more...)
Early studies using affinity fractionation schemes showed that only about one-third of the chains in a heparin preparation actually bind with high affinity to antithrombin. Comparing the sequence of the bound chains with those that did not bind failed to reveal any substantial differences in composition, consistent with the later discovery that the binding site consists of only five sugar residues (Figure 38.2) (the average heparin chain is about 50 sugar residues). This observation can be extended to virtually all GAG-binding proteins, inferring that the binding sites represent a very small segment of the chains.
Crystals of antithrombin were prepared and analyzed by X-ray diffraction to 2.6-Å resolution. The docking site for the heparin pentasaccharide is formed by the apposition of helices A and D, which both contain critical arginine and lysine residues at the interface. The sequence in the D helix (124AKLNCRLYRKANKSSKLVSANR145) places many of the positively charged residues on one face of the helix, in proximity to the arginine residues in the A helix (41PEATNRRVW49) (Figure 38.2). The pentasaccharide is sufficient to activate antithrombin binding toward factor Xa, but it will not facilitate the inactivation of thrombin. For this to occur, a larger oligosaccharide of at least 18 residues is needed. As mentioned above, thrombin also contains a heparin-binding site, and the larger heparin oligosaccharide is thought to act as a template for the formation of a ternary complex with thrombin and antithrombin. In contrast to antithrombin, thrombin shows little oligosaccharide specificity. As might be expected, adding high concentrations of heparin actually inhibits the reaction, because the formation of binary complexes of heparin and thrombin or heparin and antithrombin predominate. This important principle of “activation at low concentrations and inhibition at high concentrations” also occurs in other systems in which ternary complexes form (Chapters 29 and 30).
Heparin is a pharmaceutical formulation produced by partial fractionation of natural GAGs derived primarily from porcine intestines or bovine lungs (Chapter 17). Mast cells are known to produce a highly sulfated version of HS that resembles heparin; highly sulfated, iduronic acid–rich heparin oligosaccharides are also present in HS isolated from other tissues as well, especially the skin. Although heparin has proven to be of great therapeutic use, its role in vivo remains unclear. Heparin and CS are often found in storage granules along with biogenic amines, proteases, and other proteins, possibly enabling efficient storage. Mast cells degranulate in response to specific antigen stimulation, resulting in release of stored heparin, histamine, and proteases. When this occurs, local anticoagulation might occur, but localized coagulation defects have not been described in animals bearing mutations that alter mast cells or heparin. A small percentage of endothelial cell HS contains antithrombin-binding sequences as well. However, these binding sites appear to be located on the abluminal side of blood vessels, and mice lacking the central 3-O-sulfated GlcNS unit, a hallmark of the antithrombin-binding sequence (Figure 38.2), do not show any systemic coagulopathy after birth. Nevertheless, antithrombin deficiency causes massive disseminated coagulopathy. Perhaps these findings indicate that lower-affinity binding sequences are sufficient to activate antithrombin. This system illustrates an important caveat: one cannot necessarily ascribe functions to endogenous proteoglycans based on the effects of GAGs added in vitro to experimental systems.
FGF–HEPARIN INTERACTIONS ENHANCE STIMULATION OF FGF RECEPTOR SIGNAL TRANSDUCTION
A large number of growth factors can be purified based on their affinity for heparin. The heparin-binding family of FGFs has grown to more than 22 members and includes the prototype FGF2, otherwise known as basic fibroblast growth factor. FGF2 has a very high affinity for heparin (Kd ∼ 10−9 m) and requires 1.5–2 m NaCl to elute from heparin-Sepharose. FGF2 has potent mitogenic activity in cells that express one of the FGF signaling receptors (FGFRs; four FGFR genes are known and multiple splice variants exist). Cell-surface HS binds to both FGF2 and FGFR, facilitating the formation of a ternary complex. Both binding and the mitogenic response are greatly stimulated by heparin or HS, which promotes dimerization of the ligand–receptor complex.
The costimulatory role of HS (and heparin) in this system is reminiscent of the heparin/antithrombin/thrombin story. Indeed, the minimal binding sequence for FGF2 also consists of a pentasaccharide. However, this pentasaccharide is not sufficient to trigger a biological response (mitogenesis). For this to occur, a longer oligosaccharide (10-mer) containing the minimal sequence and additional 6-O-sulfo groups are needed to bind FGFR. The sequence that binds to both FGF2 and FGFR is prevalent in heparin but rare in HS. The requirement for this rare binding sequence reduces the probability of finding this particular arrangement in naturally occurring HS chains. Thus, some preparations of HS are inactive in mitogenesis, and those containing only one-half of the bipartite binding sequence are actually inhibitory.
The structure of FGF2 cocrystallized with a heparin hexasaccharide has since been obtained (Figure 38.3). The heparin fragment ([GlcNS6Sα1-4IdoA2Sα1-4]3) was helical and bound to a turn-rich heparin-binding site on the surface of FGF2. Only one N-sulfo group and the 2-O-sulfo group from the adjacent iduronic acid are bound to the growth factor in the turn-rich binding domain, and the next GlcNS residue is bound to a second site, consistent with the minimal binding sequence determined with oligosaccharide fragments. No significant conformational change in FGF2 occurs on heparin binding, consistent with the idea that heparin primarily serves to dimerize FGF2 and juxtapose components of the FGF signal-transduction pathway. The crystal structure of acidic FGF (FGF1) has also been solved and shows similar sequences on its surface. However, the oligosaccharide sequence that binds with high affinity to FGF1 contains 6-O-sulfo groups.
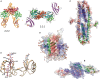
FIGURE 38.3.
Crystal and nuclear magnetic resonance (NMR) solution structures of GAG–protein complexes. (A) Crystal structure of the 2:2:2 FGF2:FGFR1:heparin complex (side view) and a 2:2:1 complex. (B) Structure of the dimeric V-C1 domains of RAGE (receptor (more...)
The cocrystal structure of the complex of (FGF2-FGFR)2, first solved in the absence of heparin/HS ligand, showed a canyon of positively charged amino acid residues, suggestive of an unoccupied heparin-binding site. Subsequently, the heparin–oligosaccharide-containing complex was solved after introduction of heparin oligosaccharides, suggesting a 2:2:2 complex of FGF2:FGFR:HS (Figure 38.3). Another important feature of this complex is the orientation of the nonreducing ends of the HS chains that terminate in an N-sulfoglucosamine residue, which arises by endolytic cleavage of chains by the enzyme heparanase (Chapter 17). The structure of the FGF–FGFR–HS complex is not without controversy; structural analysis of complexes formed in solution and purified by gel filtration has suggested a very different structure consisting of a 2:2:1 complex (Figure 38.3).
OTHER ATTRIBUTES OF GAG–PROTEIN INTERACTIONS
In some cases, the interaction of GAG chains with proteins may depend on metal cofactors. For example, L- and P-selectins have been shown to bind to a subfraction of HS chains and heparin in a divalent-cation-dependent manner. This observation raises the possibility that other examples of cation-dependent interactions with GAG chains may exist. GAG binding to L-selectin helps in leukocyte rolling. Furthermore, the interaction can be pharmacologically inhibited by exogenous heparin at clinically acceptable doses, and also by chemically modified derivatives that lack anticoagulant activity.
CS proteoglycans in the central nervous system (CNS) influence cell migration and axon pathfinding and regulate neurite outgrowth. The interaction of rare, highly sulfated disaccharide sequences in CS chains with morphogens and growth factors impact CNS development and play roles in CNS pathology.
HS proteoglycans are often expressed in a spatially and temporally limited fashion. The temporary placement of an HS proteoglycan at a specific tissue site might or might not coincide with the presence of its appropriate protein ligand. Furthermore, if the binding partner has no access to the HS proteoglycan, it cannot interact—adding an additional level of specificity. Recent studies show that the fine structure of HS chains also changes during development, thus enabling or disabling specific associations between ligands and receptors.
Gradients of morphogens, factors that determine cell fates based on concentration, also determine the patterns of cell and tissue organization during development (Chapters 25–27). The mechanism of morphogen gradient formation is controversial, but interestingly, virtually all morphogens can interact with heparin and HS. These interactions can affect transport of ligands, receptor interactions, endocytosis, and degradation, which together may have a role in determining the robustness of the gradient. The GAG chains of proteoglycans also offer a linear domain over which proteins can diffuse. By limiting the space available to these proteins from the three-dimensional space of extracellular fluids and the extracellular matrix to one-dimensional space along the chains, the chance of encounters among heparin-binding proteins, such as FGF and its receptor (FGFR), may be enhanced. Thus, the critical role of HS proteoglycans may be in controlling the kinetics of protein–protein interactions rather than the thermodynamics of such encounters.
ACKNOWLEDGMENTS
The authors appreciate helpful comments and suggestions from Kristian Saied-Santiago, Eathen Ryan, Patience Sanderson, and Kristin Stanford.
FURTHER READING
- Li W, Johnson DJ, Esmon CT, Huntington JA. 2004. Structure of the antithrombin–thrombin–heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat Struct Mol Biol 11: 857–862. [PubMed: 15311269]
- Mohammadi M, Olsen SK, Goetz R. 2005. A protein canyon in the FGF–FGF receptor dimer selects from an a la carte menu of heparan sulfate motifs. Curr Opin Struct Biol 15: 506–516. [PubMed: 16154740]
- Duchesne L, Octeau V, Bearon RN, Beckett A, Prior IA, Lounis B, Fernig DG. 2012. Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol 10: e1001361. [PMC free article: PMC3398970] [PubMed: 22815649]
- Kamhi E, Joo EJ, Dordick JS, Linhardt RJ. 2013. Glycosaminoglycans in infectious disease. Biol Rev Camb Philos Soc 88: 928–943. [PubMed: 23551941]
- Thacker BE, Xu D, Lawrence R, Esko JD. 2014. Heparan sulfate 3-O-sulfation: A rare modification in search of a function. Matrix Biol 35: 60–72. [PMC free article: PMC4039620] [PubMed: 24361527]
- Xu D, Esko JD. 2014. Demystifying heparan sulfate–protein interactions. Annu Rev Biochem 83: 129–157. [PMC free article: PMC7851832] [PubMed: 24606135]
- Deshauer C, Morgan AM, Ryan EO, Handel TM, Prestegard JH, Wang X. 2015. Interactions of the chemokine CCL5/RANTES with medium-sized chondroitin sulfate ligands. Structure (London, England: 1993) 23: 1066–1077. [PMC free article: PMC4456249] [PubMed: 25982530]
- Mizumoto S, Yamada S, Sugahara K. 2015. Molecular interactions between chondroitin-dermatan sulfate and growth factors/receptors/matrix proteins. Curr Opin Struct Biol 34: 35–42. [PubMed: 26164146]
- Pomin VH, Mulloy B. 2015. Current structural biology of the heparin interactome. Curr Opin Struct Biol 34: 17–25. [PubMed: 26038285]
- Smith PD, Coulson-Thomas VJ, Foscarin S, Kwok JC, Fawcett JW. 2015. “GAG-ing with the neuron”: The role of glycosaminoglycan patterning in the central nervous system. Exp Neurol 274: 100–114. [PubMed: 26277685]
- GAG-BINDING PROTEINS ARE COMMON
- METHODS FOR MEASURING GAG–PROTEIN BINDING
- CONFORMATIONAL AND SEQUENCE CONSIDERATIONS
- HOW SPECIFIC ARE GAG–PROTEIN INTERACTIONS?
- ANTITHROMBIN–HEPARIN: A PARADIGM FOR STUDYING GAG-BINDING PROTEINS
- FGF–HEPARIN INTERACTIONS ENHANCE STIMULATION OF FGF RECEPTOR SIGNAL TRANSDUCTION
- OTHER ATTRIBUTES OF GAG–PROTEIN INTERACTIONS
- ACKNOWLEDGMENTS
- FURTHER READING
- Review Proteins That Bind Sulfated Glycosaminoglycans.[Essentials of Glycobiology. 2022]Review Proteins That Bind Sulfated Glycosaminoglycans.Xu D, Prestegard JH, Linhardt RJ, Esko JD. Essentials of Glycobiology. 2022
- Review Proteins that Bind Sulfated Glycosaminoglycans.[Essentials of Glycobiology. 2009]Review Proteins that Bind Sulfated Glycosaminoglycans.Esko JD, Linhardt RJ. Essentials of Glycobiology. 2009
- The chemical modification of glycosaminoglycan structure by oxygen-derived species in vitro.[Biochim Biophys Acta. 1995]The chemical modification of glycosaminoglycan structure by oxygen-derived species in vitro.Moseley R, Waddington R, Evans P, Halliwell B, Embery G. Biochim Biophys Acta. 1995 Jun 9; 1244(2-3):245-52.
- Review Specificity of glycosaminoglycan-protein interactions.[Curr Opin Struct Biol. 2018]Review Specificity of glycosaminoglycan-protein interactions.Kjellén L, Lindahl U. Curr Opin Struct Biol. 2018 Jun; 50:101-108. Epub 2018 Feb 9.
- A sulfated glycosaminoglycan array for molecular interactions between glycosaminoglycans and growth factors or anti-glycosaminoglycan antibodies.[Anal Biochem. 2013]A sulfated glycosaminoglycan array for molecular interactions between glycosaminoglycans and growth factors or anti-glycosaminoglycan antibodies.Takada W, Fukushima M, Pothacharoen P, Kongtawelert P, Sugahara K. Anal Biochem. 2013 Apr 15; 435(2):123-30. Epub 2013 Jan 16.
- Proteins That Bind Sulfated Glycosaminoglycans - Essentials of GlycobiologyProteins That Bind Sulfated Glycosaminoglycans - Essentials of Glycobiology
Your browsing activity is empty.
Activity recording is turned off.
See more...