

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings R, Esko J, et al., editors. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1999.


Essentials of Glycobiology.
Show detailsPrimary contributions to this chapter were made by J.D. Marth (HHMI, University of California at San Diego).
THE BIOSYNTHESIS OF ASPARAGINE (N)-linked oligosaccharides (N-glycans) is described in this chapter, with an emphasis on vertebrate systems. Included is an overview of the mechanisms in N-glycan formation, processing, control of protein folding, and structural diversification. Also presented is a biological overview of N-glycan structure-function relationships that have been defined by genetic lesions in intact vertebrates.
Background
The biosynthesis of N-glycans was elucidated in the 1960s and 1970s by using cell-free systems and lectin-resistant cell lines. A surprising finding was the requirement for production of a lipid-linked oligosaccharide precursor structure that is transferred en bloc to nascent proteins in the ER. N-glycan addition was noted to occur on asparagines in the sequence context Asn-X-Ser/Thr. All eukaryotic cells produce N-glycans and have conserved the earliest biosynthetic steps involving the synthesis of this dolichol-oligosaccharide precursor as well as several subsequent processing reactions in the ER. These conserved processing reactions include cycles of glucose removal and readdition, which have been found to participate in protein-folding mechanisms. Another unexpected finding in N-glycan biosynthesis is the extent of structural diversification in the Golgi apparatus from high-mannose N-glycans into a large repertoire of hybrid and complex N-glycan subtypes that are secreted or positioned at the vertebrate cell surface. Of the organisms studied, including those within the largest phylum Arthropoda, only vertebrates appear to produce a diverse and dynamic repertoire of hybrid and complex N-glycan subtypes (see Chapter 3). This emanates from the presence of a large number of enzymes encoded in the vertebrate genome, which include glycosyltransferases and glycosidases that reside in the ER and Golgi apparatus (Chapter 17). Variations in glycan structures among plants, invertebrates, and various microbes are described elsewhere (see Chapters 20, 21, and 36).
The molecular cloning of vertebrate glycosyltransferase and glycosidase genes involved in N-glycan biosynthesis began in the 1980s and continues to the present. The encoded enzymes show remarkable substrate specificity and their unique expression patterns contribute to the variation of N-glycan structures noted among particular cell types. Observations that N-glycan structures change in a variety of cellular and organismic events, including embryogenesis, cell activation, and cancer (see Chapters 34 and 35), have led to many hypotheses regarding N-glycan function. With the isolation and manipulation of genes that regulate the N-glycan diversification pathways in the intact organism, investigations on the physiologic functions of N-glycans have become possible and productive (see Chapters 5, 32, and 33).
Synthesis of the Dolichol Oligosaccharide Precursor (1–12)
There are many excellent reviews that describe pathways in N-glycan biosynthesis. The first events involve assembling an oligosaccharide precursor structure linked to the lipid Dol-P (Figure 7.1). The majority of the dolichol lipid exists in cellular compartments other than the ER and is either esterified or in the form of free dolichol, indicating that most of the dolichol lipid is not participating in the biosynthesis of N-glycans but is involved in the formation of cellular membranes. Evidence from multiple systems indicates that if cellular levels of Dol-P are reduced by treatment with compactin, exogenous cholesterol, or coenzyme Q, then N-glycosylation is significantly impaired. Abolition of N-glycosylation in intact cells and embryos has been observed following treatment with tunicamycin, an analog of UDP-GlcNAc that blocks the first step in dolichol oligosaccharide precursor formation by inhibition of GlcNAc-1-phosphotransferase activity.

Figure 7.1
Structure of the Dol-P lipid used in constructing the dolichol oligosaccharide precursor in N-linked oligosaccharide biosynthesis. The number of isoprene repeats (n) varies between 15 and 19.
The dolichol oligosaccharide precursor consists of dolichol lipid bearing a pyrophosphate linkage to an oligosaccharide composed of 14 specific monosaccharide linkages. The structure has been conserved among all eukaryotes (see Chapter 6). The cellular topology of the biosynthetic pathway is also remarkable. Several enzymes operating in its biosynthesis have not yet been isolated and characterized, although their activities appear evident by inference to precursor structures experimentally defined (see below). Cytosolic and luminal fractions have both been found to contain various intermediates in dolichol oligosaccharide biosynthesis, suggesting that certain assembly reactions occur at one or the other side of the membrane. Various ER and Golgi sugar nucleotide transporters exist that serve to transport various monosaccharide donors across the membrane bilayer for glycan biosynthesis by luminal glycosyltransferases. Recent molecular cloning and characterization of these transporters indicate that they are essential in the formation of various glycans bearing the relevant monosaccharide (see Chapter 6). In the production of the dolichol oligosaccharide precursor, however, these transporters are not necessary.
Biochemical analyses of dolichol oligosaccharide intermediates have provided a relatively clear outline of the details regarding assembly of the dolichol oligosaccharide precursor (Figure 7.2). Evidence from multiple studies involving protease and glycosidase sensitivity indicates that the initial linkages involving the two core GlcNAc monosaccharides (inhibited by tunicamycin) and the first five mannose residues occur on the cytosolic side of the ER membrane. The monosaccharide donors include UDP-GlcNAc and GDP-Man for the first seven linkage reactions that occur on the cytosolic side of the ER membrane. In the first two steps, GlcNAc residues are added to the Dol-P lipid by GlcNAc-1-phosphotransferase and then by a GlcNAc-transferase. Next, five mannose residues are added sequentially using GDP-Man as donor (step III). By a mechanism not fully understood, the Man5GlcNAc2-Dol precursor must “flip” across the membrane bilayer to become oriented in the lumen of the ER. Next, four mannoses are added in rapid succession using the Dol-P-Man donor (step IV). The assembly of the dolichol oligosaccharide precursor is completed with the addition of three glucose residues donated by Dol-P-Glc (step V). It appears that Dol-P-Man and Dol-P-Glc are also formed on the cytoplasmic side of the ER and then are flipped across the bilayer to become available to enzymes in the lumen of the ER. The completed dolichol lipid-linked oligosaccharide precursor is now ready for transfer to asparagine residues on nascently translated proteins.

Figure 7.2
Biosynthesis of the dolichol oligosaccharide precursor.
Transfer of the Dolichol Oligosaccharide Precursor to Nascent Proteins (13–17)
A multisubunit protein complex in the ER membrane of eukaryotes transfers the lipid-linked oligosaccharide precursor to asparagine residues on nascently translated proteins. This complex is termed the oligosaccharyltransferase (OST). The composition of the OST complex remains under investigation. At least nine nonidentical subunits comprise the heteromeric OST complex in yeast, where most data have been gathered from mutational studies. Members of the yeast OST complex (homologous proteins in higher eukaryotes in parentheses) include Ost1p (ribophorin I), Stt3p, Wbp1p (OST48), Ost3p, Ost6p, Swp1p (ribophorin II), Ost2p (DAD1), Ost5p, and Ost4p. Ost1p is essential for OST activity. Proteins associated with mammalian OST include DAD1 (defender against apoptotic cell death), which was initially isolated as a negative regulator of programmed cell death (apoptosis). All OST subunits are transmembrane proteins, with one to eight transmembrane domain regions. In a mechanism not fully understood, the OST complex binds to the lipid-linked oligosaccharide and transfers it to nascently translated proteins by cleavage of the high-energy GlcNAc-P bond, releasing Dol-P-P in the process (Figure 7.3).
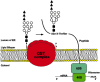
Figure 7.3
The OST complex residing in the ER membrane transfers the dolichol oligosaccharide precursor to asparagine residues on nascently translated proteins. In yeast, at least nine distinct subunits comprise the complex, including Ost1p, Stt3p, Wbp1p, Ost3p, (more...)
Early studies of peptides modified by N-glycosylation resolved that the minimal sequence requirement surrounding the acceptor asparagine is Asn-X-Thr/Ser, where X is any amino acid except for proline. (In some rare cases, Asn-X-Cys is also utilized for N-glycosylation.) Recent studies of a model rabies virus glycoprotein suggest that the identity of the amino acid following the Ser/Thr residues can affect the efficiency of glycosylation. Only about 30% of potential N-glycosylation sites appear to be used among all polypeptides bearing this consensus sequence, although this percentage can vary significantly among specific glycoproteins. Studies with model glycoproteins indicate that transfer of the dolichol oligosaccharide precursor occurs approximately 30 amino acids from the active ribosome, consistent with results involving protease treatment of microsomal fractions and spacing requirements for transfer in the luminal compartment of the ER.
Initial Steps in N-Glycan Processing and Control of Protein Folding (18–29)
Following the covalent attachment of the oligosaccharide from the dolichol oligosaccharide precursor to asparagine residues, a series of processing reactions occurs. The first several steps appear to be conserved among all eukaryotic cells and are now known to play key roles in regulating vertebrate glycoprotein folding and lysosomal trafficking.
Glucosidases I and II act first on the protein-linked oligosaccharide precursor to remove all three glucoses sequentially. These glycosidases are present in the lumen of the ER with glucosidase I acting specifically on the single α1–2-linked terminal glucose. Where it has been examined (e.g., using tunicamycin), the rate-limiting step in N-glycosylated protein secretion appears to be due in large part to the time a glycoprotein spends in the ER. Removal of the first two outer glucoses occurs rapidly and is followed minutes later by removal of the third. Removal of glucose residues is associated with protein folding mechanisms, and this process contributes to the ER retention time of a given glycoprotein (Figure 7.4). Improperly folded proteins are reglucosylated by an α-glucosyltransferase located in the lumen of the ER, which acts on the Man9GlcNAc2-Asn oligosaccharide of improperly folded proteins. The glucosyltransferase may function as a sensor for improperly folded proteins; however, the nature of this sensor is presently unresolved. Re-glucosylated N-glycans are retained in the ER where they are either refolded into a proper conformation, or they are deglucosylated and degraded.
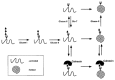
Figure 7.4
Calnexin functions during glycoprotein folding in the ER. With removal of glucose residues by glucosidase (Glcase) I and II, the glycoprotein either is properly folded and ready for further processing or is reglucosylated by a glucosyltransferase (Glc-T). (more...)
An understanding of how glucose-bearing N-glycan precursors modulate protein folding began with the discovery of molecular chaperones that bind to newly translated proteins in the ER. One such chaperone, termed calnexin, was first identified in association with the signal sequence receptor (SSRα) and also with newly translated major histocompatibility complex class I heavy chain. Subsequent studies indicated that calnexin selectively binds to newly synthesized proteins that are incompletely folded. Moreover, it was determined that calnexin is a lectin and binds specifically to glycoproteins in the secretory pathway bearing N-glycans, but not to nonglycosylated secreted proteins. In studies to refine the lectin-binding specificity, the presence of the innermost glucose-linked α3 to mannose was found to be crucial.
The major points in the stepwise mechanism by which glucosidase I, glucosidase II, calnexin, and α-glucosyltransferase act are relatively clear. The initiating event is the processing of the N-glycan precursor by glucosidases I and II to yield Glc1Man9GlcNAc2. Studies using ribonuclease B as a model glycoprotein indicate that calnexin binds independently of protein conformation by virtue of the single remaining α1–3-linked glucose on the Glc1Man8GlcNAc2-Asn oligosaccharide structure and that this binding is in a dynamic state of equilibrium. Calnexin appears to be acting purely as a lectin and may be best described as a retention factor in the protein-folding pathway involving molecular chaperones. In this model, calnexin binding does not prevent protein folding, but rather acts as a “docking” molecule during the protein-folding process.
A second lectin with partially overlapping specificity has been identified and termed calreticulin, which exists as a soluble lectin in the lumen of the ER. Both calnexin and calreticulin appear to promote the proper folding of N-glycans in part by inhibiting inappropriate or premature protein oligomerization and by reducing N-glycan degradation. Interestingly, the specificity of N-glycans bound appears somewhat different between calnexin and calreticulin; however, the biological relevance of this is presently unclear. A few glucose-containing N-glycan precursors have been occasionally observed in the Golgi of some cells, possibly having escaped regulation by this pathway. However, an endo-mannosidase activity is able to act on these precursors, thereby generating the Man8GlcNAc2-Asn oligosaccharide structure that can then continue in the N-glycan processing and secretory pathways (see below). In cells treated with glucosidase inhibitors castanospermine and 1-deoxynojirimycin, or deficient in either glucosidase I or II, calnexin does not associate with glycan substrates, and increased misfolding of certain proteins has been observed in some cases. It has been suggested that only a subset of glycoproteins is conformationally affected by loss of calnexin function. It is also possible that only a subset of cellular glycoproteins may be influenced during folding in the ER by the role of glucosylation and deglucosylation as outlined above.
N-Glycan Processing and Trafficking Involving Terminal Mannose Linkages (30–34)
Following glucose trimming and release from the ER, N-glycans become available for glycosidase reactions in the ER and Golgi. These N-glycans are referred to as the high-mannose subtype, indicating that they terminate in unsubstituted mannose residues. Following the production of the Man8GlcNAc2-Asn structure in the ER, this N-glycan is found in the cis-Golgi where two avenues exist in the processing pathway (Figure 7.5). On certain glycoproteins, the Man8GlcNAc2-Asn N-glycan is modified by a GlcNAc-phosphotransferase, and the GlcNAc residues are subsequently removed yielding Man-6-P. Although the precise location in the ER/Golgi of these reactions is not presently established, this modification is the key structural determinant in targeting glycoproteins to the lysosomal compartment and forms the basis of the first human malady to be recognized as a defect in glycosylation (I-cell disease, Chapter 23).

Figure 7.5
Processing of vertebrate N-linked oligosaccharides in the ER and Golgi. Most N-glycan processing takes place through the action of an ER-localized α-mannosidase I followed by a Golgi-resident isozyme. Some that have escaped into the Golgi with (more...)
In most cases, however, distinct α-mannosidase enzymes located in the ER and Golgi sequentially process the high-mannose N-glycans (Figure 7.5). These enzymes are termed class I α-mannosidases and include α-mannosidase 1A and 1B which act specifically on α1–2-linked mannose residues. Although each is capable of generating the Man5GlcNAc2-Asn structure from the Man9-containing precursor in vitro, differences in their subcellular distributions exist. N-glycan processing in mammals is initially similar to that in yeast in that a Man8GlcNAc2-Asn structure is generated from the Man9-GlcNAc2-Asn structure in the ER by removal of an α1–2-linked mannose on the middle chain. Next, and in contrast with vertebrate cells, yeast add mannose to this structure to produce high-mannose N-glycans with up to 15 mannose residues. In vertebrates, cell surface and secreted high-mannose N-glycans (Man9 and smaller) can normally be found, generally at low levels, as most often processing has resulted in a Man5GlcNAc2-Asn glycan that becomes a substrate in the Golgi for the diversification of extracellular N-glycans (see below).
An exception to the above processing reactions exists as the result of a unique endo-mannosidase in vertebrates that acts on glucosylated N-glycan precursors in the Golgi that have escaped glucosidase action in the ER. The product of the endo-mannosidase reaction is Man8–3GlcNAc2-Asn, and this is the enzyme that may account for the inability of glucosidase inhibitors to completely block the production of further processing events in N-glycan diversification.
The Diversification of N-Glycans (35–42)
An understanding of substrate specificities regarding the glycosyltransferases and glycosidases acting in N-glycan biosynthesis has been obtained during the last two decades by analyses of lectin-resistant cell lines deficient in specific glycosyltransferases and glycosidases, as well as from the use of cell-free systems that process N-glycans in vitro. These specificities involve the oligosaccharide moieties from the previous enzymatic step. The processed high-mannose Man5GlcNAc2-Asn N-glycan serves as a substrate for the diversification of N-glycans in the Golgi. Extracellular N-glycans in vertebrates exist as high-mannose, hybrid, or complex subtypes (Figure 7.6). Hybrid structures are defined as those with both substituted (GlcNAc linkage) and unsubstituted mannose residues. Complex N-glycans refer to those in which both the α3- and α6-linked mannose residues are substituted with GlcNAc moieties. When total N-glycans are analyzed from various cells, most vertebrate extracellular N-glycans are found to be of the complex subtype.

Figure 7.6
Vertebrate N-glycan diversification in the Golgi as shown generates three N-glycan subtypes: high-mannose, hybrid, and complex. Most secreted and cell surface N-glycans are of the complex type and are generated by one of two possible routes. α1–6 (more...)
The first enzyme needed in building diverse N-glycan structures is termed GlcNAcT-I and is encoded by a single gene in mammals (Mgat1). GlcNAcT-I adds a GlcNAc in β1–2 linkage to produce a hybrid N-glycan structure that is the substrate for α-mannosidase II activity. α-Mannosidase II acts specifically on the GlcNAc1Man5GlcNAc2-Asn hybrid N-glycan in the medial Golgi to remove the α1–3-linked and α1–6-linked mannose residues as depicted (Figure 7.6). The resulting processed hybrid N-glycan product (GlcNAc1Man3GlcNAc2-Asn) is the specific substrate for the Mgat2-encoded enzyme GlcNAcT-II that catalyzes the conversion of hybrid to complex N-glycans. It was initially thought that the route to complex N-glycans always required α-mannosidase II processing; however, studies of α-mannosidase-II-deficient mice have indicated the presence of an alternate pathway independent of α-mannosidase II function. The candidate enzyme in this alternate pathway is a distinct α-mannosidase found enriched in Golgi fractions (termed α-mannosidase III) that acts on the processed high-mannose N-glycan Man5GlcNAc2-Asn, producing Man3GlcNAc2-Asn. This latter N-glycan is also found to be an excellent substrate for GlcNAcT-I in vitro, yielding GlcNAc1Man3GlcNAc2-Asn by this alternate route.
N-glycans of the hybrid and complex type may exist with two or more GlcNAc-bearing branches that are referred to as antennae. In forming multi-antennary N-glycan structures, GlcNAc residues may be added to the trimannosyl core by six different GlcNAc transferases (I–VI) (Figure 7.7). Up to five branches have been observed on N-glycans of some vertebrate glycoproteins. GlcNAcT-I and GlcNAcT-II must both act to produce a complex N-glycan. All GlcNAc residues except that added by GlcNAcT-III can be extended with additional monosaccharide linkages. Hybrid N-glycans can be mono- or biantennary; the latter have two branches on the α1–3-linked mannose as a result of GlcNAcT-I and GlcNAcT-IV action. Some reactions preclude others, so that only a subset of linkages performed by the GlcNAc transferases can occur on any one N-glycan structure. This may be due to competition between enzymes for the same substrate. For example, GlcNAcT-III can act on the hybrid N-glycan GlcNAc1Man5GlcNAc2-Asn, and if it does, α-mannosidase II cannot cleave the two outer mannose residues; thus, the N-glycan is forever of the “unprocessed hybrid” subtype. GlcNAcT-IV branching is inhibited by GlcNAcT-III action but appears optimal on N-glycans bearing the GlcNAcT-V branch. GlcNAcT-V requires the prior activity of GlcNAcT-II. GlcNAcT-III and GlcNAcT-V are sometimes exclusive, as the action of GlcNAcT-III inhibits the action of GlcNAcT-V. GlcNAcT-VI action is uncommon, but it may require prior branching by GlcNAcT-II and GlcNAcT-V.
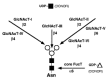
Figure 7.7
UDP-GlcNAc transferases isolated and characterized in N-glycan branching and the GDP-α1–6 fucosyltransferase involved in core fucosylation in animal cells. Not all reactions shown occur on any given N-glycan as some glycosyltransferases (more...)
Core fucosylation is a common structural feature among N-glycans. After processing reactions in the ER and early Golgi, many vertebrate N-glycans are modified by a fucosyltransferase that adds fucose in an α1–6 linkage to the GlcNAc residue that is linked to asparagine (Figures 7.6 and 7.7). This modification is found on hybrid and complex N-glycan subtypes following GlcNAcT-I action and is inhibited by the bisecting GlcNAc added by GlcNAcT-III. Another core fucosylation reaction commonly occurs in plant and invertebrate N-glycans in which fucose is added in α1–3 linkage also to the asparagine-linked GlcNAc residue (see Chapter 20).
Multiple N-glycosylation sites on the same protein may contain different glycan structures, a finding referred to as microheterogeneity. It appears that factors other than protein sequence may influence N-glycan diversification. Such factors may include sugar nucleotide metabolism, transport rates in the ER and Golgi, and the localization of glycosyltransferases in the “assembly line” model of their action in the Golgi which can determine which enzymes encounter glycan substrates first. As the N-glycan transits through the medial- and trans-Golgi, it becomes available to glycosyltransferases increasingly localized toward the end of the assembly line and which therefore control more distal structural modifications (e.g., the sialyltransferases and sulfotransferases in the trans-Golgi; see Chapter 15). Therefore, varied expression levels of glycosyltransferases can affect the repertoire of N-glycan structures produced in a given cell type. Some glycosyltransferase and glycosidase genes contain promoter regions bearing transcription-factor-binding elements that function in growth regulatory and oncogene transformation pathways, including motifs used by the ets gene family (see Chapters 34 and 35). Another mechanism controlling N-glycan diversity may include cellular regulators of glycosyltransferase activity. For example, α-lactalbumin in milk has been found to alter the acceptor specificity of a β1–4 galactosyltransferase to recognize glucose instead of N-acetylglucosamine (see Chapter 17).
The General Biology of N-Glycans (43–70)
Variation in N-glycan diversity occurs during vertebrate embryogenesis, cell activation, morphogenesis, cell cycle entry, and cellular transformation by oncogenes (see Chapters 34 and 35). In early studies to define N-glycan function, tunicamycin treatment or site-directed mutagenesis of the Asn-X-Ser/Thr motif was accomplished, precluding all N-glycan formation. It is now known that the oligosaccharide precursor and core structure can be necessary early in protein folding in the ER, thus precluding an analysis of specific linkages in N-glycan function. Nevertheless, those proteins still secreted may exhibit alterations in half-life in serum (e.g., erythropoietin), and such proteins may also show aberrant or defective binding properties. An interesting example of such a role for an N-glycan involves a sulfated GalNAc residue present on hormones including lutropin, thyroid-stimulating hormone, and pro-opiomelanocortin (see Chapter 16). This unique N-glycan structure controls glycoprotein half-life. A receptor for sulfated GalNAc is located in the liver endothelium and rapidly removes glycoproteins bearing this N-glycan from circulation. This is an example of a glycan structure on an extremely restricted subset of cellular glycoproteins that appears focused to provide a specific function. Most N-glycan functions have been difficult to detect in cultured animal cells, however, as lectin-resistant cell lines that are deficient in specific glycosyltransferases and glycosidases are commonly viable and do not provide good clues of glycan function (see Chapter 31). Therefore, studies of enzyme deficiencies in intact organisms have become a focus for understanding N-glycan function.
Both naturally occurring and experimentally induced mutations in N-glycan biosynthesis have been identified and studied in vertebrates (Table 7.1). The first recognized defect involved absence of the Man-6-P signal resulting in loss of lysosomal trafficking, and a severe syndrome termed I-cell disease (see Chapter 23). Some naturally occurring variations are species-specific, such as the α1–3Gal terminal structure. Its induced deficiency causes cataracts in mice and generates a major xenotransplantation barrier in humans. The carbohydrate-deficient glycoprotein syndromes (CDGS) comprise a number of relatively severe genetic diseases in humans (see Chapter 32). These syndromes are caused by incomplete or failed production in early steps of N-glycan biosynthesis. As a result, glycoproteins exist with fewer Asn-X-Ser/Thr sequons bearing N-glycan moieties. Several CDGS types exist and have been termed types I–V, with subtypes being present (e.g., Ia and Ib, see Table 7.1). All CDGS typing thus far has been based on the isoelectric focusing pattern of serum transferrin obtained in the clinic. A unique transferrin pattern representing an abnormal N-glycan profile can be characteristic of more than one specific enzyme deficiency. The etiology and nomenclature of CDGS type I variants are still evolving. All cases of CDGS type I have been found to be deficiencies in enzymes operating early in the production of the dolichol oligosaccharide precursor, and the syndromes differ somewhat in their clinical presentation. This may be due to the nature of the structural defect in the precursor and how well it is able to be attached and processed in the ER and early Golgi. However, all CDGS type I cases present as childhood diseases with failure to thrive. Interestingly, phosphomannose isomerase deficiency (often termed type 1b) is clinically cured by mannose ingestion. The defect in CDGS type II involves loss of Mgat2 gene function with an absence of GlcNAcT-II activity and complex N-glycans. Few patients presently have been identified and they are more severely affected than CDGS type I individuals. Symptoms are many and include mental retardation, locomotor dysfunction, and susceptibility to infection. Genetic diseases of glycosylation in humans continue to be found as medical awareness increases.
Table 7.1
Enzyme deficiencies and effects in vertebrate N-glycan biosynthesis.
Experimentally induced glycosyltransferase and glycosidase deficiencies in the intact organism are providing much information on N-glycan function (see Chapter 33). Mouse embryos deficient in Mgat1-gene-encoded GlcNAcT-I die between embryonic days 9 and 10 (equivalent to the end of the first trimester in human gestation time). In comparison, the GlcNAcT-I-deficient Lec 1 CHO cell line is viable and proliferates normally in vitro. Morphogenic abnormalities were present in embryos lacking GlcNAcT-I, including defects in neural tube formation, situs inversus of the heart, and impaired vascularization. Two examples of embryonic lethality exist in models of N-glycan biosynthetic deficiency as of this writing. The other is a deficiency in GlcNAc-1-phosphotransferase which is required to build the dolichol oligosaccharide precursor and without which N-glycosylation cannot occur. Both mutations inhibit early biosynthetic steps where no overlap exists with another isozyme, and thus the glycan product cannot be made by an alternate pathway. With other studies, the presence of alternate pathways may be evident. Studies of mice lacking α-mannosidase II revealed that this enzyme is essential for complex N-glycan biosynthesis only in erythroid cell types where a dyserythropoiesis develops. Such studies may aid in understanding the etiology of some cases of human CDA type II where this enzyme is sometimes deficient.
Hematopoiesis, immune function, and inflammation responses are frequently affected in vertebrates lacking specific extracellular N-glycan linkages. For example, loss of FucT-VII results in a profound leukocytosis with deficits in inflammatory responses and lymphoid homing to the lymph nodes (see Chapter 26). This appears to reflect the essential role of selectins and their carbohydrate ligands in regulating these processes. Genetic deficiency of the ST6Gal-I sialyltransferase ablated formation of the Sia6LacNAc trisaccharide (Siaα2-6Galβ1–4GlcNAc-) and resulted in an immunodeficiency with defective B-lymphocyte activation and attenuated antibody production upon immunization. Such model systems provide a means to investigate how glycans are capable of modulating biological processes and will likely lead to an understanding as to why the hundreds of glycosyltransferase and glycosidase genes have been conserved throughout vertebrate phylogeny.
Future Directions
As N-glycan biosynthesis is becoming relatively well resolved, studies of induced and sporadic deficiencies of N-glycan biosynthesis in intact organisms will provide a rich source of information on the physiologic processes in which N-glycans are involved. Such studies may further disclose the presence of alternate biosynthetic pathways, or additional isozymes, present in various cell types. N-glycan function may also encompass aspects of pathogen-host interactions during infection and the immune responses that normally follow (see Chapters 3, 28, 33, and 36). Addressing the mechanism of how N-glycans regulate physiology will involve identifying those glycoproteins bearing the targeted or modified glycan structure, and assessing the function of these altered glycoconjugates. Recent studies indicate that a restricted set of glycoproteins may be generated in an organism from any one glycosyltransferase, especially if that enzyme is part of an isozyme family and is specifically expressed in certain cell types. Structural analyses of oligosaccharides associated with mutant genotypes and phenotypes will be necessary in fully characterizing N-glycan structure-function relationships. Understanding the role of N-glycans in physiology is of increasing relevance to cellular biologists in various disciplines who study glycoproteins yet lack information regarding the function of the attached glycans.
References
- 1.
- Abeijon C, Mandon E C, Hirschberg C B. Transporters of nucleotide sugars, nucleotide sulfate and ATP in the Golgi apparatus (review). Trends Biochem. Sci. 1997;22:203–207. [PubMed: 9204706]
- 2.
- Hirschberg C B, Robbins P W, Abeijon C. Transporters of nucleotide sugars, ATP and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annu. Rev. Biochem. 1998;67:49–69. [PubMed: 9759482]
- 3.
- Hirschberg C B, Snider M D. Topography of glycosylation in the rough endoplasmic reticulum and Golgi apparatus. Annu. Rev. Biochem. 1987;56:63–87. [PubMed: 3304145]
- 4.
- Hubbard S C, Ivatt R J. Synthesis and processing of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1981;50:555–583. [PubMed: 7023366]
- 5.
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1985;54:631–664. [PubMed: 3896128]
- 6.
- Liu T, Stetson B, Turco S J, Hubbard S C, Robbins P W. Arrangement of glucose residues in the lipid-linked oligosaccharide precursor of asparaginyl oligosaccharides. J. Biol. Chem. 1979;254:4554–4559. [PubMed: 220251]
- 7.
- Robbins P W, Hubbard S C, Turco S J, Wirth D F. Proposal for a common oligosaccharide intermediate in the synthesis of membrane glycoproteins. Cell. 1977;12:893–900. [PubMed: 202393]
- 8.
- Robbins P W. Genetic regulation of asparagine-linked oligosaccharide synthesis. Biochem. Soc. Trans. 1991;19:642–645. [PubMed: 1783191]
- 9.
- Rosner M R, Hubbard S C, Ivatt R J, Robbins P W. N-asparagine-linked oligosaccharides: Biosynthesis of the lipid-linked oligosaccharides. Methods Enzymol. 1982;83:399–408. [PubMed: 7098942]
- 10.
- Rush J S, Waechter C J. Transmembrane movement of a water-soluble analogue of mannosylphosphoryldolichol is mediated by an endoplasmic reticulum protein. J. Cell Biol. 1995;130:529–536. [PMC free article: PMC2120537] [PubMed: 7622555]
- 11.
- Snider M D, Robbins P W. Transmembrane orientation of protein glycosylation. Mature oligosaccharide-lipid is located on the luminal side of microsomes from Chinese hamster ovary cells. J. Biol. Chem. 1982;257:6796–6801. [PubMed: 7085604]
- 12.
- Struck D.K. and Lennarz W.J. 1980. The function of saccharide-lipids in synthesis of glycoproteins. In The biochemistry of glycoproteins and proteoglycans (ed. Lennarz W.J.), pp. 35–83. Plenum Press, New York.
- 13.
- Fu J, Ren M, Kreibach G. Interactions among subunits of the oligosaccharyltransferase complex. J. Biol. Chem. 1997;272:29687–29692. [PubMed: 9368036]
- 14.
- Kelleher D J, Gilmore R. DAD1, the defender against apoptotic cell death, is a subunit of the mammalian oligosaccharyltransferase. Proc. Natl. Acad. Sci. 1997;94:4994–4999. [PMC free article: PMC24619] [PubMed: 9144178]
- 15.
- Mellquist J L, Kasturi L, Spitalnik S L, Shakin-Eshleman S H. The amino acid following an Asn-X-Ser/Thr sequon is an important determinant of N-linked core glycosylation efficiency. Biochemistry. 1998;37:6833–6837. [PubMed: 9578569]
- 16.
- Silberstein S, Gilmore R. Biochemistry, molecular biology, and genetics of the oligosaccharyltransferase. FASEB J. 1996;10:849–858. [PubMed: 8666161]
- 17.
- Kelleher D J, Gilmore R. The Saccharomyces cerevisiae oligosaccharyltransferase is a protein complex composed of Wbp1p, Swp1p, and four additional polypeptides. J. Biol. Chem. 1994;269:12908–12917. [PubMed: 8175708]
- 18.
- Grinna L S, Robbins P W. Glycoprotein biosynthesis. Rat liver microsomal glucosidases which process oligosaccharides. J. Biol. Chem. 1979;254:8814–8818. [PubMed: 479161]
- 19.
- Hammond C, Braakman I, Helenius A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. 1994;91:913–917. [PMC free article: PMC521423] [PubMed: 8302866]
- 20.
- Hebert D N, Foellmer B, Helenius A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell. 1995;81:425–433. [PubMed: 7736594]
- 21.
- Labriola C, Cazzulo J J, Parodi A J. Retention of glucose units added by the UDP-Glc:glycoprotein glucosyltransferase delays exit of glycoproteins from the endoplasmic reticulum. J. Cell Biol. 1995;130:771–779. [PMC free article: PMC2199956] [PubMed: 7642696]
- 22.
- Ora A, Helenius A. Calnexin fails to associate with substrate proteins in glucosidase-deficient cell lines. J. Biol. Chem. 1995;270:26060–26062. [PubMed: 7592804]
- 23.
- Ou W -J, Cameron P H, Thomas D Y, Bergeron J J M. Association of folding intermediates of glycoproteins with calnexin during protein maturation. Nature. 1993;364:771–776. [PubMed: 8102790]
- 24.
- Parodi A J, Mendelzon D H, Lederkremer G Z. Transient glucosylation of protein-bound Man9GlcNAc2, Man8GlcNAc2, and Man7GlcNAc2 in calf thyroid cells. A possible recognition signal in the processing of glycoproteins. J. Biol. Chem. 1983;258:8260–8265. [PubMed: 6863289]
- 25.
- Saunier B, Kilker R D Jr.,, Tkacz J S, Quaroni A, Herscovics A. Inhibition of N-linked complex oligosaccharide formation by 1-deoxynojirimycin, an inhibitor of processing glucosidases. J. Biol. Chem. 1982;257:14155–14161. [PubMed: 6216253]
- 26.
- Sousa M, Parodi A J. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glucosyltransferase. EMBO J. 1995;14:4196–4203. [PMC free article: PMC394502] [PubMed: 7556060]
- 27.
- Tatu U, Helenius A. Interactions between newly synthesized glycoproteins, calnexin and a network of resident chaperones in the endoplasmic reticulum. J. Cell Biol. 1997;136:555–565. [PMC free article: PMC2134297] [PubMed: 9024687]
- 28.
- Turco S J, Robbins P W. The initial stages of processing of protein-bound oligosaccharides in vitro. J. Biol. Chem. 1979;254:4560–4567. [PubMed: 438206]
- 29.
- Zapun A, Petrescu S M, Rudd P M, Dwek R A, Thomas D Y, Bergeron J M. Conformation-independent binding of monoglucosylated ribonuclease B to calnexin. Cell. 1997;88:29–38. [PubMed: 9019402]
- 30.
- Dairaku K, Spiro R G. Phylogenetic survey of endomannosidase indicates late evolutionary appearance of this N-linked oligosaccharide processing enzyme. Glycobiology. 1997;7:579–586. [PubMed: 9184840]
- 31.
- Moremen K W, Trimble R B, Herscovics A. Glycosidases of the asparagine-linked oligosaccharide processing pathway. Glycobiology. 1994;4:113–125. [PubMed: 8054711]
- 32.
- Reitman M L, Varki A, Kornfeld S. Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5′-diphosphate-N-acetylglucosamine:glycoprotein N-acetylglucosaminylphosphotransferase activity. J. Clin. Invest. 1981;67:1574–1579. [PMC free article: PMC370727] [PubMed: 6262380]
- 33.
- Spiro M J, Bhoyroo V D, Spiro R G. Molecular cloning and expression of rat liver endo-α-mannosidase, an N-linked oligosaccharide processing enzyme. J. Biol. Chem. 1997;272:29356–29363. [PubMed: 9361017]
- 34.
- Tulsiani D R, Hubbard S C, Robbins P W, Touster O. α-d-mannosidases of rat liver Golgi membranes. Mannosidase II is the GlcNAcMan5-cleaving enzyme in glycoprotein biosynthesis and mannosidases Ia and IB are the enzymes converting Man9 precursors to Man5 intermediates. J. Biol. Chem. 1982;257:3660–3668. [PubMed: 7061502]
- 35.
- Beyer T.A. and Hill R.L. 1982. Glycosylation pathways in the biosynthesis of nonreducing terminal sequences in oligosaccharides of glycoproteins. In The glycoconjugates (ed. Horowitz M. and Pigman W.), vol. III, pp. 25–45. Academic Press, New York.
- 36.
- Brisson J -R, Carver J P. The relation of three-dimensional structure to biosynthesis in the N-linked oligosaccharides. Can. J. Biochem. Cell Biol. 1983;61:1067–1078. [PubMed: 6627107]
- 37.
- Brockhausen I, Hull E, Hindsgaul O, Schachter H, Shah R N, Michnick S W, Carver J P. Control of glycoprotein synthesis: Detection and characterization of a novel branching enzyme from hen oviduct, UDP-N-acetylglucosamine:GlcNAcβ1–6 (GlcNAcβ1–2)Manβ-R (GlcNAc TO Man) β-4-N-acetylglucosaminyltransferase VI. J. Biol. Chem. 1989;264:11211–11221. [PubMed: 2525556]
- 38.
- Harpaz N, Schachter H. Control of glycoprotein synthesis: Processing of asparagine-linked oligosaccharides by one or more rat liver golgi α-d-mannosidases dependent on the prior action of UDP-N-acetylglucosamine:α-d-mannoside β2-N-acetylglucosaminyltransferase I. J. Biol. Chem. 1980;255:4894–4902. [PubMed: 6445359]
- 39.
- Lis H, Sharon N. Protein glycosylation: Structural and functional aspects. Eur. J. Biochem. 1993;218:1–27. [PubMed: 8243456]
- 40.
- Narasimhan S. Control of glycoprotein synthesis: UDP-GlcNAc:glycopeptide β4-N-acetylglucosaminyltransferase III, an enzyme in hen oviduct which adds GlcNAc in β1–4 linkage to the β-linked mannose of the trimannosyl core of N-glycosyl oligosaccharides. J. Biol. Chem. 1982;257:10235–10242. [PubMed: 6213618]
- 41.
- Nishikawa Y, Pegg W, Paulsen H, Schachter H. Control of glycoprotein synthesis: Purification and characterization of rabbit liver UDP-N-acetylglucosamine:α-3-d-mannoside β-1,2-N-acetylglucosaminyltransferase I. J. Biol. Chem. 1988;263:8270–8281. [PubMed: 2967294]
- 42.
- Schachter H. Biosynthetic controls that determine the branching and microheterogeneity of protein-bound oligosaccharides. Biochem. Cell Biol. 1986;64:163–181. [PubMed: 3521675]
- 43.
- Asano M, Furukawa K, Kido M, Matsumoto S, Umesaki Y, Kochibe N, Iwakura Y. Growth retardation and early death of β-1,4-galactosyltransferase knockout mice with augmented proliferation and abnormal differentiation of epithelial cells. EMBO J. 1997;16:1850–1857. [PMC free article: PMC1169788] [PubMed: 9155011]
- 44.
- Brew K, Vanaman T C, Hill R L. The role of α-lactalbumin and the A protein in lactose synthetase: A unique mechanism for the control of a biological reaction. Proc. Natl. Acad. Sci. 1968;59:491–497. [PMC free article: PMC224699] [PubMed: 5238979]
- 45.
- Burda P, Borsig L, de Rijk-van Andel J, Wevers R, Jaeken J, Carhon H, Berger E G, Eabi M. A novel carbohydrate-deficient glycoprotein syndrome characterized by a deficiency in glucosylation of the dolichol-linked oligosaccharide. J. Clin. Invest. 1998;102:647–652. [PMC free article: PMC508925] [PubMed: 9710431]
- 46.
- Charuk H M, Tan J, Bernardini M, Haddad S, Reithmeier R A, Jaeken J, and Schachter H. Carbohydrate-deficient glycoprotein syndrome type II: An autosomal recessive N-acetylglucosaminyltransferase II deficiency different from typical hereditary erythroblastic multinuclearity, with a positive acidified-serum lysis test (HEMPAS). Eur. J. Biochem. 1995;230:797–805. [PubMed: 7607254]
- 47.
- Chui D, Oh-Eda M, Liao Y -F, Panneerselvam K, Lal A, Marek K W, Freeze H H, Moremen K W, Fukuda M N, Marth J D. α-mannosidase-II deficiency results in dyserythropoiesis and unveils an alternate pathway in oligosaccharide biosynthesis. Cell. 1997;90:157–167. [PubMed: 9230311]
- 48.
- Crookston J H, Crookston M C, Burnie K L, Francombe W H, Dacie J V, Davis J A, Lewis S M. Hereditary erythroblastic multinuclearity associated with a positive acidified-serum test: A typical congenital dyserythropoietic anaemia. Br. J. Haematol. 1969;17:11–26. [PubMed: 5807784]
- 49.
- Demetriou M, Nabi I R, Coppolino M, Dedhar S, Dennis J W. Reduced contact-inhibition and substratum adhesion in epithelial cells expressing GlcNAc-transferase V. J. Cell Biol. 1995;130:383–392. [PMC free article: PMC2199932] [PubMed: 7615638]
- 50.
- Fukuda M N. Congenital dyserythropoietic anaemia type II (HEMPAS) and its molecular basis. Baillieres Clin. Haematol. 1993;6:493–511. [PubMed: 8043936]
- 51.
- Fukuda M N, Masri K A, Dell A, Luzzatto L, Moremen K W. Incomplete synthesis of N-glycans in congenital dyserythropoietic anemia type II caused by a defect in the gene encoding α-mannosidase II. Proc. Natl. Acad. Sci. 1990;87:7443–7447. [PMC free article: PMC54763] [PubMed: 2217175]
- 52.
- Granovsky M, Fata J, Khokha R, Dennis J W. N-acetylglucosaminyltransferase V null mice show hypersensitivity to T cell mitogens and intestinal hyperplasia. Glycobiology. 1998;8:A156.
- 53.
- Hennet T, Chui D, Paulson J C, Marth J D. Immune regulation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. 1998;95:4504–4509. [PMC free article: PMC22519] [PubMed: 9539767]
- 54.
- Hill R L, Brew K. Lactose synthetase. Adv. Enzymol. Relat. Areas Mol. Biol. 1975;43:411–490. [PubMed: 812340]
- 55.
- Ioffe E, Stanley P M. Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc. Natl. Acad. Sci. 1994;91:728–732. [PMC free article: PMC43022] [PubMed: 8290590]
- 56.
- Jaeken J, Carchon H, Stibler H. The carbohydrate-deficient glycoprotein syndromes: pre-Golgi and Golgi disorders? Glycobiology. 1993;3:423–428. [PubMed: 8286854]
- 57.
- Jaeken J, Schachter H, Carchon H, De Cock P, Coddeville B, Spik G. Carbohydrate deficient glycoprotein syndrome type II: A deficiency in Golgi localized N-acetyl-glucosaminyltransferase II. Arch. Dis. Child. 1994;71:123–127. [PMC free article: PMC1029941] [PubMed: 7944531]
- 58.
- Kim S, Metha D, Srikrishna G, Murch S, Freeze H. Carbohydrate deficient glycoprotein spectrum disorders: Additional glycosylation defects and potential therapy. Glycobiology. 1998;8:A153.
- 59.
- Körner C, Lehle L, von Figura K. Abnormal synthesis of mannose 1-phosphate derived carbohydrates in carbohydrate-deficient glycoprotein syndrome type I fibroblasts with phosphomannomutase deficiency. Glycobiology. 1998;8:165–171. [PubMed: 9451026]
- 60.
- Körner C, Knauer R, Holzbach U, Hanefeld F, Lehle L, Figura K. Carbohydrate-deficient glycoprotein syndrome V: Deficiency of dolichol-P-Glc: Man9GlcNAc2-PP-dolichylglucosyltransferase. Proc. Natl. Acad. Sci. 1998;95:13200–13205. [PMC free article: PMC23759] [PubMed: 9789065]
- 61.
- Lu Q, Hasty P, Shur B D. Targeted mutation in β1,4-galactosyltransferase leads to pituitary insufficiency and neonatal lethality. Dev. Biol. 1997;181:257–267. [PubMed: 9013935]
- 62.
- Maly P, Thall A D, Petryniak B, Rogers G E, Smith P L, Marks R M, Kelly R J, Gersten K M, Cheng G Y, Saunders T L, Camper S A, Camphausen R T, Sullivan F X, Isogai Y, Hindsgaul O, Von Andrian U H, Lowe J B. The α(1,3)fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell. 1996;86:643–653. [PubMed: 8752218]
- 63.
- Manzella S M, Hooper L V, Baenziger J U. Oligosaccharides containing β1–4-linked N-acetylgalactosamine, a paradigm for protein-specific glycosylation. J. Biol. Chem. 1996;271:12117–12120. [PubMed: 8647799]
- 64.
- Manzella S M, Dharmesh S M, Baenziger J U. Developmental regulation of a pregnancy-specific oligosaccharide structure, Neuα2,6Galβ1,4GlcNAc, on select members of the rat placental prolactin family. J. Biol. Chem. 1997;272:4775–4782. [PubMed: 9030532]
- 65.
- Marek K W, Vijay I, Marth J D. A recessive deletion in the GlcNAc-1-phosphotransferase gene results in peri-implantation embryonic lethality. Glycobiology. 1999;9:1263–1271. [PubMed: 10536042]
- 66.
- Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman J J, Van Schaftingen E. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat. Genet. 1997;16:88. [PubMed: 9140401]
- 67.
- Metzler M, Gertz A, Sarkar M, Schachter H, Schrader J W, Marth J D. Complex asparagine-linked oligosaccharides are required for morphogenic events during post-implantation development. EMBO J. 1994;13:2056–2065. [PMC free article: PMC395055] [PubMed: 8187759]
- 68.
- Priatel J J, Sarkar M, Schachter H, Marth J D. Isolation, characterization, and inactivation of the mouse Mgat3 gene: The bisecting N-acetylglucosamine in asparagine-linked oligosaccharides appears dispensable for viability and reproduction. Glycobiology. 1997;7:45–56. [PubMed: 9061364]
- 69.
- Tan J, Dunn J, Jaeken J, Schachter H. Mutations in the mgat2 Gene Controlling Complex N-glycan Synthesis Cause Carbohydrate-deficient Glycoprotein Syndrome Type Ii, An Autosomal Recessive Disease with Defective Brain Development. Am. J. Hum. Genet. 1996;59:810–817. [PMC free article: PMC1914797] [PubMed: 8808595]
- 70.
- Tearle R G, Tange M G, Zannettino Z L, Katarelos M, Shinkel T A, Van Denderen B J, Lonie A J, Lyons I, Nottle M B, Cox T, Becker C, Peura A M, Wigley P L, Crawford R J, Robins A J, Pearse M J, d'Apice A J. The alpha-1,3-galactosyltransferase knockout mouse. Implications for xenotransplantation. Transplantation. 1996;61:13–19. [PubMed: 8560551]
- Background
- Synthesis of the Dolichol Oligosaccharide Precursor
- Transfer of the Dolichol Oligosaccharide Precursor to Nascent Proteins
- Initial Steps in N-Glycan Processing and Control of Protein Folding
- N-Glycan Processing and Trafficking Involving Terminal Mannose Linkages
- The Diversification of N-Glycans
- The General Biology of N-Glycans
- Future Directions
- References
- N-Glycans - Essentials of GlycobiologyN-Glycans - Essentials of Glycobiology
- VP5 [Epizootic hemorrhagic disease virus 2]VP5 [Epizootic hemorrhagic disease virus 2]gi|1706837236|gb|QDO66962.1|Protein
Your browsing activity is empty.
Activity recording is turned off.
See more...