

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology. 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2009.


Essentials of Glycobiology. 2nd edition.
Show details
Pure glycans of defined structure are essential research tools in glycobiology. Unlike proteins and nucleic acids, which can be obtained in homogeneous forms using biological methods such as recombinant expression and polymerase chain reaction (PCR), glycans produced in biological systems are heterogeneous. Furthermore, the quantities that can be obtained from biological systems are rather small. Chemical synthesis can be used to obtain homogeneous glycans in larger quantities than are available from most cellular production systems. Enzymes can be used together with chemical methods to further simplify the process of glycan synthesis. Chemical synthesis can be further employed to incorporate glycans into homogeneous glycoproteins. This chapter summarizes the chemical and enzymatic synthetic methods to produce glycans and glycoproteins.
CHEMICAL SYNTHESIS OF GLYCANS
Controlling Regiochemistry
The inherent chemical properties of oligosaccharides make their synthesis more complex than the other major classes of biomolecules (i.e., oligonucleotides and oligopeptides). The fundamental challenge of glycan synthesis is the requirement for modifying one specific hydroxyl group in the presence of many others. Therefore, glycan synthesis is characterized by the manipulation of various protecting groups, chemical moieties that mask the hydroxyl groups and prevent them from reacting with other chemical reagents. Hydroxyl-protecting groups are selectively added and removed from glycan structures, allowing for chemical alteration of the exposed hydroxyl groups. Subsequently, in a typical glycan synthetic scheme, the exposed hydroxyl group serves as a point for further elaboration. The selective exposure of one hydroxyl group allows for regioselective addition of another monosaccharide unit. Synthetic schemes of this type are commonly applied to the generation of O- and N-linked glycans (see Chapters 8 and 9) as well as proteoglycans (see Chapter 16) and glycosphingolipids (see Chapter 10).
The choice of protecting groups and the sequence of protecting group installation are essential for a synthetic route to be successful. The most common protecting groups include persistent protecting groups, such as benzyl ethers that stay in place through many synthetic steps, and temporary protecting groups, such as esters that are removed during intermediate steps in a synthesis. A wealth of information exists on the chemical generation of glycans and various protecting group manipulations used in the context of glycan synthesis. The reviews listed at the end of the chapter provide a comprehensive overview of this subject.
Controlling Stereochemistry
A glycosidic linkage is formed through the activation of a glycosyl donor to create a reactive electrophilic species that couples with the glycosyl acceptor’s hydroxyl group. This coupling reaction results in either the formation of an α- or β-linkage (see Chapter 2). A synthetic challenge in glycan synthesis is the stereospecific formation of glycosidic bonds (Figure 49.1A; Chapter 2). There are a variety of methods available to generate stereospecific glycosidic linkages. The yield and the stereochemical outcome of these reactions depend on the steric and electronic nature of the glycosylating agent (termed the glycosyl donor) and the nucleophilic hydroxyl group on the glycosyl acceptor. Good reaction yields are considered to range from 60% to 95%, and the ratios of the linkages at the anomeric position (α:β) vary from complete selectivity (all α or all β) to equal mixtures. A current method used to control the stereochemistry at the anomeric center involves the use of certain protecting groups, such as ester moieties, on the 2-hydroxyl group (Figure 49.1B). Under glycosylation reaction conditions, these “neighboring protecting groups” form a cyclic oxonium ion intermediate that shields one face of the molecule, leading exclusively to the formation of a “trans” glycosidic linkage (i.e., where the anomeric substituent and the C-2 hydroxyl group are on opposite sides of the ring, as in β-glucosides). The opposite anomeric stereochemistry, termed a cis glycosidic bond, is more difficult to construct with high specificity because neighboring group participation is not possible. In the absence of such protecting group participation, mixtures of anomers are often formed.
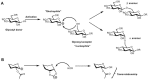
FIGURE 49.1
(A) Stereospecific formation of glycosidic bonds as either an α or β linkage. (LG) Leaving group. (B) Formation of a cyclic oxonium ion intermediate leading to the formation of a β-glycosidic linkage.
A Representative Chemical Glycan Synthesis
The synthesis of the tumor-associated glycan antigen Lex-Ley is one example of the construction of a complex glycan. Figure 49.2 illustrates the synthetic scheme followed for the preparation of this nonasaccharide and depicts the transformations required in a typical glycan synthetic procedure. These synthetic routes are often time consuming and technically challenging. Indeed, the synthesis of Lex-Ley took many months and the overall yield was less than 1%. This synthetic scheme also highlights a major difficulty with glycan synthesis—that is, isolating glycans via chromatography after individual coupling steps. Because of the low yields and complex purifications, traditional methods are not amenable to rapid and efficient synthesis of glycans.

FIGURE 49.2
Various transformations in a typical complex glycan synthesis. (P) Protecting group; (R) P or H; (R′) nitrogen-protecting group; (R*) unique hydroxyl-protecting group and fucosylation site.
AUTOMATED METHODOLOGY FOR GLYCAN SYNTHESIS
Unlike proteins and nucleic acids, for which automated synthesizers are commercially available, the glycans cannot yet be synthesized using a general commercial system. However, a number of methodologies are currently being developed by synthetic chemists to address the inherent challenges in glycan chemistry that have prohibited automation. Two different approaches to the automation of glycan assembly are presented in this chapter: (1) the one-pot solution-phase synthesis and (2) the solid-phase synthesis method.
One-pot Glycan Synthesis
The one-pot approach uses a computer program to automate glycan synthesis. This strategy exploits differences in the anomeric reactivity of a large set of diverse thioglycoside building blocks. The thioglycoside possesses an –SR group at the anomeric center, rather than the natural –OR group. These derivatives are stable under ambient conditions, but they can be converted to reactive leaving groups using reagents that interact with the thioether functionality (i.e., N-iodosuccinimide and dimethylthiosulfonium triflate).
More than 400 thioglycoside analogs with a wide spectrum of protecting groups on the monosaccharide hydroxyl groups have been prepared and characterized with respect to their relative reactivity. This study produced a table of relative reactivity values (RRVs) that could be programmed into a computer. A computer program, Optimer, allows the user to input the desired glycan structure, and, in return, the program displays an appropriate combination of building blocks needed to generate that glycan. The program also dictates the stereochemistry as α- or β-directing. Once Optimer provides the synthetic blueprint, the chemist proceeds to follow the procedure by sequential addition of the building blocks to a single reaction vessel. Typically, the building blocks are added starting from the most reactive and ending with the least reactive. The final glycan is globally deprotected and purified.
The assembly of the cancer antigen Globo H, a hexasaccharide, illustrates the power of this streamlined approach (Figure 49.3). The key advantage to using a one-pot approach is the rapid planning and ordering of the synthetic steps. In addition, a programmable one-pot synthesis eliminates intermediate purification steps utilized in traditional synthetic schemes. Depending on the glycan, the entire process of one-pot synthesis can take from minutes to days. However, the solution one-pot approach requires the availability of a large number of monosaccharide building blocks. These building blocks are not yet commercially available, and expertise in chemical synthesis is required to generate them.

FIGURE 49.3
Synthesis of complex glycan structures using one-pot methodology. (PG) Protecting group; (LG) leaving group.
Solid-phase Glycan Synthesis
A major breakthrough in peptide synthesis was the development of solid-phase synthetic methodology. This development in synthetic chemistry made peptides available to biologists and facilitated studies of their structures and functions. Likewise, the availability of a general solid-phase method for glycan synthesis could vastly accelerate access to homogeneous material for biological studies. Progress toward this end is well under way.
The first automated glycan synthesizer was constructed by modifying a peptide synthesizer, and the synthetic process closely resembles protocols established for the generation of polypetides (Figure 49.4). The first monosaccharide is attached through its reducing end via a cleavable linker to a polystyrene bead. Upon removal of a temporary protecting group, the hydroxyl group to be modified in the coupling reaction is exposed. Addition of a glycosyl donor together with an activator, typically a reagent that generates a good leaving group at the anomeric position, produces the new glycosidic linkage. An excess amount of the glycosyl donor can be used to drive the reaction to completion. Side products, reagents, and unreacted starting material are washed away while the desired glycan remains covalently attached to the polymer support. Next, another protecting group is selectively removed to provide another site for subsequent monosaccharide addition.
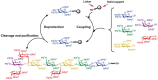
FIGURE 49.4
Solid-phase glycan synthesis using a peptide synthesizer to generate the carbohydrate antigen Lex-Ley. (PG) Protecting group; (LG) leaving group.
The assembly of the tumor-associated antigen Lex-Ley using a peptide synthesizer and glycosyl phosphate building blocks illustrates the utility of the solid-phase approach (Figure 49.4). In contrast to the solution-phase synthesis of Lex-Ley, which took months (see above), solid-phase synthesis of the nonasaccharide took less than 2 days, including cleavage of fully protected material from the solid support. Removal of the protecting groups required an additional 3 days.
The major advantage of solid-phase synthesis is that excess reagents can be used to drive each reaction to completion without the worry of purifying these reagents from the biopolymer, which is attached to solid support. Similar to the one-pot synthetic approach, only one purification step is required (after the polymer is liberated from the solid phase), which is often the most time-consuming aspect of glycan assembly. The bottleneck of automated solid-phase synthesis is the need for protected monosaccharide building blocks that are not yet commercially available. This issue will need to be addressed in order for solid-phase glycan synthesis to achieve the generality and accessibility of solid-phase peptide synthesis.
ENZYMATIC SYNTHESIS OF GLYCANS
Nature has addressed the challenges of glycan synthesis by evolving enzymes that couple monosaccharides with exquisite regio- and stereospecificity. These enzymes can be harnessed by chemists to catalyze the formation of glycosidic bonds in a synthetic context (see Chapter 5 for an introduction to glycosyltransferases). In addition to glycosyltransferases, glycosidases whose natural function is the cleavage of glycosidic bonds (see Chapters 5 and 41) may be manipulated to “run in reverse“ and function as glycosylating enzymes.
Employing glycosyltransferases and glycosidases in a synthetic scheme has strengths and weaknesses that are in many ways complementary to those of chemical synthesis. Chemical synthesis allows for the preparation of diverse natural and unnatural structures but requires the extensive use of protecting groups and the preparation of specialized precursor compounds. In contrast, enzymes are typically much less flexible and/or available but do not require protecting groups or elaborate precursors, and they construct glycosidic linkages with perfect regio- and stereochemical control. Many successful applications of enzymes in glycan production have utilized “chemoenzymatic” syntheses, relying on a hybrid of chemical and enzymatic steps that typically begins with chemical synthesis and ends with enzymatic extension.
Synthesis of Glycans Using Glycosyltransferases
The majority of glycosyltransferases used in glycan synthesis catalyze the transfer of a glycosyl donor to a sugar or amino acid acceptor. These transferases generally utilize nucleotide diphosphate sugar donors (e.g., UDP-GlcNAc), with occasional exceptions such as sialyl-transferases, which require CMP-sialic acids. The glycosyltransferases are subdivided into two classes termed retaining and inverting (see Chapter 5). Retaining glycosyltransferases transfer the donor with retention of stereochemistry at the anomeric position, whereas inverting glycosyltransferases transfer the donor with inversion at the anomeric position. A representative example is MurG, the inverting GlcNAc transferase responsible for the final step in bacterial cell wall biosynthesis (Figure 49.5; see Chapter 20). Notable aspects of this glycosylation event are the regiospecificity (only a single hydroxyl group is glycosylated) and complete control of anomeric stereochemistry (only the β-anomer is produced).

FIGURE 49.5
Example of an inverting GlcNAc transferase.
Figure 49.6 shows a comparison of the enzymatic and chemical synthesis of tetra- and pentasaccharide cell-surface epitopes from Neisseria meningitidis (a causative agent of meningitis). The enzymatic synthesis of the pentasaccharide (Figure 49.6A) relies on the use of three recombinant enzymes: a GlcNAc transferase, a fusion protein combining UDP-Glc epimerase and Gal transferase activities, and a fusion protein combining CMP-Neu5Ac synthase and transferase activities. A lactose derivative was carried through three sequential reactions, all giving remarkable yield. The GlcNAc transfer (96% yield) was followed by in situ epimerization of UDP-Glc to UDP-Gal, Gal transfer (96% yield), in situ synthesis of CMP-Neu5Ac from CTP and Neu5Ac, and subsequent sialylation producing the final pentasaccharide (97% yield).

FIGURE 49.6
A comparison of the enzymatic (A) and chemical (B) syntheses of tetra- and pentasaccharide glycans. (PG) Protecting group; (LG) leaving group.
For comparison, a chemical synthesis of the tetrasaccharide began with the same lactose derivative (Figure 49.6B). Conversion to a suitably protected lactose-based glycosyl acceptor in three steps gave reasonable (55%) yield. Subsequent coupling with a lactose-based glycosyl donor (available in nine steps from lactose) generated the protected tetrasaccharide in modest (40%) yield. The fully deprotected tetrasaccharide was obtained after two further steps (66% yield). In this example, the tetrasaccharide was not sialylated because of difficulties with the final chemical transformations. This is a significant limitation because sialic acid appears as a terminal sugar of many mammalian glycans. However, the synthesis of complex glycans with terminal sialyl residues can be readily achieved using sialyltransferases. Overall, the enzymatic procedure was far superior to the chemical counterpart.
The potential advantages of glycan synthesis using glycosyltransferases are immediately obvious. The reactions give high yields, with complete regio- and stereospecificity, without the need for installation and removal of numerous protecting groups. However, glycosyl-transferases have a number of significant limitations. First is the issue of availability. All of the enzymes used in the above synthesis were discovered, cloned, overexpressed, and purified in the laboratory and would not otherwise be readily available. However, although obtaining enzymes can be problematic, many glycosyltransferases are currently available from commercial sources and a growing number of academic labs. A second limitation is the regio- and stereospecificity of the enzymes. Glycosyltransferases generally carry out one reaction nearly perfectly, but they are unable to tolerate even minor variations in donor or acceptor (one exception will be noted below). This means that every novel synthetic transformation requires a new enzyme to be isolated, cloned, and purified. In contrast, although chemical methods are seldom as selective or high yielding, they may be applied to the preparation of a wide range of glycans. A third limitation of glycosyltransferases is that their nucleotide sugar substrates are often unstable and/or very expensive (e.g., CMP-Neu5Ac). Although this problem can be addressed by in situ nucleotide sugar generation, such reactions increase the complexity of the enzymatic process. Finally, many glycosyl-transferases are strongly inhibited by the nucleotide generated following sugar transfer to the acceptor. This problem can be addressed by using excess nucleotide sugar, enzymatic degradation of the nucleotide by an added phosphatase, or nucleotide recycling in which the nucleotide is enzymatically converted back into the nucleotide sugar.
Generating large amounts of a desired glycan with enzymatic reactions is also problematic. Although chemical processes can be carried out on gram or even multiton scales, enzymes are scarce and/or too expensive to be feasible alternatives. However, the issue is ameliorated to some extent because most research applications require only small amounts (10–100 mg) of the glycan of interest, and once the challenges associated with enzyme identification and overexpression have been addressed, enzymatic syntheses can be viable on a large scale. This is illustrated by the preparation of a trisaccharide related to the N. meningitidis epitopes described above, which was carried out on an approximately 100-g scale in a manner that allowed reisolation and reuse of the enzyme (Figure 49.7). The cost and stability of the nucleotide sugar were addressed by in situ regeneration of the requisite CMP-Neu5Ac, and the complexity of the system was reduced by use of a fusion protein containing two enzyme activities. Smaller-scale experiments with the same fusion enzyme demonstrated some tolerance for variation of both the donor and acceptor components of the reaction. Additionally, Galβ1-4GlcNAcβ-(Fl), Galβ1-4Glcβ-(Fl), and Galα1-4Galβ1-4Glcβ-(Fl) (where Fl is a fluorescent tag) were all found to be effective substrates for glycosylation. In addition, the enzyme was found to transfer Neu5Propyl and Neu5Gc as effectively as Neu5Ac. So far, enzymatic synthesis has proved to be the only viable and cost-effective approach for the commercial-scale preparation of sialyl Lewisx (Chapter 6).

FIGURE 49.7
Example of multigram enzymatic synthesis.
Synthesis of Glycans Using Glycosidases
Glycosidases provide an alternative to the use of glycosyltransferases for the enzymatic synthesis of glycans. Glycosidases usually cleave glycans into smaller fragments or monosaccharides (see Chapter 5). They are separated into two categories: endoglycosidases, which cleave internal glycosidic bonds of glycans, and exoglycosidases, which cleave of the nonreducing terminal sugar of a glycan. A representative example of an exoglycosidase is the β-N-acetylhexosaminidase involved in a late stage of hyaluronan degradation (Figure 49.8).

FIGURE 49.8
Example of an exoglycosidase.
In general, glycosidases are more stable and readily available than glycosyltransferases and are also more tolerant to variations in substrate structure. To use glycosidases for synthetic purposes, their normal function must be reversed. All enzymatic processes are (formally) equilibrium processes. It is, in principle, possible to force a glycosidase to run in reverse by exposing the enzyme to a large excess of the reaction products and allowing the system to reach equilibrium. However, because the reverse reaction is invariably endothermic, the equilibrium will always favor the cleavage products. Replacing the anomeric hydroxyl group of the glycosyl donor fragment with a good leaving group, such as para-nitrophenol (PNP), shifts the equilibrium further toward the glycosylation product. Figure 49.9 illustrates the glycosidase-mediated synthesis of two sialyl-Tn antigen epitopes. Beginning with the methyl ester of GalNAcα-N-Ac-threonine, treatment with Gal-β-PNP in the presence of the β-galactosidase from bovine testes provides the corresponding disaccharide in modest yield (22%) with complete regio- and stereoselectivity. Subsequent treatment with the PNP derivative of sialic acid (Neu5Ac α-PNP) in the presence of Vibrio cholerae sialidase provided the α2-6-linked sialylated product in low yield but with excellent regio- and stereoselectivity (only trace amounts of isomeric products could be detected). Likewise, Salmonella typhimurium sialidase provided the α2-3-linked trisaccharide, again in low yield, but with high regio- and stereoselectivity.

FIGURE 49.9
The glycosidase-mediated synthesis of two sialyl-Tn antigen epitopes.
Oligosaccharide synthesis with glycosidases retains three of the advantages associated with glycosyltransferases—that is, good-to-excellent regio- and stereoselectivities without extensive protecting group manipulation. Additional benefits include greater stability and accessibility of glycosidases and greater tolerance to variations in substrate structure. Although the yields of glycosidase-based reactions are lower than glycosyltransferase-catalyzed processes, there are transformations for which the requisite glycosyltransferase is not available. In such cases, the use of glycosidases can complement transferase-based methods for synthesis.
SYNTHESIS OF GLYCOCONJUGATES
In nature, glycans are often found in the context of glycoproteins. The heterogeneity of these protein-associated glycans has interfered with protein structure elucidation and complicated attempts to determine the effects of the glycan on protein function. Therapeutic glycoproteins, the fastest growing class of pharmaceutical reagents, are reliant on their glycans for pharmacokinetic properties and stability. The biological and biophysical study of glycoproteins could benefit tremendously from the availability of homogeneously glycosylated material. With the development of improved methods for glycan synthesis, chemists have recently taken on the challenge of synthesizing whole glycoproteins using combinations of chemical and enzymatic methods.
Solid-phase Glycopeptide Synthesis
Short polypeptides (<50 residues) can be readily prepared by solid-phase peptide synthesis (SPPS). Glycans can be incorporated through the glycosylation of amino acid building blocks prior to synthesis of the polypeptide biopolymer. The glycosylated amino acid is often referred to as a “cassette.” The hydroxyl groups of the glycan component are typically protected as acetyl or benzoyl esters, which are easily removed with base after the completion of peptide assembly. Cassette strategies have been widely used in the synthesis of glycopeptides from mucin-type glycoproteins (Chapter 9) and can be complemented by both the one-pot and solid-phase glycan synthesis methodologies.
As mentioned earlier in the chapter, glycosyltransferases are powerful tools for the construction of defined carbohydrate structures. The enzymatic transfer of individual monosaccharides to preformed glycopeptides is an excellent alternative to the synthesis of large glycosyl–amino acids. After glycopeptide synthesis using a glycosyl–amino acid cassette, the glycan is deprotected and then elaborated with glycosyltransferases. The synthesis of a PSGL-1 fragment (see Chapter 31) is an elegant example of using both chemical and enzymatic methodologies to generate a complex glycopeptide (Figure 49.10). The peptide backbone was generated following solid phase peptide synthetic procedures with the N-acetyl-galactosidase residue incorporated using a cassette strategy. Following the cleavage and deprotection of the glycopeptide, the appropriate glycosyltransferases were used to elaborate the glycan to generate the desired hexasaccharide. Sulfation was also carried out enzymatically to sulfate the tyrosine amino acid residues, generating the final desired compound. Thus, by employing a synthetic cassette strategy in tandem with enzymatic elaboration, a complex glycoconjugate was synthesized that would otherwise be prohibitively difficult to generate.
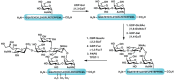
FIGURE 49.10
The chemoenzymatic synthesis of a PSGL-1 glycopeptide fragment. PAPS, 3′-phosphoadenosine 5′-phosphosulfate; TPST-1; tyrosylprotein sulfotransferase 1.
Synthesis of Glycopeptides by Native Chemical Ligation
The synthesis of larger glycoproteins (i.e., >50 amino acid residues) cannot be achieved using this linear synthetic strategy. The field of protein chemistry has provided a convergent approach to the synthesis of larger proteins, termed native chemical ligation (NCL) (Figure 49.11). This technique involves the condensation of two unprotected peptide fragments, one bearing a carboxy-terminal thioester and the other an amino-terminal cysteine residue. The reaction is mild, selective, and compatible with the presence of glycans on the fragments. Thus, SPPS with glycosyl–amino acid cassettes can be used to generate glycopeptide fragments, which are then assembled by NCL into larger glycoproteins.
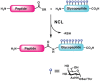
FIGURE 49.11
Native chemical ligation (NCL).
The Semi-synthesis of Glycopeptides by Expressed Protein Ligation
The synthesis of proteins larger than 20 kD in size using native chemical ligation is extremely difficult given the limitations on the size of the fragments that are made by SPPS. To access glycoproteins beyond this size, expressed protein ligation (EPL) has been artfully employed (Figure 49.12). This method generates recombinant proteins with carboxyterminal thioesters using the natural phenomenon of intein-mediated protein splicing. Thus, a recombinant protein bearing a carboxy-terminal thioester can be coupled by the NCL reaction to a synthetic glycopeptide adorned with an amino-terminal cysteine residue. Alternatively, a recombinant protein encoding an amino-terminal cysteine residue can be ligated to a synthetic glycopeptide functionalized with a carboxy-terminal thioester. This modular combination of recombinant technology and chemical synthesis has produced full-length glycoproteins with homogeneous glycans.

FIGURE 49.12
General approach to glycoprotein synthesis using expressed protein ligation (EPL). (A) Assembly of glycoproteins bearing glycans near the amino terminus. (B) Assembly of glycoproteins bearing glycans near the carboxyl terminus.
FURTHER READING
- Hanessian S. Preparative carbohydrate chemistry. Marcel Dekker; New York: 1997.
- Wellings DA, Atherton E. Standard Fmoc protocols. Methods Enzymol. 1997;289:44–67. [PubMed: 9353717]
- Koeller KM, Wong C.-H. Synthesis of complex carbohydrates and glycoconjugates: Enzyme-based and programmable one-pot strategies. Chem Rev. 2000;100:4465–4493. [PubMed: 11749355]
- Vocadlo DJ, Withers SG. Glycosidase-catalyzed oligosaccharide synthesis. Carbohydrates Chem Biol. 2000;2:723–844.
- Marcaurelle LA, Bertozzi CR. Recent advances in the chemical synthesis of mucin-like glyco-proteins. Glycobiology. 2002;12:69R–77R. [PubMed: 12107076]
- Seeberger PH. Automated carbohydrate synthesis to drive chemical glycomics. Chem Commun. 2003;2003:1115–1121. [PubMed: 12778696]
- Pratt MR, Bertozzi CR. Synthetic glycopeptides and glycoproteins as tools for biology. Chem Soc Rev. 2005;34:58–68. [PubMed: 15643490]
- Werz DB, Seeberger PH. Carbohydrates as the next frontier in pharmaceutical research. Chem Eur J. 2005;11:3194–3206. [PubMed: 15798968]
- Liu L, Bennett CS, Wong C.-H. Advances in glycoprotein synthesis. Chem Commun. 2006;2006:21–33. [PubMed: 16353085]
- Bennett CS, Wong CH. Chemoenzymatic approaches to glycoprotein synthesis. Chem Soc Rev. 2007;36:1227–1238. [PubMed: 17619683]
- Faijes M, Planas A. In vitro synthesis of artificial polysaccarides by glycosidases and glycosynthases. Carbohydr Res. 2007;342:1581–1594. [PubMed: 17606254]
- Seeberger PH. Automated oligosaccharide synthesis. Chem Soc Rev. 2008;37:19–28. [PubMed: 18197330]
- Review Chemical Synthesis of Glycans and Glycoconjugates.[Essentials of Glycobiology. 2015]Review Chemical Synthesis of Glycans and Glycoconjugates.Seeberger PH, Overkleeft HS. Essentials of Glycobiology. 2015
- Review Chemical Synthesis of Glycans and Glycoconjugates.[Essentials of Glycobiology. 2022]Review Chemical Synthesis of Glycans and Glycoconjugates.Seeberger PH, Overkleeft HS. Essentials of Glycobiology. 2022
- The logic of automated glycan assembly.[Acc Chem Res. 2015]The logic of automated glycan assembly.Seeberger PH. Acc Chem Res. 2015 May 19; 48(5):1450-63. Epub 2015 Apr 14.
- Review Chemoenzymatic Synthesis of Glycans and Glycoconjugates.[Essentials of Glycobiology. 2022]Review Chemoenzymatic Synthesis of Glycans and Glycoconjugates.Overkleeft HS, Seeberger PH. Essentials of Glycobiology. 2022
- Review Advances in the Chemical Synthesis of Carbohydrates and Glycoconjugates.[Adv Biochem Eng Biotechnol. 2021]Review Advances in the Chemical Synthesis of Carbohydrates and Glycoconjugates.Malik A, Seeberger PH, Varón Silva D. Adv Biochem Eng Biotechnol. 2021; 175:201-230.
- Chemical and Enzymatic Synthesis of Glycans and Glycoconjugates - Essentials of ...Chemical and Enzymatic Synthesis of Glycans and Glycoconjugates - Essentials of Glycobiology
- Essentials of GlycobiologyEssentials of Glycobiology
Your browsing activity is empty.
Activity recording is turned off.
See more...