

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings RD, Esko JD, et al., editors. Essentials of Glycobiology. 2nd edition. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2009.


Essentials of Glycobiology. 2nd edition.
Show details
Altered glycosylation is a universal feature of cancer cells, and certain glycan structures are well-known markers for tumor progression. This chapter discusses some glycan biosynthetic pathways that are frequently altered in cancer cells, the correlation between altered glycosylation and clinical prognosis, the genetic basis of some of these changes, and in vitro and in vivo studies that indicate the pathological importance of these pathways.
HISTORICAL BACKGROUND
Like normal cells during embryogenesis, tumor cells undergo activation and rapid growth, adhere to a variety of other cell types and cell matrices, and invade tissues. Embryonic development and cellular activation in vertebrates are typically accompanied by changes in cellular glycosylation profiles. Thus, it is not surprising that glycosylation changes are also a universal feature of malignant transformation and tumor progression. The earliest evidence came from observing that plant lectins (e.g., wheat germ agglutinin) showed enhanced binding to and agglutination of tumor cells. Next, it was found that in vitro transformation was frequently accompanied by a general increase in the size of metabolically labeled glycopeptides produced by trypsinization of surface molecules from cancer cells. With the advent of monoclonal antibody technology in the late 1970s, investigators in search of a “magic bullet” against cancer cells found that many of their “tumor-specific” antibodies were directed against glycan epitopes, especially those borne on glycosphingolipids. In most cases, further studies showed that these epitopes were “onco-fetal antigens”; that is, they were also expressed in embryonic tissues and, in a few cell types, in the normal adult. Significant correlations between certain types of altered glycosylation and the prognosis of tumor-bearing animals or patients increased interest in these changes. In several instances, in vitro cellular assays and in vivo animal studies have further supported the view that these changes are critical to aspects of tumor cell behavior. This chapter outlines the most common changes that have been described and their potential relevance.
GLYCOSYLATION CAN BE ALTERED IN VARIOUS WAYS IN MALIGNANCY
Glycan changes in malignant cells take a variety of forms. Examples have been found of loss of expression or excessive expression of certain structures, the persistence of incomplete or truncated structures, the accumulation of precursors, and, less commonly, the appearance of novel structures. Alterations in early branch points in the normal pathways of biosynthesis can markedly affect the relative amount of one class of structure while allowing the dominance of another. However, this is not simply the random consequence of disordered biology in tumor cells. It is striking that of all the possible glycan biosynthetic changes, only a limited subset of changes are frequently correlated with malignant transformation and tumor progression. Given that cancer is a “microevolutionary” process in which only the fittest cells in a genetically heterogeneous population survive, it is reasonable to suggest that these specific glycan changes are selected for during tumor progression. The commonest of these changes are discussed below, including consideration of the likely biosynthetic mechanisms and the possible biological consequences.
ALTERED BRANCHING OF N-GLYCANS
Classic reports of increased size of tumor cell–derived glycopeptides have now been convincingly explained by an increase in β1–6 branching of N-glycans (Figure 44.1), which results from enhanced expression of UDP-GlcNAc:N-glycan GlcNAc transferase V (GlcNAcT-V; see Chapter 8). The change in expression of this enzyme seems to result from increased transcription of its gene (also called MGAT5) and can be induced by various mechanisms, including viral and chemical carcinogenesis. Such responses have been linked to specific features of the 5′-promoter region of MGAT5. Cell lines with increased GlcNAcT-V expression show an increased frequency of metastasis in animal models, and spontaneous revertants for loss of enzyme activity lose this metastatic phenotype. Clinical specimens of some human tumors show increased staining with the plant lectin L-phytohemagglutinin (L-PHA), which preferentially recognizes branched N-glycans bearing the β1–6 branched GlcNAcT-V product (see Chapter 45). Transfection of MGAT5 cDNA into cultured cells causes a visually obvious transformed phenotype associated with colony formation in soft agar, increased cell spreading, enhanced invasiveness through membranes, and tumorigenic behavior by previously nontumorigenic cells. Most convincingly, MGAT5-deficient mice show a striking reduction in the growth and metastasis of breast tumors induced by a viral oncogene. Conversely, metabolic inhibition of N-glycan processing by the plant alkaloid swainsonine (which blocks α-mannosidase II, thereby preventing complete processing of N-glycans and abrogating addition of the β1–6 branch) gives some reversal of tumorigenic behavior.

FIGURE 44.1
The increased size of N-glycans that occurs upon transformation can be explained by an elevation in GlcNAc transferase-V (GNT-V) activity, which catalyzes the β1–6 branching of N-glycans. This, in turn, may lead to enhanced expression (more...)
Taken together, all of these data indicate that GlcNAcT-V plays an important part in the biology of cancer. Indeed, by conventional criteria, such a suite of characteristics is typical of a true “oncogene.” What remains to be clarified are the precise mechanism(s) by which these biochemical and structural changes result in the biological outcomes observed. Some possibilities (Figure 44.1) include (1) an increase in poly-N-acetyllactosamine-containing glycans (potentially recognized by galectins) that are preferentially found on this β1–6 branch; (2) alterations in the cell-surface half-life of growth factor receptors caused by changes in galectin-mediated lattice formation; (3) increased outer-chain polyfucosylation and sialyl Lewisx production (potentially recognized by the selectins); and (4) a general biophysical effect of the branching itself on membrane protein structure. In the last instance, it is suggested that the β1–6 branch has a conformation very different from other outer antennae of N-glycans, tending to exist in a “broken wing” conformation, perhaps directly promoting the association of the glycan chain with the nearby polypeptide surface. In this manner, β1–6 branching may affect the physical properties and functional behavior of glycosylated cell-surface adhesion molecules, such as the integrins, and signaling receptors, such as the T-cell receptor and cytokine receptors.
Enhanced expression of another glycosyltransferase affecting N-glycan structure UDP-GlcNAc:N-glycan GlcNAc transferase III (GlcNAcT-III), which catalyzes the addition of the bisecting GlcNAc branch, has been reported in certain tumors, such as rat hepatomas (Figure 44.1). However, deliberate overexpression of GlcNAcT-III in other tumor cell types caused suppressed function of growth factor receptors and altered cellular morphology, and actually reduced the rate of tumor metastasis. Mice genetically deficient in GlcNAcT-III show a reduced rate of chemical hepatocellular carcinogenesis, despite the fact that (unlike the case in rats) GlcNAcT-III is not up-regulated in mouse hepatomas. The data suggest that an independent glycoprotein factor with bisecting GlcNAc residues facilitates tumor progression in the mouse liver. Overall, although GlcNAcT-III expression appears to affect the biology of tumors, the results are not as clear-cut and consistent as those seen with GlcNAcT-V.
ALTERED EXPRESSION AND GLYCOSYLATION OF MUCINS
Mucins are large glycoproteins with a “rod-like” conformation, which carry many clustered glycosylated serines and threonines in tandem repeat regions (see Chapter 9). Overexpression of mucins in carcinomas has been described for many years, starting with the classic studies of episialin (now known as MUC-1) on mouse tumor cells. Most epithelial mucin polypeptides belong to the MUC family. In the normal polarized epithelium, mucins are expressed exclusively on the apical domain, toward the lumen of a hollow organ. Likewise, soluble mucins are secreted exclusively into the lumen. However, loss of correct topology in malignant epithelial cells (Figure 44.2) allows mucins to be expressed on all aspects of the cells, and soluble mucins can then enter the extracellular space and body fluids such as the blood plasma. The simultaneous expression of both membrane-bound and secreted forms of mucins by many carcinoma cells confounds discussion of their pathophysiological roles, because the two forms of mucins could have opposing effects. Regardless, the secreted mucins often appear in the bloodstream of patients with cancer and can be detected by monoclonal antibodies. In cancers of epithelial origin (carcinomas) in particular, mucins appear to be the major carriers of altered glycosylation. A critical pathophysiological role of mucins in malignancy is suggested by the inhibitory effects of preincubation with GalNAcα-benzyl or acetylated GlcNAcβ1–3Galβ-naphthalenemethanol, which blocks O-glycosylation or the assembly of outer structures on mucins (see Chapters 9 and 50). Apart from specific interactions of the O-glycans with endogenous lectins (see below), the rod-like structures of the mucins and their negative charge are thought to repel intercellular interactions and sterically prevent other adhesion molecules such as cadherins and integrins from carrying out their functions. Thus, in some instances, mucins may act as “anti-adhesins” that can also promote displacement of a cell from the primary tumor during the initiation of metastasis. Evidence suggests that they might also physically block interactions between blood-borne carcinoma cells and the host cytolytic cells such as natural killer cells. In addition, mucins may mask presentation of antigenic peptides by major histocompatibility complex (MHC) molecules.
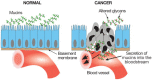
FIGURE 44.2
Loss of normal topology and polarization of epithelial cells in cancer results in secretion of mucins into the bloodstream. The tumor cells invading the tissues and bloodstream also present such mucins on their cell surfaces.
Another abnormal feature of carcinoma mucins is incomplete glycosylation. One common consequence with the O-glycans is the expression of Tn and T antigens (Figure 44.3; see also Chapter 9). Because such structures occur infrequently, in normal tissues, it is thought that they may provoke immune responses in the patient. Indeed, a correlation exists between the expression of the T (Galβ1–3GalNAc-α1-O-Ser/Thr) and Tn (GalNAc-α1-O-Ser/Thr) antigens, the spontaneous expression of antibodies directed against them, and the prognosis of patients with carcinomas. The most extreme form of underglycosylation results in expression of “naked” mucin polypeptides. Clinical trials are under way to deliberately provoke or enhance these immune responses by injecting patients with synthetic peptide antigens, sometimes bearing Tn or sialyl-Tn (Siaα2-6GalNAc-α1-O-Ser/Thr) structures. It is of note that the best immune responses to the glycosylated antigens are seen when they are presented in arrays, exactly as seen on the mucins. This is possibly because of multivalent binding of the antigen to the surface immunoglobulin “antigen receptor” of B cells, giving maximum cellular activation.

FIGURE 44.3
Incomplete glycosylation in the O-linked pathway results in expression of the Tn antigen, the sialylated Tn antigen (a “dead-end” structure), or the T antigen (Thomsen–Friedenreich antigen or unmodified core 1 structure). Multiple (more...)
As for the mechanism causing excessive sialyl-Tn to appear on tumor cells, current evidence does not support simple overexpression of an ST6GalNAc Golgi enzyme that can produce this structure. The more likely explanation appears to be a mutation of the gene encoding Cosmc, a chaperone required for the expression of the β1–3 galactosyltransferase that synthesizes the Galβ1–3GalNAc-α1-O-Ser/Thr core-1 unit of O-glycans. As the gene for Cosmc is on the X chromosome (Xq24), a single mutation would be sufficient to eliminate expression. In this scenario, sialyl-Tn accumulation would then result as a side effect of the loss of ability to make the core-1 O-glycan and its extensions along with expression of the specific ST6GalNAc sialyltransferase (Figure 44.3). Given the frequency of sialyl-Tn accumulation by cancer cells, it is likely that its expression confers some as yet unknown advantage to the tumor cells. Conversely, an increase of UDP-galactose transporter has been implicated in the induction of T-antigen expression.
CHANGES IN SIALIC ACID EXPRESSION IN MALIGNANCY
The classic observation of increased wheat germ agglutinin binding to animal tumor cells is likely explained by an overall increase in cell-surface sialic acid content, which in turn reduces attachment of metastatic tumor cells to the matrix, and may help protect them from recognition by the alternative pathway of complement activation (by recruiting the factor H controlling protein). The increase in sialylation is often manifested as specific increases in α2–6-linked sialic acids attached to outer N-acetyllactosamine (Galβ1–4GlcNAc units) or to inner GalNAc-α1-O-Ser/Thr units on O-glycans. There is some evidence that the overexpression of Siaα2–6Galβ1–4GlcNAc units on N-glycans may enhance β1-integrin action. As discussed above, sialyl-Tn expression may well be a “side effect” of decreased O-glycan extension, and it is currently a target for immunotherapy.
Apart from the amount and linkage of sialic acids, there can also be significant changes in their modifications. Sialic acid 9-O-acetylation either can be up-regulated (e.g., the ganglioside epitope 9-O-acetylated GD3 [Figure 44.4] is increased in melanoma cells in species ranging from humans to fish) or may be decreased (e.g., on the O-glycans of colon carcinomas). Some tumor cell types have also been reported to express small amounts of de-N-acetyl gangliosides (Figure 44.4), wherein the N-acetyl group typical of common sialic acids is removed, exposing a free amino group. Again, the pathological significance of these molecules is uncertain. Some studies have suggested that O-acetylated gangliosides may protect cells from apoptosis, and that de-N-acetylgangliosides act by stimulating tyrosine phosphorylation of the epidermal growth factor (EGF) receptor. As described in Chapter 32, a subgroup of I-type lectins called Siglecs specifically recognize many structural features of sialic acids and are found on innate immune cells. It is possible that altered sialylation of tumor cells affects interactions with some Siglecs. Such interactions could be potentially beneficial to the tumor cell by sending an inhibitory signal to innate immune cells.

FIGURE 44.4
Some pathways for expression of gangliosides in human neuroectodermal tumors. The heavy arrows indicate pathways that are up-regulated. The dashed arrows indicate pathways that have not yet been formally proven. O-Acetylation of the terminal sialic acid (more...)
Another interesting phenomenon is the aberrant expression of N-glycolylneuraminic acid (Neu5Gc) in human tumor cells. This sialic acid differs from the usual N-acetylneuraminic acid (Neu5Ac) by the addition of a single oxygen atom (Figure 44.5). Adult humans do not express significant levels of Neu5Gc on their normal cells, and they mount an immune response to this epitope when infused with Neu5Gc-containing animal serum. This is because the hydroxylase enzyme responsible for creating Neu5Gc is mutated in humans (see Chapter 14). As there is no definitive alternate pathway for Neu5Gc production, reports of aberrant Neu5Gc expression in human tumors are best explained by uptake of Neu5Gc from dietary sources (or from fetal bovine serum in the case of cultured human cancer cells). Regardless of the mechanism, this re-expression of Neu5Gc may explain why some patients with cancer spontaneously develop so-called “Hanganutziu–Deicher” antibodies that are directed against gangliosides that contain Neu5Gc. The presumed selective advantage to tumor cells of accumulating Neu5Gc in the face of an immune response needs explanation. One possibility under study is that the resulting weak immune response is actually beneficial to tumor growth, by enhancing chronic inflammation and angiogenesis.

FIGURE 44.5
N-Acetylneuraminic acid (Neu5Ac) and N-glycolylneuraminic acid (Neu5Gc). The latter is a nonhuman sialic acid that accumulates in tumor cells, apparently from dietary origins. A negatively charged carboxylate occurs at the C-1 position of Neu5Gc and linkages (more...)
SIALYLATED LEWIS STRUCTURES AND SELECTIN LIGANDS ON CANCER CELLS
Immunohistochemical studies on tumor specimens have shown that Lewisx and Lewisa structures (see Chapters 13 and 45) are frequently overexpressed in carcinomas, being carried on O-glycans as well as on N-glycans and glycosphingolipids. Indeed, sialyl Lewisx and sialyl Lewisa were first identified as tumor antigens. The expression of these antigens by epithelial carcinomas correlates with tumor progression, metastatic spread, poor prognosis in humans, and metastatic potential in mice. These sialylated fucosylated structures also form critical components of most natural ligands for the selectins (see Chapter 31). Thus, it was reasonable to postulate that tumor cells gain a selective advantage by presenting pathological selectin ligands that mediate interactions with endogenous selectins. Indeed, calcium-dependent selectin ligands on carcinoma cells have been demonstrated, and mucin-like tumor antigens binding to E-selectin were directly demonstrated in the blood of colon carcinoma patients. Likewise, overexpression of E-selectin in the transgenic mouse liver induced redirection of the metastatic patterns of syngeneic carcinomas that normally colonize the lung.
Tumor metastasis is also attenuated in mice with P-selectin or L-selectin deficiency. These and other studies indicate that interactions between tumor-derived mucins and selectin molecules play a part in the metastatic cascade of some carcinoma cells (Figure 44.6). This ties in with the classic observation that cancer cells entering the bloodstream form complex thromboemboli with platelets and leukocytes, which are thought to facilitate arrest at ectopic sites, assist interactions with the endothelium, and help in evasion of the immune system. Current data suggest that this phenomenon can be explained by interactions between platelet and/or endothelial P-selectin and carcinoma mucins. Thus, carcinoma cells show a reduced metastatic rate in P-selectin-deficient mice, which can be explained at least in part by a lack of P-selectin-dependent rosetting of platelets on the tumor cells. With regard to L-selectin, one of the mechanisms appears to involve leukocyte interactions with fucosyltransferase-7 (FucT-7)-dependent endothelial ligands, which are induced at the site of tumor embolization in the vasculature. Of practical relevance is the finding that the glycosaminoglycan heparin is a potent inhibitor of P- and L-selectin interactions. Although the physiological relevance of selectin interactions with heparan sulfate is still being explored (see Chapter 31), the pharmacological effects of heparin on selectin interactions are thought to explain some prior reports of benefits of heparin therapy in cancer. Another strategy under development involves small-molecule inhibitors of selectin ligand formation (Chapter 49).

FIGURE 44.6
Potential interactions that could occur between tumor cells and selectins. Several of these have been formally proven to be important in tumor biology. See text for discussion. (Modified from Stevenson J.L., et al. 2005. Clin. Cancer Res. 11: 7003–7011.) (more...)
A related issue is that cancer patients sometimes develop thromboemboli and hypercoagulable states (Trousseau’s syndrome), which are frequently associated with mucin-producing carcinomas. In this regard, secreted mucins from tumor cells have been shown to initiate a similar form of platelet-rich microthrombosis in mice, in a P- and L-selectin-dependent manner. Notably, as with tumor metastasis, this process can be blocked by heparin but not by other mechanisms that directly interfere with the fluid-phase coagulation pathway.
“INCOMPLETE SYNTHESIS” AND ALTERED EXPRESSION OF BLOOD GROUP–RELATED STRUCTURES
In addition to changes in Lewisx- and Lewisa-related structures mentioned above, loss of normal AB blood group expression (accompanied by increased expression of the underlying H and Lewisy structures) (see Chapter 13) is associated with a poorer clinical prognosis of carcinomas in several studies. There are some other instances where glycan determinants strongly expressed on normal cells are lost upon transformation. For example, the Sda antigen (also called Cad), a blood group glycan structure abundantly expressed in the left half of the colon, is lost in colon carcinoma. Sulfation of the C-3 position of terminal galactose residues is also reduced in cancers. Sialyl-6-sulfo Lewisx and disialyl Lewisa, which are more complex structures derived from the same synthetic pathway as sialyl Lewisx/a, are also expressed preferentially on normal colonic epithelial cells but are reduced in colon cancer cells. All of these changes can be considered as biosynthetically related to the enhanced production of sialyl Lewisx/a in cancers. DNA methylation and histone deacetylation, the epigenetic mechanisms for suppression of normal gene transcription commonly observed in cancers, are proposed to lie behind these glycan alterations.
As mentioned earlier, synthesis of a variety of complex glycans in normal cells can be impaired in malignant cells, leading to the accumulation of less complex precursor structures in cancers, a concept summarized as “incomplete synthesis.” The loss of sialyl-6-sulfo Lewisx, disialyl Lewisa, 3′-sulfation, Sda antigen, and normal AB blood group determinants in cancers are good examples supporting this concept. Another example mentioned earlier is the accumulation of T, Tn, and sialyl-Tn and reduction of more complex structures in cancer cell O-glycans.
There are also rare instances in which a tumor may present a “forbidden” blood group structure (i.e., expression of a B blood group antigen in an A-positive patient; see Chapter 13 for details of the structure of ABO blood group antigens). The genetic basis for such a change remains unexplained, because the “B” transferase would theoretically require four independent amino acid changes to convert it into an “A” transferase. Regardless of the underlying mechanism, tumor regression has been noted in a few such cases, presumably mediated by the naturally occurring endogenous antibodies directed against the illegal structure. The appearance of P antigen in cancers of rare P blood type individuals is another example.
TRANSCRIPTIONAL REGULATION OF ALTERED GLYCAN EXPRESSION IN MALIGNANT CELLS
Unlike proteins, which are generally encoded by a single gene, a glycan determinant is produced by the concerted action of several related genes, making it difficult to elucidate the genetic regulatory mechanism for expression of some cell-surface glycans. With the availability of the total human genome sequence, application of DNA microarray and other molecular biological techniques to glycosyltransferases and related genes has facilitated accumulation of knowledge on the transcriptional background of cancer-associated glycan alteration. Several examples have been elucidated that indicate a direct association between glycan alteration and the genetic mechanism for malignant transformation of cells. One example mentioned earlier is transcriptional induction of MGAT5 (the gene for GlcNAc transferase-V) by v-src, H-ras, and v-fps. A binding site for a transcription factor ets-1 was shown to be present in the 5′-regulatory region of the gene. Another example is enhanced expression of sialyl Lewisx on adult T-cell leukemia cells, which are known to exhibit extremely strong tissue infiltrative activity, likely mediated by selectins. The etiologic agent for this leukemia is a retrovirus called HTLV-1. The transcriptional activator protein, Tax, encoded by the virus, binds to the 5′-regulatory region of the gene for fucosyltranserase-7, the rate-limiting enzyme of sialyl Lewisx synthesis in leukocytes, and constitutively activates its transcription (Figure 44.7). This is another good example of the direct association of malignant transformation and glycan alteration.

FIGURE 44.7
An example of a direct association of glycan alteration and malignant transformation is shown. The oncogenic protein Tax (encoded by HTLV-1, the etiologic virus for adult T-cell leukemia) induces transcription of the gene for fucosyltransferase VII ( (more...)
Under the poorly oxygenated conditions found in locally advanced tumors, hypoxia-resistant cancer cells survive by the principle of natural selection, acquiring hypoxia tolerability through a transcription factor HIF (hypoxia inducible factor), the nuclear translocation of which is facilitated by inactivation of tumor suppressors such as VHL and p53. Recently, HIF was shown to induce transcription of several genes for glycan synthesis, leading to the significant alteration of glycan profiles, including enhanced sialyl Lewisx/a expression in cancer cells. Tumor hypoxia may also affect the expression of Neu5Gc in human tumor cells mentioned earlier by influencing transcription of genes for the lysosomal sialic acid transporter.
ALTERED EXPRESSION AND SHEDDING OF GLYCOSPHINGOLIPIDS
Many of the “tumor-specific” monoclonal antibodies raised against cancer cells are reactive with the glycan portion of glycosphingolipids. Some of these structures are highly enriched in specific types of tumors (e.g., Gb3/CD77 in Burkitt’s lymphoma and GD3 in melanomas). Several types of tumors (particularly those of neuroectodermal origin such as melanoma and neuroblastoma) are characterized by the synthesis of very high levels of sialylated glycosphingolipids (gangliosides; see Chapter 10). Some of these (e.g., GD2) are not normally found at high levels in extraneural gangliosides and they are therefore considered targets for both passive immunotherapy (monoclonal antibody infusion) and active immunotherapy (immunization with purified glycolipid preparations). In some cases, gangliosides are also the major carriers of modified sialic acids (see above). Many in vitro studies suggest that some gangliosides have effects upon growth control, and it is suggested (but not proven) that this also may be the case in vivo. Some investigators have noted strong immunosuppressive effects that correlate with the large quantities of gangliosides “shed” from the cell surface by some tumors. It is not known whether this type of immunosuppression is a purely pathological phenomenon with no natural counterpart. Regardless, the levels of free gangliosides found in the body fluids of some patients with these tumors are high enough that such immunosuppression is likely to be medically relevant.
LOSS OF GLYCOPHOSPHOLIPID ANCHOR EXPRESSION
A complete loss of expression of glycosylphosphatidylinositol (GPI)-anchored proteins is seen in some cases of malignant and premalignant states involving the hematopoietic system. This results from acquired somatic mutations of hematopoietic stem cells in the PIGA gene (required for an early step in the biosynthesis of the GPI anchor precursor; see Chapter 11). Like the Cosmc gene discussed above, the PIGA gene is also on the X chromosome (Xp22.1). The consequence is a marked or complete loss of cell-surface expression of several GPI-anchored proteins on the progeny of a single hematopoietic stem cell clone. Because this mutation arises from a single stem cell, one would imagine that its progeny would not be easily detected. However, this clone often gradually replaces all of the others, giving rise to a syndrome called paroxysmal nocturnal hemoglobinuria or PNH (see Chapter 11). Although the mutation itself does not confer a malignant phenotype, the affected clone appears to be prone to become malignant (i.e., give rise to a leukemia). The mechanism(s) predisposing to the subsequent transforming event are unknown. It is also possible that the preexisting PNH cells are simply unmasked by the loss of normal cells due to an unrelated insult to the bone marrow.
CHANGES IN POLY-N-ACETYLLACTOSAMINE EXPRESSION AND GALECTIN FUNCTION
Increased expression of galectins (especially galectin-3) has also been associated with tumor progression. A molecular significance of this correlation is proposed to be the interactions of galectins with poly-N-acetyllactosamines on matrix proteins such as laminin, which aids cellular invasion. Because poly-N-acetyllactosamines are also expressed on cancer mucins and are enriched on the β1–6-branched glycans of tumor N-glycans (see above and Figure 44.1), this molecular interaction could mediate homotypic adhesion of carcinoma cells as well. Galectin recognition may also explain how adding cell-surface galactose to tumor mutants lacking the Golgi UDP-Gal transporter enhances metastasis. Another possibility is the formation of cell-surface lattices involving multiple galectin and poly-N-acetyllactosamine molecules. Overall, it remains to be elucidated exactly how galectin–poly-N-acetyllactosamines interactions alter the biology of cancer.
CHANGES IN HYALURONAN
Hyaluronan is a very large negatively charged polysaccharide composed of the repeating disaccharide [GlcAβ1–3GlcNAcβ1–4]n. It differs from other glycosaminoglycans in that it is nonsulfated and exists as a free polymer, rather than being covalently linked to a protein. Furthermore, it is synthesized and extruded from the cell directly at the plasma membrane, rather than being processed through the ER-Golgi pathway (see Chapter 15).
Many classes of malignant tumors express high levels of hyaluronan. In carcinomas, hyaluronan is usually enriched in the tumor-associated stroma (i.e., connective tissue elements and blood vessels). This stroma is more or less prominent depending on tumor type; for example, it is usually prominent in breast cancer. However, hyaluronan is also often localized immediately around the tumor cell surface. In normal tissues, hyaluronan serves at least three functions, which may also contribute to tumor progression. First, it increases levels of tissue hydration, which can facilitate movement of cells through tissues. Second, it is intrinsic to the assembly of extracellular matrices through specific interactions with other macromolecules, and thus it participates in tumor cell–matrix interactions that facilitate or inhibit tumor cell survival and invasion. Finally, hyaluronan interacts with several types of cell-surface receptors, especially CD44 and the receptor for hyaluronan-mediated motility (RHAMM/CD168). Hyaluronan–CD44 interactions are often crucial to tumor malignancy and are a current target for novel therapies.
Various alternatively spliced isoforms of CD44 are often elevated in cancer cells. However, this is not universal among different tumor types, leading to some confusion regarding the role of CD44 in cancer. Regardless, it is now clear that activation of hyaluronan–CD44 signaling is much more important in cancer progression than actual levels of CD44. In normal adult tissues, hyaluronan appears to be relatively inert with respect to cell signaling and behavior. However, during embryonic development, during tissue healing and regeneration, and in various pathological situations, hyaluronan-induced signaling via interaction with CD44 becomes activated. The consequences of this signaling are dramatic because they are essential to or promote cell behaviors such as proliferation, survival, migration, and invasion, which are also key elements of the malignant phenotype. Hyaluronan–CD44 interaction at the tumor cell surface is required for the constitutive activation of some well-known oncogenes, especially the receptor tyrosine kinase, ErbB2, which is amplified or mutated in a large number of carcinomas. Accordingly, hyaluronan–CD44 interaction promotes downstream intracellular pathways that are also hallmarks of cancer, such as the phosphatidylinositol–3-kinase/AKT and mitogen-activated protein (MAP) kinase pathways. It has been demonstrated that antagonists of hyaluronan–CD44 interaction cause inactivation of these pathways in malignant cells in culture and can inhibit tumor growth and metastasis in animal models. An exciting new development is the finding that hyaluronan stimulates multidrug resistance and that its antagonists sensitize resistant cancer cells to chemotherapeutic drugs. Evidently, stimulation of receptor tyrosine kinase activities by hyaluronan leads to increased phosphatidylinositol–3-kinase levels, which in turn stimulate new hyaluronan synthesis, thus setting up a positive feedback loop that amplifies anti-apoptotic pathways and expression of ABC-type mul-tidrug transporters (Figure 44.8).

FIGURE 44.8
Effects of hyaluronan (HA) on oncogenic signaling. Endogenous HA–CD44 interaction is required for the formation of constitutive signaling complexes containing CD44, activated receptor tyrosine kinases (RTKs), phosphatidylinositol–3-kinase (more...)
Despite convincing evidence for the involvement of hyaluronan-induced signaling in malignant cell behavior, there are several unsolved conundrums. For example, the mechanism of signaling activation is not understood. Two possibilities under consideration are alterations in CD44 status and effects of size on the signaling properties of hyaluronan. A current theme in pathophysiology is that emergent activities arise from breakdown products of extracellular matrix macromolecules, for example, the derivation of pro- and anti-angiogenic fragments from physiological proteolysis of collagens and proteoglycans. Some studies suggest a similar phenomenon for the mechanism of hyaluronan action and that hyaluronan activity may depend on its partial degradation by hyaluronidases. In agreement with this, both hyaluronan and hyaluronidase levels are elevated in several cancers and hyaluronan oligosaccharides are angiogenic. However, the mechanisms by which these breakdown products act are not understood. Despite all these remaining questions, it is clear that hyaluronan is an important therapeutic target in cancer.
CHANGES IN SULFATED GLYCOSAMINOGLYCANS
Numerous sulfated proteoglycans contribute to tissue structure and function during development and adult homeostasis. These proteoglycans contain core proteins that are characteristically decorated with covalently attached negatively charged glycosaminoglycan side chains—namely, chondroitin sulfate, dermatan sulfate, keratan sulfate, and heparan sulfate (HS) (see Chapter 16). Although the content and distribution of many proteoglycans are altered during tumorigenesis, only HS proteoglycans have been implicated in tumor pathogenesis in a widespread and convincing manner. A large number of HS and heparin variants comprise a closely related group of glycosaminoglycans derived from a common precursor, but varying in their glycan composition, especially with respect to number and location of sulfate groups. In addition, HS is a component of several proteoglycans derived from different core protein families (e.g., syndecans, glypicans, and perlecan) (see Chapter 16).
Each of these HS proteoglycan families is closely associated with positive or negative aspects of tumor progression (or both). For example, early work suggested that decreased levels of HS proteoglycans are associated with tumor progression and that HS suppresses tumorigenic properties. However, mutant CHO cells that do not produce HS and human tumor cells engineered to make less perlecan or glypican proteoglycans lose their tumorigenicity. In contrast, human mutations affecting HS polymerization result in osteochondromas and loss of glypican-3 causes an overgrowth syndrome. Even a specific proteoglycan can have tumor-promoting and -inhibitory properties. For example, while HS side chains of perlecan can promote angiogenesis, the carboxy-terminal domain of the core protein (endorepellin) is potently anti-angiogenic. More recent studies illustrate a rather complex situation in which the type and degree of processing of the HS side chains by heparanases and other enzymes or the processing of the proteoglycan core proteins by proteases can cause the various HS proteoglycans to have positive or negative effects on tumorigenesis. Despite this complexity, recent data have opened up the possibility of novel therapeutic interventions, based on the nature of HS processing and the activities of different heparanase products.
The major functions of HS proteoglycans relevant to tumor formation are (1) to promote cell–cell and cell–matrix interactions that are important in tissue assembly and (2) to bind a wide array of bioactive factors such as FGF-2 (fibroblast growth factor-2), VEGF (vascular endothelial growth factor), and numerous members of the interleukin, Wnt, and chemokine and growth factor families. Various studies have shown that the former activity can serve to build either inhibitory barriers or permissive pathways for cell invasion and that the latter activity can either sequester factors away from their receptors or present them efficaciously to these receptors. Thus, it is easy to visualize how HS proteoglycans could have opposite effects on cellular functions under varying cell and tissue arrangements. Syndecan-1 is particularly illustrative with respect to the multiple important roles of HS, especially in promoting metastasis and angiogenesis. Early work suggested that syndecan-1 is important in maintaining the normal differentiated state of epithelia and that low levels of tumor-associated syndecan-1 correlate with malignancy. However, recent work also highlights the importance of enzymatic processing by proteases and heparanase. First, a high level of proteolytically shed HS-rich ectodomain of syndecan-1 is present in the sera of patients with myeloma and some types of carcinoma and is a predictor of poor prognosis. Second, high levels of syndecan-1 ectodomain also accumulate in the tumor microenvironment, where it plays an important role in activation of chemokine and growth factor signaling and consequently tumor cell behavior. This conclusion is strongly supported by studies in which cells that are genetically engineered to produce large amounts of syndecan-1 ectodomain generate increased tumor growth, angiogenesis, and metastasis in animal models. Especially significant is the finding that this ectodomain promotes metastasis to bone tissue, which is characteristic of myeloma and certain carcinomas (e.g., of breast or prostate) in human patients. The mechanism of ectodomain shedding appears to involve members of the matrix metalloproteinase and ADAM/ADAM-TS protease families.
HS side chains of several HS proteoglycans are cleaved to fragments containing 10–20 sugar moieties by vertebrate heparanase (as opposed to smaller HS saccharides produced by bacterial eliminases, i.e., heparinases and heparitinases). Heparanase is elevated in numerous types of cancer and increased heparanase activity can lead to induction of angiogenesis and metastasis. Among the protumorigenic molecular and cellular consequences of this type of HS cleavage are (1) disassembly of basement membranes allowing penetration by tumor cells; (2) blood vessel remodeling required for angiogenesis; (3) release of HS-bound angiogenic factors, growth factors, and chemokines; and (4) increased bioactivity of the heparanase products compared to intact HS. Another mechanism of HS modification, the removal of sulfate moieties by endosulfatases, may alter HS activities and thus affect tumor growth. These studies have led to increased interest in HS as a therapeutic target in cancers (see below).
CLINICAL SIGNIFICANCE
Diagnostics
Although many tumor-specific monoclonal antibodies have been generated by immunization with tumor-derived glycosphingolipids, the appearance of corresponding glycan epitopes in the bloodstream is probably caused by the spillover of mucin glycoproteins (see Figure 44.2) bearing similar terminal glycan structures. The latter are of well-established value for detecting and monitoring the growth status of tumors (e.g., assays for CA19–9 in pancreatic cancers, CA125 and sialyl-Tn in ovarian carcinoma, and sialyl Lewisx-related glycans in lung and breast cancers allow a physician to monitor the amount of tumor remaining in a patient after surgery or chemotherapy). However, these circulating mucins also pose a potential problem for the therapeutic use of monoclonal antibodies directed against these antigens, because they represent a “sink” that would absorb any infused antibody, preventing it from reaching the tumor cell surface. In recent times, the failure of proteomics to deliver reliable cancer biomarkers has rekindled interest in glycomic profiling of serum and other body fluid glycoproteins for this purpose. Also being reinvestigated are classic findings of glycan-specific antibodies associated with cancer, such as the heterophile antibodies against the nonhuman sialic acid, Neu5Gc. Another potential advance lies in improving the specificity of known cancer biomarkers by defining glycoforms uniquely expressed by cancer cells.
Therapy
Although altered glycosylation is of clear value in diagnosis, its role in specific therapies is less clear. As mentioned above, attempts are being made at specific passive and active immunotherapy against certain tumor-associated gangliosides and against incompletely glycosylated mucins. Some trials using humanized antiganglioside antibodies have shown promising results. With regard to the increased β1–6 branching of N-glycans, clinical studies of the metabolic inhibitor swainsonine have not yet yielded definitive results.
Cimetidine, a histamine H2 antagonist, suppresses E-selectin expression on vascular beds and reportedly improves patient prognosis after surgical therapy. Recent studies have also suggested that the potent effects of heparin in attenuating tumor metastasis might be due not to its anticoagulant properties, but rather to its ability to block P- and L-selectin binding to tumor and/or host ligands (see Chapter 31). In this regard, it is disconcerting that the recent move to supplant the traditional unfractionated heparins with very low-molecular-weight heparins and heparinoids may be associated with a loss of selectin-inhibitory properties. Several compounds have also been shown to antagonize the potentially protumorigenic activities of HS. For example, low-molecular-weight heparins may inhibit tumor progression not only by blocking selectins, but also by blocking heparanase activity or by interfering with constitutive HS activities. Other inhibitors of heparanase, such as suramin, laminarin sulfate, and the sulfated phosphomannopentaose PI-88, also inhibit angiogenesis and metastasis, but some of these compounds may also potentially work by blocking selectins. Some of these compounds may act by competitively inhibiting HS binding and function. Recent advances in our understanding of HS structure and function are likely to promote further development of efficient HS-based anticancer therapies. Low-molecular-weight oligosaccharides of hyaluronan could also be useful therapeutically, because they inhibit the prooncogenic influences of constitutive polymeric hyaluronan, especially drug resistance and signaling events induced by hyaluronan–CD44 interaction. Disaccharides that can enter the cell and act as decoys to divert glycosylation pathways are also showing promise (see Chapter 50). Further studies of all these possibilities are needed.
FURTHER READING
- Hakomori S, Kannagi R. Glycosphingolipids as tumor-associated and differentiation markers. J Natl Cancer Inst. 1983;71:231–251. [PubMed: 6576183]
- Feizi T. Demonstration by monoclonal antibodies that carbohydrate structures of glycoproteins and glycolipids are onco-developmental antigens. Nature. 1985;314:53–57. [PubMed: 2579340]
- Hakomori S. Tumor associated glycolipid antigens, their metabolism and organization. Chem. Phys. Lipids. 1986;42:209–233. [PubMed: 2435423]
- Fukuda M. Possible roles of tumor-associated carbohydrate antigens. Cancer Res. 1996;56:2237–2244. [PubMed: 8625291]
- Kim YS, Gum J, Brockhausen I. Mucin glycoproteins in neoplasia. Glycoconj J. 1996;13:693–707. [PubMed: 8909996]
- Kim YJ, Varki A. Perspectives on the significance of altered glycosylation of glycoproteins in cancer. Glycoconj J. 1997;14:569–576. [PubMed: 9298689]
- Dennis JW, Granovsky M, Warren CE. Glycoprotein glycosylation and cancer progression. Biochim. Biophys. Acta. 1999;1473:21–34. [PubMed: 10580127]
- Hakomori S. Antigen structure and genetic basis of histo-blood groups A, B and O: Their changes associated with human cancer. Biochim. Biophys. Acta. 1999;1473:247–266. [PubMed: 10580143]
- Varki NM, Varki A. Heparin inhibition of selectin-mediated mechanisms in the hematogenous phase of carcinoma metastasis: Rationale for clinical trials in humans. Semin Thromb Hemost. 2002;28:53–66. [PubMed: 11885026]
- Kannagi R. Molecular mechanism for cancer-associated induction of sialyl Lewis X and sialyl Lewis A expression—The Warburg effect revisited. Glycoconj J. 2004;20:353–364. [PubMed: 15229399]
- Kannagi R, Izawa M, Koike T, Miyazaki K, Kimura N. Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci. 2004;95:377–384. [PubMed: 15132763]
- Sanderson RD, Yang Y, Suva LJ, Kelly T. Heparan sulfate proteoglycans and heparanase—Partners in osteolytic tumor growth and metastasis. Matrix Biol. 2004;23:341–352. [PubMed: 15533755]
- Toole BP. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer. 2004;4:528–539. [PubMed: 15229478]
- Fuster MM, Esko JD. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer. 2005;5:526–542. [PubMed: 16069816]
- Kannagi R, Yin J, Miyazaki K, Izawa M. Current relevance of incomplete synthesis and neo-synthesis for cancer-associated alteration of carbohydrate determinants. Biochim. Biophys. Acta. 2008;1780:525–531. [PubMed: 17980710]
- HISTORICAL BACKGROUND
- GLYCOSYLATION CAN BE ALTERED IN VARIOUS WAYS IN MALIGNANCY
- ALTERED BRANCHING OF N-GLYCANS
- ALTERED EXPRESSION AND GLYCOSYLATION OF MUCINS
- CHANGES IN SIALIC ACID EXPRESSION IN MALIGNANCY
- SIALYLATED LEWIS STRUCTURES AND SELECTIN LIGANDS ON CANCER CELLS
- “INCOMPLETE SYNTHESIS” AND ALTERED EXPRESSION OF BLOOD GROUP–RELATED STRUCTURES
- TRANSCRIPTIONAL REGULATION OF ALTERED GLYCAN EXPRESSION IN MALIGNANT CELLS
- ALTERED EXPRESSION AND SHEDDING OF GLYCOSPHINGOLIPIDS
- LOSS OF GLYCOPHOSPHOLIPID ANCHOR EXPRESSION
- CHANGES IN POLY-N-ACETYLLACTOSAMINE EXPRESSION AND GALECTIN FUNCTION
- CHANGES IN HYALURONAN
- CHANGES IN SULFATED GLYCOSAMINOGLYCANS
- CLINICAL SIGNIFICANCE
- FURTHER READING
- Review Glycosylation Changes in Cancer.[Essentials of Glycobiology. 2015]Review Glycosylation Changes in Cancer.Varki A, Kannagi R, Toole B, Stanley P. Essentials of Glycobiology. 2015
- Review Glycosylation Changes in Cancer.[Essentials of Glycobiology. 2022]Review Glycosylation Changes in Cancer.Bellis SL, Reis CA, Varki A, Kannagi R, Stanley P. Essentials of Glycobiology. 2022
- Review Functional impact of tumor-specific N-linked glycan changes in breast and ovarian cancers.[Adv Cancer Res. 2015]Review Functional impact of tumor-specific N-linked glycan changes in breast and ovarian cancers.Guo H, Abbott KL. Adv Cancer Res. 2015; 126:281-303. Epub 2015 Feb 7.
- Review Cell surface protein glycosylation in cancer.[Proteomics. 2014]Review Cell surface protein glycosylation in cancer.Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Proteomics. 2014 Mar; 14(4-5):525-46.
- Review Altered glycan structures: the molecular basis of congenital disorders of glycosylation.[Curr Opin Struct Biol. 2005]Review Altered glycan structures: the molecular basis of congenital disorders of glycosylation.Freeze HH, Aebi M. Curr Opin Struct Biol. 2005 Oct; 15(5):490-8.
- Glycosylation Changes in Cancer - Essentials of GlycobiologyGlycosylation Changes in Cancer - Essentials of Glycobiology
- Glycans in Development and Systemic Physiology - Essentials of GlycobiologyGlycans in Development and Systemic Physiology - Essentials of Glycobiology
- Same, Connectivity for PubChem Compound (Select 6870646) (8)PubChem Compound
- receptor tyrosine-protein kinase erbB-4 isoform X8 [Homo sapiens]receptor tyrosine-protein kinase erbB-4 isoform X8 [Homo sapiens]gi|530369984|ref|XP_005246434.1|Protein
- Gene Links for GEO Profiles (Select 132477897) (1)Gene
Your browsing activity is empty.
Activity recording is turned off.
See more...