

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Varki A, Cummings R, Esko J, et al., editors. Essentials of Glycobiology. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1999.


Essentials of Glycobiology.
Show detailsPrimary contributions to this chapter were made by J.B. Lowe (HHMI, University of Michigan Medical School, Ann Arbor) and J.D. Marth (HHMI, University of California at San Diego).
THIS CHAPTER DESCRIBES GLYCANS that are found attached to the various core components of N-glycans, O-glycans, glycolipids, and proteoglycans. Although these glycan structures can be unique to various core subtypes, many are found on more than one class of glycan. In postcore glycan biosynthesis, subterminal and terminal monosaccharide linkages existing at “outer” positions can establish the function(s) of the glycoconjugate.
Background
Glycan chain modifications found at outer or terminal positions generally result from the actions of one or more glycosyltransferases that modify glycan core acceptors, or precursors. Such acceptors are the product of biosynthetic processes discussed in Chapters 7, 8, and 9 and correspond to N-linked, O-linked, or lipid-linked structures shown in Figures 16.1 through 16.3. Most precursors are typically expressed by all cell types in a mammalian organism. Such structures include the multiantennary N-linked glycan precursor, the linear (unbranched) or biantennary O-linked glycan precursors, and linear (unbranched) ceramide-linked precursors. As noted for N-linked glycans in Chapter 7, but also for O-linked and lipid-linked glycans, other more complicated precursors can result from tissue- or cell-type-specific biosynthetic processes that yield, for example, tetra-antennary N-glycans (Figure 16.1). These more elaborate precursors are generally susceptible to the modification processes discussed here and in Chapter 17, leading to structural diversification at the termini of glycan chains.

Figure 16.1
N-glycan synthesis leads to “core” structures (theoretical multi-antennary structures in these examples; see Chapter 7 for details) that may be modified subsequently on specific GlcNAc residues by glycosylation reactions that may be tissue-specific, (more...)

Figure 16.3
Glycolipid synthesis leads to “core” structures (a neolactosylceramide structure, in this example; see Chapter 9 for details) that may be modified on specific GlcNAc residues by tissue-specific or developmentally regulated glycosylation (more...)

Figure 16.2
O-glycan synthesis leads to “core” structures (a “Core 2” structure, in this example; see Chapter 8 for details) that may be modified subsequently on specific residues by glycosylation reactions that may be tissue-specific, (more...)
Regulated Glycosylation of Constitutively Expressed Precursors to Terminal Chain Structures (1–7)
As implied by Figures 16.1 through 16.3, regulated biosynthetic processes determine the precise nature of the terminal modifications of such precursors. In contrast to precursor chain synthesis, these processes are generally regulated in a tissue- or cell-lineage-specific manner. Many terminal glycosylation reactions are regulated during embryogenesis and in the postnatal period as part of the normal developmental program (see Chapter 34). Changes in terminal glycan structures have also been observed in association with malignant transformation (see Chapter 35). Functional correlates for a number of such changes in outer chain structures have been identified and are discussed throughout this book. Most researchers in the field will agree that the majority of regulated changes observed in terminal glycosylation do not yet have a defined functional correlate. As discussed in Chapter 17, the tissue- and/or lineage-specific regulation of outer chain biosynthesis is largely a function of careful regulation of expression of the glycosyltransferases responsible for outer chain modifications. Generally speaking, the specific portfolio of terminally acting glycosyltransferases expressed in any given cell type will determine the nature of the outer chain structures expressed by that cell type.
Type-2 Chains (1–3,8–12)
The core precursors representative of N-linked, O-linked, and lipid-linked glycans are typically modified with GlcNAc residues (see Figures 16.1 through 16.3). With a few exceptions to be noted later in this chapter, virtually all subsequent modifications characterized as terminal first require modification of one or more of these GlcNAc residues by the addition of a galactose moiety in β1–4 linkage. The resulting structure has been termed a “type-2” chain; its component disaccharide is the lactosamine unit (Figure 16.4). Expression of this structure and the corresponding β1–4 galactosyltransferase(s) can therefore be considered to be rather constitutive, in contrast to most other outer chain modifications discussed here. As also illustrated in this chapter, lactosamine-terminated structures on N-linked, O-linked, and lipid-linked glycans serve as acceptor substrates for a large variety of subsequent modifications to the now subterminal GlcNAc, as well as to the terminal galactose moiety itself. An alternative to type-2 lactosamine production occurs when the “lacdiNAc” glycan structure is generated by the action of a β1–4 GalNAc transferase (GalNAcβ1–4GlcNAc; not shown, see discussion below). Aside from its role as a precursor to subsequent outer chain glycosylations, the lactosamine unit is known to be recognized by the S-type lectins (see Chapter 27). The function of such interactions in the context of the intact mammalian organism remains open to conjecture. As related in Chapter 33, efforts have been made to uncover the biological role(s) for type-2 chains through the generation and analysis of mice deficient in a β1–4 galactosyltransferase locus thought to be solely responsible for type-2 chain biosynthesis. However, it is now apparent that several different β1–4 galactosyltransferase loci can contribute to β1–4 galactosylation and type-2 chain expression, which may act to distinguish the functions of this structure among various physiologic systems of whole animals.

Figure 16.4
Modification of exposed GlcNAc moieties by galactosylation. Modification by β1–4-linked galactose residues (top) is generally found in all mammalian tissues. This reaction is catalyzed by β1–4 galactosyltransferases(s) (more...)
Type-1 Chains (4,13,14)
The GlcNAc residues that terminate N-linked, O-linked, and lipid-linked glycan core precursors may also be modified by galactose in β1–3 linkage (Figure 16.4). The resulting structure has been termed a “type-1” chain; its component disaccharide is the neolactosamine unit. In humans, expression of type-1 chains is mostly restricted to epithelia, in the gastrointestinal or reproductive tracts, for example. Such chains represent precursors to a variety of terminal structures, through modification by glycosyltransferases that can utilize structures with the Galβ1–3GlcNAc linkage and that modify either the terminal galactose moiety, or the now subterminal GlcNAc moiety. As discussed below, these include sialylated structures, and blood-group-active structures generated by fucosylation and by modification with terminal galactose or GlcNAc moieties. Expression of the type-1 chains, and the corresponding β1–3 galactosyltransferase, represents examples of structures and enzymes that are regulated in a tissue-specific manner, as is the case for many outer chain modifications. Although the type-1 structure functions as a precursor to subsequent outer chain glycosylations, specific roles for this disaccharide have not been clearly defined.
Polylactosamines (15–28)
Glycoproteins and glycolipids frequently bear glycans that include linear polymers of the type-2 lactosamine unit. These structures are termed polylactosamines. Polylactosamine biosynthesis is directed by the alternative actions of one or more β1–4 galactosyltransferases and one or more β1–3 GlcNAcT (Figure 16.5). They have been described on a variety of glycoproteins and may be components of N-glycans, O-glycans, and glycolipids. It is clear that some glycoproteins or glycolipids are preferentially modified by polylactosamine chains, relative to others. These observations imply that the enzymatic machinery responsible for polylactosamine biosynthesis is capable of discriminating between distinct glycoprotein or glycolipid molecules that are otherwise identical with respect to the exposed GlcNAc and galactose residues that serve as proximal modification points for polylactosamine chain biosynthesis. Polylactosamines have been noted to preferentially reside on multiantennary N-glycans, especially the β1–6 branch, whose synthesis is under control of GlcNAcT-V (Figure 16.6). Similarly, polylactosamine chains on O-chains associated with mucin-type glycoproteins are often preferentially displayed on the β1–6 branch whose synthesis is directed by a Core 2 β1–6 GlcNAcT (Figure 16.6). Polylactosamine length is also under apparent regulatory control, with N-linked polylactosamines generally found to be longer than O-linked chains. Chapter 17 discusses the mechanisms that might account for these regulatory events. As will be noted, polylactosamines serve as precursors for subsequent modifications, such as fucosylation and sialylation. The linear nature of these chains, and the hydrophilic character of their component disaccharide unit, predicts that they will maintain an extended linear conformation. This implies that polylactosamines may serve as scaffolds for the presentation of specific terminal glycan structures, including sialylated and/or fucosylated structures, whose functions require them to be displayed at or above the surface of the cell's glycocalyx. Polylactosamines are recognized with high affinity by mammalian S-type lectins (see Chapter 27), in a way that implies recognition of internal structures, independent of the presence of terminal galactosyl moieties. Chapter 27 discussed the physiological relevance of interactions between polylactosamines and S-type lectins.

Figure 16.5
Polylactosamine synthesis.

Figure 16.6
Polylactosamine chains.
β1–6GlcNAc Branching Structures (29–33)
Polylactosamines are subject to further glycosyltransferase-dependent arborizing modifications. Such branching modifications include addition of N-acetylglucosamine to internal galactose residues, in β1–6 linkage, through the actions of β1–6 GlcNAcTs (Figure 16.7). Branched and nonbranched polylactosamines correspond to the antigens of the Ii-blood group system. The I- and i-blood group systems were discovered through an investigation of a cold-dependent agglutinating antibody (cold agglutinin) in a patient with acquired hemolytic anemia. The relationship between cold agglutinin disease, anti-I or anti-i antibodies, and infectious agents is discussed in more detail in Chapter 37. In the instance of the anti-I antibody relevant to the discovery of the Ii-blood group system, the antibody reacted with the red cells of all but rare potential red cell donors. Nonreactive donors were classified as having the i-blood group, whereas reactive donors were assigned to the I-blood group. β1–6-branched polylactosamine structures correspond to the I-blood group antigen, a precursor to the ABO blood group antigens on erythrocytes, whereas linear polylactosamine chains correspond to the so-called i-blood group antigen. Acceptor site specificity studies indicate that two distinct β1–6 GlcNAcTs yield different types of β1–6-branched products, starting with linear polylactosamine precursors (Figure 16.7).

Figure 16.7
Polylactosamine-derived β1–6 GlcNAc branching structures associated with I-antigen expression. Linear polylactosamine chains, displayed by asparagine-linked, threonine/serine-linked or lipid-linked moieties (R) (see Figure 16.6), may be (more...)
Branching I- and i-reactive glycan chains are expressed by many human cells and tissues. The i-antigen is abundantly expressed on the surface of red cells taken from the human embryo, erythrocytes in cord blood, or during times of altered erythropoiesis. Such cells are relatively deficient in expression of I-antigen. However, during the first 18 months of life, I-antigen reactivity on red cells reaches adult levels, and i-antigen reactivity declines to very low levels. This developmentally regulated pattern is presumed to be consequent to developmental regulation of a cognate I β1–6 GlcNAcT locus. Rare individuals have been described who never express the I-antigen on their red cells, but who maintain levels of red cell i-antigen expression. It is not known if other tissues in such individuals are also deficient in I-antigen. Persons with this phenotype (i-phenotype) are presumed to be homozygous for null alleles at the β1–6 GlcNAcT locus responsible for synthesis of the β1–6-branching structures associated with I-antigen expression. However, the molecular basis for this rare phenotype and the identity of the relevant β1–6 GlcNAcT, among those currently identified by molecular cloning studies, remain to be defined. Individuals with the i-phenotype have no obvious pathophysiological phenotype associated with absence of red cell I-antigen expression.
The A, B, and H Blood Group Structures (34–71)
In humans, linear polylactosamines and their β1–6-branched variants are subject to tissue-specific modifications that form glycans of the ABO blood group system (Figures 16.8 to 16.11). This system was discovered early in the 20th century by Landsteiner and colleagues. Although they were not aware of the underlying glycan basis, their work revealed that humans could be divided into different classes according to the presence or absence of serum constituents that would agglutinate red cells isolated from other humans. We now know that these serum constituents are antibodies and that their cognate antigens correspond to glycans whose structures are genetically polymorphic. These glycan structural polymorphisms are determined by allelic glycosyltransferases with different functional properties.

Figure 16.8
Type-1 A, B, and O(H) blood group structures.

Figure 16.11
Type-4 A, B, and O(H) blood group structures.
A, B, and O(H) blood group antigens are oligosaccharide moieties based on the type-1 and type-2 precursors described above and on so-called type-3 and type-4 glycan precursors (Figures 16.8 to 16.11). The H blood group structure, in its unmodified form, results in the O blood type grouping (see below). The A, B, and H antigens are formed on these precursors by the sequential action of distinct glycosyltransferases, encoded by three genetic loci (the ABO, H, and Secretor loci) (Figure 16.12). The pathway of ABO blood group antigen synthesis begins with the modification of precursor glycans by α1–2 fucosyltransferases. These enzymes form the blood group H determinant, represented by the disaccharide unit Fucα1–2Galβ1-. The human genome encodes two different α1–2 fucosyltransferases, corresponding to the products of the H and the Secretor (Se) blood group loci. The H-α1–2 fucosyltransferase is expressed in erythrocyte precursors and utilizes type-2 and type-4 precursors to form type-2 and type-4 H antigens on red cells (Figures 16.9, 16.11, and 16.12). The Se α1–2 fucosyltransferase is expressed in epithelial cells and utilizes type-1 and type-3 precursors to form type-1 and type-3 H determinants (Figures 16.8, 16.10, and 16.12) in epithelia lining the lumen of the gastrointestinal, respiratory, and reproductive tracts and salivary glands, for example.
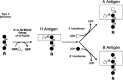
Figure 16.12
Summary of A, B, and O(H) blood group structures and their synthesis.
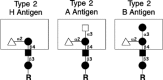
Figure 16.9
Type-2 A, B, and H blood group structures.
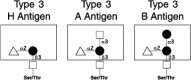
Figure 16.10
Type-3 A, B, and O(H) blood group structures.
A or B blood group determinants are subsequently formed from type-1, -2, -3, or -4 H-active glycans by codominant glycosyltransferases encoded by the ABO blood group locus. The blood group A molecule is formed by an α1–3 GalNAcT corresponding to the A allele of the ABO locus (Figure 16.12). The blood group B allele encodes an α1–3 galactosyltransferase that forms the blood group B determinant (Figure 16.12). O alleles encode functionally inert polypeptides that do not further modify H-active precursors and therefore represent null alleles at this locus. Thus, individuals capable of constructing A molecules exclusively (blood group A) will have genotypes AA or AO, whereas blood group B individuals will have the genotypes BB or BO. Individuals capable of expressing both A and B antigens (AB blood group) maintain the genotype AB. Blood group O individuals do not express either A or B antigen, leaving their H antigens unmodified, and are homozygous for the (null) O allele (genotype OO).
The ABO antigens are expressed on the surfaces of red cells and many other tissues, including the vascular endothelium, and by a variety of epithelia. In this form, ABO blood group molecules are displayed by integral membrane proteins and membrane-associated glycolipids. Some tissues also synthesize water-soluble forms of these molecules, as glycans on secreted glycoproteins, on glycosphingolipids, and on free oligosaccharides. As discussed below, the ability to express soluble ABH-active blood group molecules is a genetically determined trait that is a function of an individual's alleles at the Se locus.
On each human red blood cell, approximately 1–2 million ABH determinants are displayed by the anion transport protein, also known as Band 3. This represents approximately 80% of the total complement of red cell ABH determinants. Another 5 × 105 ABH determinants localize to the red cell glucose transport protein (Band 4.5). Both of these integral membrane proteins display ABH antigens on a single N-linked, branched polylactosamine whose terminal branches can display several ABH determinants. Small numbers of ABH antigens are also expressed by other red cell glycoproteins. Each red cell also expresses approximately 5×105 glycolipid-based ABH determinants. Many of these glycolipids correspond to A, B, and H antigen-modified polylactosamines linked to ceramide and have been termed polyglycosylceramides or macroglycolipids. A, B, and H determinants based on type-4 chains (Figure 16.11) are also represented in human red cell glycolipids.
ABH determinants expressed by the epidermis are primarily constructed from type-2 precursor chains. Mucins derived from the gastric mucosa and from ovarian cyst fluid express type-3 A, B, and H antigens. As noted above, the epithelia lining the digestive, respiratory, urinary, and reproductive tracts express type-1 oligosaccharides, as do the epithelia of some salivary and other exocrine glands. These tissues are responsible for synthesis of soluble forms of the ABH determinants, which are therefore largely represented by type-1 molecules. Expression of the A, B, and H determinants in such secretory tissues is a function of the Se locus-encoded α1–2 fucosyltransferase, since the H locus-encoded α1–2 fucosyltransferase is not expressed in these tissues. Humans homozygous for null alleles at the Se locus are incapable of synthesizing H determinants in these tissues and do not elaborate soluble forms of the H (or A or B) determinants in saliva or in other tissues, even though such tissues in these individuals continue to synthesize the soluble precursors used by the Se locus-encoded α1–2 fucosyltransferase. The term nonsecretor is used to describe the phenotype of such persons and refers to the fact that soluble blood group H, A, and B substances cannot be detected in their saliva.
Serological procedures used in the context of characterizing red cells for use in transfusion have identified variants of A and B blood group determinants. These are known as A or B subgroups. The A and B antigens of these subgroups are close structural variants of the A and B antigens, but they typically yield weak reactivity with serological typing reagents used to characterize A and B antigens on red cells. For example, the lectin Dolichos biflorus will agglutinate the red cells from most blood group A individuals (known as A1 individuals), but it will not agglutinate red cells from the rather less common A2 subgroup individuals. A antibodies may also be prepared that react with A1, but not A2, cells, for example.
The biochemical basis for the serological discrepancies between A1 and A2 cells is partly accounted for by the greater number of A-active molecules on A1 cells, relative to A2 cells. The molecular structures of A1 and A2 subgroup antigens are also distinct (Figure 16.13). The structural differences between A1 and A2 cells are accounted for by differences in the catalytic activities of the allelic blood group A transferases corresponding to A1 and A2 alleles, with the A1 transferase exhibiting a higher specific activity in vitro. It is clear that the variant antigenic reactivities of the other A and B subgroups can be accounted for by variant A and B transferase alleles, although the structural basis for these differing reactivities is not yet known.

Figure 16.13
Structural differences between the A1 and A2 subgroup antigens. The type-2 A structure representative of the A2 phenotype (left) may be synthesized by A1 or A2 subgroup transferases. The type-2 A structure is then modified by a β1–3 galactosyltransferase (more...)
The heritable red cell antigenic polymorphisms determined by the ABO locus have important medical implications. Early in life, the immune system in an individual generates IgM class antibodies directed against the ABO oligosaccharide antigens that are absent from that individual's red cells. The antibodies likely represent an immune response to oligosaccharide antigens synthesized by bacterial and fungal organisms in the environment, and whose structures are similar or identical to those of the A and B blood group molecules. For instance, type-O individuals do not make A and B determinants and therefore maintain relatively high titers of circulating IgM antibodies (termed isoagglutinins) that react with A and B blood group molecules. Similarly, blood group B individuals maintain circulating IgM class anti-A isoagglutinins, but they do not make isoagglutinins against the blood group B determinant, which is, in these individuals, a “self” antigen. Sera taken from individuals typed as blood group A contain IgM class anti-B antibodies, but not anti-A antibodies. Finally, individuals with the AB blood group do not make either anti-A or anti-B IgM class isoagglutinins. Anti-H antibodies are not made in most individuals because a substantial fraction of the H structures is not converted to A and/or B determinants, even in those with a functional A or B transferase allele.
These IgM isoagglutinins are able to efficiently trigger the complement cascade and circulate in human plasma at titers sufficient to cause complement-dependent lysis of transfused erythrocytes that display the corresponding antigen. Such acute antibody- and complement-dependent red cell lysis is associated with the clinical manifestations of an immediate, or acute, transfusion reaction, which can include hypotension, shock, acute renal failure, and death from circulatory collapse. This problem is avoided by ensuring that the ABO type of the transfused red cells is compatible with the patient's ABO type. Specifically, this means choosing red cells that are deficient in the ABO antigens which are also lacking in the recipient (i.e., an A recipient may receive red cells from another A person, or from an O person, but not from a person typed as B or from a person typed as AB). Practically speaking, this is accomplished in blood banks through procedures known as typing and cross-matching. In the typing procedure, units of red cell products, typed previously for the A and B antigens, are chosen that match the patient's ABO type. To ensure that these prospective red cell units are then truly “compatible” with the recipient, the patient's serum is cross-matched with each of the prospective donor red cell units. The cross-match is done by mixing an aliquot of the patient's serum with a small aliquot of each prospective red cell unit and examining the mixture under low-power magnification. The red cells of compatible units do not agglutinate with the patient's serum, whereas incompatibility is indicated by agglutinated red cells (agglutinated by antibodies in the recipient's serum). Although these procedures are used to ensure compatibility between the ABO phenotype of transfused red cells and recipient plasma, they are also used to ensure compatibility between the red cells circulating in a recipient who must be transfused with plasma. Similar ABO compatibility concerns are important in heart, kidney, liver, and bone marrow transplantation procedures. The “type and cross” procedures have virtually eliminated ABO blood group incompatibility-dependent red cell transfusion reactions in the United States. In the very rare instances where such a transfusion reaction has occurred, the cause is usually accounted for by clerical errors introduced during the processing of the patient's serum or the transfused red cells.
The cross-matching procedures discussed above helped to identify a rare ABO blood group phenotype termed the Bombay phenotype, so named because the first identified H-deficient individual lived in that city. These individuals were found to maintain red cells that were deficient in red cell H, A, and B antigens, whereas their sera contained IgM class antibodies that reacted with the red cells from virtually all donors, including O red cells (H-antigen-positive, A- and B-antigen-negative). Subsequent investigations indicate that these persons are homozygous for null alleles at the H α1–2 fucosyltransferase locus and are also nonsecretors (due to homozygosity for null alleles at the Se α1–2 fucosyltransferase locus). These persons therefore are incapable of synthesizing A, B, or H determinants in any tissue, maintain robust titers of circulating IgM class anti-H, anti-A, and anti-B antibodies, and are therefore cross-match-incompatible with the red cells of all except other Bombay (H-deficient) donors. A related phenotype, termed the para-Bombay phenotype, corresponds to nullizygosity at the H locus in individuals that maintain at least one functional Se α1–2 fucosyltransferase allele (secretor-positive).
Functions for the ABO blood group oligosaccharides are not known, and the processes that have led to the polymorphisms at the ABO locus are also mysterious. Indeed, the fact that Bombay individuals do not exhibit any remarkable pathological phenotype argues that functions for the A, B, and H antigens that may have existed earlier in evolution are no longer relevant. Although it has been proposed that polymorphism at the ABO locus may have provided a selective advantage for protection from certain infectious agents in prehistoric times, strong evidence to support this contention is not available. Thus, the nature of the selective pressures that yielded polymorphism at the ABO locus remains unclear.
In this context, however, it is worth noting that a variety of associations have been made between ABO blood group phenotype and the relative risk for a spectrum of diseases. Two such associations are discussed below; the remainder are not discussed because they generally represent modest and imperfect correlations, some of which have not withstood repetitive examination, and virtually all are without a clear mechanistic relationship.
The first possible association concerns the well-known but mechanistically mysterious correlation between an individual's ABO phenotype and plasma levels of von Willebrand factor (vWF), a glycoprotein involved in hemostasis. Recent studies in the mice indicate that the circulating half-life of vWF (and thus its level in plasma) varies according to the nature of the alleles at a specific glycosyltransferase locus, which encodes an enzyme that modifies vWF glycans. Specifically, in a strain of mice with very low levels of plasma vWF, the resulting von Willebrand's disease phenotype is a dominantly inherited trait that maps to a β1–4-linked GalNAcT locus for the Sda blood group, as discussed in more detail below. Moreover, the disease trait is associated with an allele of a β1–4 GalNAcT locus that directs expression of the enzyme to vascular endothelial cells, a major site of vWF synthesis. Expression of the enzyme in endothelial cells decorates the glycans on vWF with the corresponding β1–4GalNAc-linked modification. Glycoforms of vWF characterized by this β1–4-linked GalNAc modification are rapidly removed from the plasma by the hepatic asialoglycoprotein receptor. In contrast, mice with normal vWF levels do not express this β1–4 GalNAcT in vascular endothelium, so their vWF is not modified by β1–4-linked GalNAc moieties and is cleared less rapidly. Thus, inheritance of this form of murine von Willebrand's disease is accounted for by allelism at a glycosyltransferase locus. In humans, glycoform-dependent clearance mechanisms could also account for the generally lower vWF levels in persons of the O phenotype, since vWF is modified by the ABO blood group antigens in humans and since the A, B, and H blood group structures may be recognized with different affinities by the asialoglycoprotein receptor and other such lectins.
A second possible exceptional association is the proposed role for gastrointestinal ABO and Lewis blood group antigens in the pathogenesis of the ulcerogenic spirochete Helicobacter pylori (discussed in further detail below, in the context of the Lewis blood group antigens). Well-known associations exist between the group-A phenotype and a modest increase in the relative risk for stomach cancer (relative risk of 1.2) and between blood group O phenotype and a slight increase in the relative risk of developing peptic ulcers (relative risk of ~1.3). Both of these disorders are now clearly and causally associated with infection by H. pylori, but it remains to be determined if there is a mechanistic relationship between ABO-dependent ulcer and cancer risk associations and H. pylori infection (discussed again in the context of the Lewis blood group antigens below).
The oligosaccharides synthesized through the actions of the H and Se α1–2 fucosyltransferases are also without clear function. There are numerous reports of associations between secretor status and various diseases; most are weak associations and without clear mechanistic relationship. However, the increased relative risks for recurrent urinary tract infection in female nonsecretors and for peptic ulceration in nonsecretors generally are worthy of brief discussion. It has been implied that these associations may be partly accounted for by adhesive interactions between blood group substances expressed by the relevant epithelia and bacterial pathogens that use such substances to promote epithelial invasion. There is some support for this hypothesis in the context of peptic ulceration, since α1–2-fucosylated glycans constructed through the action of the Se α1–2 fucosyltransferase can support adhesion of H. pylori. However, as noted below in the context of the α1–3-fucosylated glycans of the Lewis blood group antigen family, the relationship between susceptibility to H. pylori infection and blood group status is substantially imperfect.
Lewis Blood Group Structures (72–94)
The Lewis blood group antigens correspond to a structurally similar set of α1–3-fucosylated glycan structures (Figure 16.14). The term Lewis refers to the family name of individuals suffering from a red blood cell incompatibility problem that helped lead to the discovery of this blood group.

Figure 16.14
Representative type-1 and type-2 Lewis structures. (Top) Type-2-based structures; (bottom) type-1-based structures. Type-1 and type-2 structures differ in the linkage of the outermost Gal (β1–3 and β1–4, respectively) and (more...)
The Lewis A antigen (Lea) is synthesized by an α1–3/1–4 fucosyltransferase encoded by the Lewis (Le) blood group locus (Figures 16.15). The Lewis B antigen (Leb) is synthesized by the concerted actions of the Lewis α1–3 fucosyltransferase and the α1–2 fucosyltransferase encoded by the Se blood group locus (Figure 16.15). The nature of the alleles at an individual's Le and Se loci determines the complement of Lewis-active oligosaccharide molecules that will be constructed in that individual (Figure 16.15). Secretor-positive individuals convert type-1 oligosaccharide precursors to type-1 H molecules. The resulting type-1 H determinants may then be used as precursors by the Lewis locus-encoded α1–3/1–4 fucosyltransferase to form the Leb structure (Figure 16.15). Individuals that construct Leb exhibit the Le(a-b+) phenotype (Figure 16.15). Nonsecretors do not synthesize type-1 H determinants in secretory epithelia, but such unsubstituted type-1 molecules can be converted to Lea-active oligosaccharides by the Lewis α1–3/1–4 fucosyltransferase. These individuals exhibit the Le(a+b-) phenotype (Figure 16.15). Individuals that are homozygous for null alleles at the Lewis locus can have two different phenotypes. Such Lewis-negative individuals who are positive for secretor status construct type-1 H determinants, but these structures remain unconverted to Leb determinants (Figure 16.15). These persons exhibit the Le(a-b-) phenotype. In contrast, in individuals who are Lewis-negative nonsecretors, type-1 precursors are not substituted by either the Lewis or the Secretor fucosyltransferases, accounting for absence of Lea, Leb, or H structures, and their Le(a-b-) phenotype.

Figure 16.15
Lewis blood group phenotypes. The structures of the Lewis blood-group-active glycolipids are determined by the presence or absence of the Se locus α1–2 fucosyltransferase and the Lewis locus α1–4 fucosyltransferase. R is (more...)
Expression of Lea and Leb molecules, and the Lewis α1–3/1–4 fucosyltransferase, is restricted largely to the same epithelia that express the Secretor α1–2 fucosyltransferase. Again, these epithelia elaborate soluble forms of these antigens that are released into secretions and body fluids. Lea and Leb antigens are also detectable on red cells, at a level of approximately 4500 and 7300 Lea molecules per cell. However, red cell precursors do not synthesize these molecules. Instead, Lewis antigens are acquired by the red cell membrane through passive adsorption of Lewis-positive glycosphingolipid molecules that circulate in plasma as lipoprotein complexes and aqueous dispersions.
Antibodies against the Lea antigens have been implicated in occasional instances of transfusion reactions, through the same mechanisms noted above for ABO incompatibility in transfusion. Anti-Leb antibodies are rarely, if ever, associated with clinical problems. The relatively benign attributes of these antibodies are accounted for by the fact that anti-Lea and anti-Leb antibodies are effectively neutralized by soluble Lea or Leb substances in transfused plasma. Moreover, transfused Lewis antigen-positive erythrocytes rapidly revert to a Lewis antigen-negative phenotype in Lewis-negative recipients after transfusion, owing to reversal of the absorptive process by which the red cells had accumulated these antigens in the donor.
There are other members of the Lewis blood group family of glycan structures. These include the Lex (SSEA-1) and Ley molecules and forms of the Lea and Lex determinants that are sialylated and or sulfated (see Figure 16.14). These structures are formed through the actions of one or more α1–3 fucosyltransferases in addition to the Lewis α1–3(4) fucosyltransferase.
As also noted for the ABO and Se loci, numerous associations have been made between the Lewis blood group phenotype and susceptibility to various diseases. As with the ABO and H-dependent associations, most of these are typically weak and generally are without clear mechanistic relationship. However, some members of the Lewis blood group antigen family have functional relevance in the context of selectin-dependent leukocyte and tumor cell adhesion processes (see Chapter 26). The relevant members include especially the sialylated and/or sulfated members represented by the sialyl Lex tetrasaccharide and its sulfated variants (Figure 16.14). These molecules provide essential contributions to the glycoproteins and glycolipids that function as selectin counter-receptors on leukocytes and probably also on tumor cells (see Chapters 26 and 35).
The Lewis blood group antigens have also been implicated in the pathogenesis of H. pylori. It is clear that this organism is a causative agent in chronic active gastritis. Infection with this organism is also associated with hypertrophic gastropathy, duodenal ulcer, gastric adenocarcinoma, and gastrointestinal lymphoma. Colonization of the stomach by H. pylori almost certainly requires adhesion to the gastric mucosal epithelium and to the mucus released by these cells. This organism can adhere to the Leb blood group antigen, at least in the limited number of strains that have been examined, implying that this oligosaccharide may function as a receptor for H. pylori in vivo. However, clinical studies that have characterized the relationship, in vivo, between gastric colonization by H. pylori and the ABO, Lewis, and Secretor status of the human host are not necessarily consistent with this hypothesis. Such studies found no correlation between H. pylori infection rate and host Leb phenotype, little or no correlation between H. pylori infection rate and ABO and Secretor phenotype, and a weak association between lymphocytic infiltration in H. pylori infection and ABO or Secretor status. The Lewis blood group adhesion hypothesis is further complicated by the fact that some strains of H. pylori can themselves elaborate expression of Leb antigen. Thus, the physiological relationship, if any, between H. pylori pathobiology and the Lewis or ABO antigens is probably much more complex than can be accounted for by simple blood group antigen-dependent adhesive processes.
P Blood Group Structures (95–106)
The P blood group antigens are glycan structures displayed by membrane-associated glycosphingolipids on red cells and on other tissues, including the urothelium (Figure 16.16). The glycans in the P blood group system are constructed by the sequential action of a series of distinct glycosyltransferases. Little is known about the enzymes or genes that direct expression of the P family of antigens, causing the nomenclature, biochemistry, and genetics of this system to be somewhat complex.

Figure 16.16
Antigens of the P blood group system.
Synthesis of the P antigens involves two different pathways, each of which begins with lactosylceramide as a common precursor (Figures 16.17 and 16.18). In one pathway, P antigen biosynthesis begins with an α1–4 galactosyltransferase (Pk transferase) that synthesizes the Pk antigen. The Pk antigen is then modified by a β1–3 GalNAcT, termed the P transferase, to form the P antigen. In the second pathway, P1 antigen biosynthesis involves three sequential glycosylation reactions, again starting with lactosylceramide. The first two enzyme reactions lead to paragloboside synthesis. Paragloboside is then used as a substrate by the P1 transferase, which forms the P1 molecule.

Figure 16.17
Pk and P antigen biosynthesis.
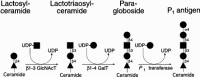
Figure 16.18
P1 antigen biosynthesis via paragloboside.
Expression of these antigens is polymorphic in humans. The most common P blood group phenotype is termed P1. These individuals express full activity of each of the enzymes in these pathways, and their red cells express both P and P1 antigens. P1 individuals also express small amounts of Pk determinants because the P transferase does not completely convert all Pk precursor determinants into P determinants. The other common blood group phenotype is P2. These individuals are apparently nullizygous at the P1 transferase locus. Red cells from P2 individuals express normal levels of P and Pk antigens, but are deficient in P1 determinants.
Three rare P blood group phenotypes have been described. The first, termed the P1k phenotype, is associated with deficiency of P transferase activity, and is presumed to be consequent to nullizygosity for the P transferase locus. P1k individuals cannot convert the Pk structure into the P structure and therefore express more than the normal numbers of Pk determinants. The parallel P1 synthesis pathway is intact in these persons, yielding normal numbers of P1 determinants. The second, termed the P2k phenotype, is found in rare individuals, who are presumed to be nullizygous at both the P transferase and the P1 transferase loci. Consequently, neither of the parallel pathways is completed, resulting in deficiencies in both P and P1 antigen expression, but increased numbers of Pk determinants. The third, termed the p phenotype, is characterized by a deficiency of all three P antigens (P, P1, and Pk). Nullizygosity at the Pk transferase and P1 transferase loci can account for this phenotype. P antigen is not synthesized, regardless of P transferase activity, because the P precursor (Pk) is not present. However, p phenotype red cells express low levels of the P antigen reactivity because the normal P transferase activity in these persons can form a glycan with P antigen reactivity, through the addition of an α 1–4-linked galactose to paragloboside derived from the other pathway (Figures 16.17 and Figure 16.18).
These considerations assume that the P, P1, and Pk transferases represent the products of three different loci. However, there are alternative genetic models that could explain the observed human phenotypes. A definitive understanding of this system will await the isolation and characterization of the genes corresponding to these enzymes. Antibodies directed against various P blood group antigens have been implicated in transfusion reactions in individuals with the p phenotype, who often maintain antibodies against P, P1, and Pk determinants. Complement-fixing, cold-reactive anti-P antibodies known as “Donath-Landsteiner” antibodies have been implicated in intravascular hemolysis observed in paroxysmal cold hemoglobinuria (see Chapter 37).
Physiological functions for the P blood group antigens are not known. However, these molecules have been assigned roles in the pathophysiology of urinary tract infections and in parvovirus infection. A role for P blood group antigens in the pathogenesis of urinary tract infections is implied by the observation that various uropathogenic strains of Escherichia coli express adhesins that bind to the Galα1–4Gal moiety of the Pk and P1 antigens. The P1 determinant is expressed on the urothelium of P1 individuals and may facilitate bacterial infection by mediating attachment of bacteria to the lining of the urinary tract. This hypothesis is supported by the observation that P1 individuals have a higher risk, relative to P2 individuals, for urinary tract infections and pyelonephritis. It is also supported by the observation that adhesion of a pyelonephritic strain of E. coli to renal tissue is mediated by a bacterial adhesin specific for the Galα1–4Gal structure and that deficiency of the adhesin severely attenuates the pyelonephritic phenotype of the organism.
The P blood group antigens have also been assigned a role as a receptor for the human parvovirus B19. This virus causes erythema infectiosum and leads to congenital anemia and hydrops fetalis following infection in utero. It is also associated with transient aplastic crisis in patients with hemolytic anemia and with cases of pure red cell aplasia and chronic anemia in immunocompromised individuals. Parvovirus B19 replication is restricted to erythroid progenitor cells; an adhesive interaction between the virion and P-antigen-active glycolipids is involved in viral infection of erythroid progenitors. Individuals with the p blood group phenotype are apparently resistant to parvovirus B19 infection.
The α1–3Gal Structure (107–116)
The Galα1–3Gal epitope is synthesized from type-2 glycolipid and glycoprotein precursors by a specific α1–3 galactosyltransferase (Figure 16.19). This structure and the corresponding α1–3 galactosyltransferase are expressed by New World primates and many nonprimate mammals but are largely absent from the cells and tissues of Old World primates which include Homo sapiens. The molecular basis for the species-specific absence of this enzyme and its oligosaccharide product involves the inactivation of the locus encoding the α1–3 galactosyltransferase in primate taxa that do not express the epitope (see Chapter 33).
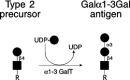
Figure 16.19
Structure and synthesis of the Galα1–3Gal antigen. The α1–3 galactosyltransferase uses unsubstituted type-2 precursors to form the Galα1–3Gal epitope. R may be glycolipid- or glycoprotein-based glycan structures. (more...)
A physiological function for the Galα1–3Gal epitope has not been identified. There have been reports that the Galα1–3Gal epitope is found in small amounts on human red cells and may participate in red cell turnover, but these observations are at odds with the apparent absence of a functional α1–3 galactosyltransferase locus in humans. Similar considerations apply to the apparent expression of the Galα1–3Gal epitope on human thyroid cells or human cancer cells. A proposed role for the Galα1–3Gal epitope as a sperm receptor on mouse oocytes is contradicted by studies showing that mice homozygous for an induced null mutation in this locus are fertile (see Chapter 33). These mice also have cataracts, but a mechanistic relationship between this phenotype and the absence of Galα1–3Gal epitopes is not available. These mice have no other recognized pathological phenotype as of this writing.
Species that lack expression of the Galα1–3Gal epitope, including especially humans, elaborate naturally occurring anti-Galα1–3Gal antibodies, by mechanisms that involve immunization through exposure to microbial antigens similar or identical to the Galα1–3Gal epitope. These anti-Galα1–3Gal antibodies present a major barrier to the use of porcine and other nonprimate organs for xenotransplantation in humans, since they bind to Galα1–3Gal epitopes on the vascular endothelium in such xenotransplants and mediate hyperacute graft rejection through complement-mediated endothelial cell cytotoxicity. Efforts are in progress to overcome this barrier through the generation of animal organ donors that have been genetically modified to express proteins that diminish complement-dependent cytotoxicity (see Chapter 33). Approaches include transgenic expression of enzymes such as the H α1–2 fucosyltransferase that may diminish Galα1–3Gal expression by diverting type-2 precursor substrates toward H antigen synthesis and away from their utilization by the α1–3 galactosyltransferase. The creation of induced null mutations in the gene encoding α1–3 GalT of such animal organ donors represents one definitive solution to the problem of Galα1–3-Gal-dependent hyperacute rejection. Unfortunately, the pluripotent embryonic stem cells required for this approach are not yet available for pigs or other large animal organ donors.
Naturally occurring anti-Galα1–3Gal antibodies have also been shown to significantly diminish the infective efficiency of recombinant retroviruses. This problem occurs because the packaging cell lines used to propagate these viruses are derived from species that express the Galα1–3Gal epitope and decorate the viral coat proteins. This problem has been solved through the generation of packaging cell lines that are deficient in the cognate α1–3 galactosyltransferase. This technical problem may reflect a natural mechanism that acts to restrict the interspecies spread of retroviral genomes (see Chapter 3).
The Forssman Antigen (117–123)
The Forssman antigen, also known as globopentosylceramide, is a glycolipid structure formed by the addition of GalNAc in α1–3 linkage to the terminal GalNAc residue of globoside (Figure 16.20). There is also evidence in the literature that Forssman-specific monoclonal antibodies can detect Forssman-reactive glycoproteins, but the nature of these molecules is not known. The Forssman antigen molecule is expressed during embryonic and adult life in rodents and other mammals (see Chapter 34), but uncertainty exists about the ability of humans to express this antigen. For example, there is evidence that humans maintain moderate titers of naturally occurring anti-Forssman antibodies in plasma, suggesting that humans do not express the Forssman antigen. In contrast, there is evidence that such antibodies are not consistently present in humans and that when generated, may contribute to the pathogenesis of the Guillain-Barre syndrome by binding to glycolipid components of peripheral nerve myelin. Similarly, evidence exists that small amounts of Forssman reactivity may be found on human gastrointestinal epithelium, by various human cultured cell lines, by pulmonary and gastointestinal tract carcinomas. These conflicting observations may be a reflection of varied specificities of the anti-Forssman monoclonal antibodies used by different investigators and by differences in epitope reactivity achieved with immunohistochemical procedures versus thin-layer chromatography/antibody overlay procedures. The function of this antigen is not known. The ability of anti-Forssman antibodies to disrupt tight junction formation, apical-basal polarization, and adhesion suggests that this molecule may participate in cell-cell adhesion and communication processes. The mechanisms that account for these observations are not defined, nor are there corollary studies that have confirmed these observations in a more physiological context.
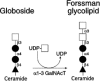
Figure 16.20
Forssman antigen biosynthesis. Globoside serves as the substrate for the Forssman α1–3 N-acetylgalactosaminyltransferase (α1–3 GalNAcT) that forms globopentosylceramide, also termed the Forssman glycolipid.
Sulfated Terminal β-linked GalNAc Structures on Pituitary Glycoproteins (124–127)
Glycans with sulfated terminal β-linked GalNAc moieties have been studied extensively in the context of pituitary glycoprotein hormones lutropin (LH), thyrotropin (TSH), and follicle-stimulating hormone (FSH). These heterodimeric glycoproteins are composed of a common α-subunit and a unique β-subunit. Each subunit is decorated with biantennary N-linked glycans. The N-glycans on TSH and LH contain an unusual structure consisting of a GalNAc moiety attached to one or both GlcNAc residues on the biantennary glycan, and additionally modified by sulfation or by α2–6-linked sialic acid (Figure 16.21). This modification contrasts with the N-glycans on FSH and on most N-glycans, where GlcNAc residues are modified by β1–4-linked galactose moieties, which are often then modified by α2–3- or α2–6-linked sialic acid. The free α-subunit common to LH, TSH, and FSH, and present as a synthetic intermediate in pituitary cells, is also modified by this determinant, as are other proteins synthesized by the pituitary and in other tissues. This structure has also been found on the O-glycans of pro-opiomelanocortin.

Figure 16.21
Structure and synthesis of N-glycans bearing terminal GalNAc and sulfate moieties, including those associated with pituitary hormones LH and FSH.
Synthesis of this sulfated GalNAc glycan determinant is controlled by a β1–4 GalNAcT activity that modifies the subterminal GlcNAc residues of the biantennary glycan chains (Figure 16.21). Expression of this enzyme is restricted to only a few cell types, including the cells in the pituitary that elaborate LH, TSH, and FSH. This β1–4-linked GalNAc moiety is then sulfated by a sulfotransferase also expressed in pituitary cells. In some tissues, including the pituitary, the β1–4-linked GalNAc moiety is subsequently modified by α2–6-linked sialic acid residues. Subterminal GlcNAc residues beneath the β1–4-linked GalNAc moiety have also been observed to be modified by α1–3-linked fucose (and see Chapter 26). In contrast, synthesis of β1–4-linked galactose-terminated biantennary chains characteristic of FSH is directed by a β1–4 galactosyltransferase (Figure 16.21), an enzyme activity found in virtually all cell types, including those of the pituitary gland.
Although a β1–4 GalNAcT and a β1–4 galactosyltransferase are both expressed in the pituitary cell, the N-glycans on LH and TSH are decorated with the unusual β1–4-linked GalNAc moiety, whereas the identical N-glycans on the nearly identical glycoprotein FSH are instead exclusively decorated with the alternate and more common β1–4-linked galactose moiety. This protein-specific glycosylation event is a consequence of interaction between the β1–4 GalNAcT and specific protein sequence motifs present on the α and β subunits of LH and TSH. This interaction causes this enzyme to increase the catalytic efficiency with which it modifies the biantennary N-glycans on LH and TSH, at the expense of modification by the competing β1–4 galactosyltransferase, which does not recognize the peptide motif. The resulting terminal β1–4-linked GalNAc moieties are subsequently susceptible to modification by sulfation or by α2–6 sialylation (Figure 16.21). In contrast, the peptide sequence motif recognized by the β1–4 GalNAcT is not present in the sequence of the β-subunit of FSH, and the recognition motif on the α-subunit of FSH is not available to the enzyme. Consequently, the biantennary N-glycans on FSH are not susceptible to modification by the β1–4 GalNAcT and are instead modified exclusively by the competing β1–4 galactosyltransferase.
These differential glycosylation events have profound consequences for the ovulatory cycle throughout vertebrate taxa. Maximal stimulation of the ovary by LH during the preovulatory surge is associated with levels of circulating LH that rise and fall in a highly pulsatile manner. The pulsatile characteristic of circulating LH levels assures maximal stimulation of the ovarian LH receptor, since sustained high LH levels of this receptor would lead to LH receptor desensitization. The pulsatile rise and fall in LH levels are due in part to pulsatile release of the hormone by the pituitary. However, it is also a consequence of a rapid clearance of the hormone from the circulation. Clearance of the hormone from the circulation is mediated by a receptor specific for the sulfated GalNAcβ1–4 GlcNAc terminus. This receptor is expressed by hepatic endothelial cells, and by Kupffer cells, and binds LH with an apparent Km of approximately 160 nm. Receptor binding is followed by internalization and lysosomal degradation. The protein sequence of the receptor is identical to a previously characterized protein termed the macrophage mannose receptor. The mechanisms that account for tissue-specific differences in this receptor's ligand recognition characteristics remain to be defined.
Similar GalNAcβ1–4-GlcNAc (LacdiNAc) termini have also been reported on N-glycans from other vertebrate sources, such as bovine milk, rat prolactin, and kidney epithelial cells, as well as in invertebrates such as snails and parasitic worms. These residues generally do not become sulfated as in pituitary hormones, but are frequently 2–6-sialylated in vertebrates. It is not clear how the GalNAcTs responsible for producing these structures are related to the enzyme acting specifically on pituitary hormones.
Sialylated Terminal β-linked GalNAc Structures (128–138)
Terminal GalNAc modifications of α2–3-sialylated structures are found on glycoproteins and on glycolipids (Figure 16.22). The former correspond to the human Sda blood group structure, and a murine oligosaccharide, termed the CT1 or CT2 antigen, first described on cytotoxic T lymphocytes (CTLs). The glycolipid-based form of this structure is known as the ganglioside GM2.
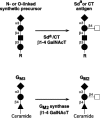
Figure 16.22
Production of the Sda or CT antigen and the glycolipid GM2.
The polypeptide-linked form of this structure was first identified as the pentasaccharide Siaα2–3(GalNAcβ1–4)Galβ1–4GlcNAcβ1–3Gal-, released from N-glycans present on Tamm-Horsfall glycoprotein isolated from human urine. This structure is formed by addition of β1–4-linked GalNAc to the galactose moiety of α2–3-sialylated type-2 chains (Figure 16.22). A β1–4 GalNAcT capable of catalyzing this reaction has been found in human kidney and urine, intestine, colon, and blood plasma. Rare humans lacking the ability to construct this determinant can form naturally occurring antibodies against it and are said to lack the corresponding (Sda) blood group determinant. Absence of this antigen in these humans has no apparent associated detrimental phenotype.
In the mouse, this structure corresponds to the antigen(s) recognized by a pair of IgM class monoclonal antibodies termed CT1 and CT2, isolated for their ability to block lysis of cellular targets by a murine CTL clone. These antibodies recognize similar but nonidentical antigens expressed by activated CTLs, but not by naive T lymphocytes. Activation-dependent display of the CT antigens correlates with inducible expression of the cognate enzyme activity. Both CT antibodies also bind to intraepithelial lymphocytes, a type of constitutively activated T-lymphocyte resident in the intestinal mucosa. The major proteins immunoprecipitated from CTL cell lines by CT antibodies belong to isoforms of the CD45 cell surface transmembrane tyrosine phosphatase required for T-cell proliferation in response to antigen. It is likely that other lymphocyte cell surface proteins are also modified with this structure. As with human cells, this glycan structure is constructed by a β1–4 GalNAcT that modifies β1–4-linked galactose substituted with α2–3-linked sialic acid on O- and N-glycans. These human and mouse β1–4 GalNAcTs can transfer GalNAc to both N- and O-glycans present on glycoproteins but not on to the glycolipid GM3 (Siaα2–3Galβ1–4Glc-Cer), even though both can efficiently use 3′-sialyllactose (Siaα2–3Galβ1–4Glc) as a substrate in vitro.
The function of this structure in humans or mice is unknown. As noted earlier in this chapter, in rare strains of mice with a dominantly inherited form of von Willebrand's disease, the corresponding β1–4 GalNAcT is aberrantly expressed in vascular endothelium. Since the blood-clotting protein vWF is also expressed by endothelial cells, expression of the enzyme at this unusual location decorates the glycans on vWF in these mice. The resulting vWF glycoform is rapidly cleared from the circulation, which accounts for the low levels of this protein in this strain of mice. Rapid clearance is mediated by the asialoglycoprotein receptor, which exhibits affinity for terminal GalNAc structures.
The glycolipid-linked form of this structure, termed GM2, is synthesized from the ganglioside GM3 (Figure 16.22). GM2 synthesis is catalyzed by a β1–4 GalNAcT (GM2 synthase) that shares primary sequence similarity with the β1–4 GalNAcT responsible for synthesis of the glycoprotein form of this structure. However, this enzyme cannot effectively utilize glycoprotein glycan precursors. The function of this structure is not known. However, it is widely expressed in the CNS and PNS, and in the adrenal gland, for example, suggesting a role in these organ systems. This possibility is supported by analysis of mice homozygous for an induced null mutation at the GM2 synthase locus. These mice exhibit modest conduction defects in the PNS and exhibit biochemical defects resulting in male sterility (and see Chapter 33). Studies are in progress to determine how GM2 contributes to homeostasis in these systems and to characterize the mechanisms responsible for these abnormal phenotypes.
α2–3-sialylated Structures (139–154)
Sialic acid is found in α2–3 linkage on many, and perhaps all, cells and tissues in vertebrates. Members of a family of at least five different α2–3 sialyltransferases (ST3Gal-I, ST3Gal-II, ST3Gal-III, ST3Gal-IV, and ST3Gal-V) are responsible for synthesis of these structures (Figure 16.23). Studies of the expression patterns of these genes indicate that STGal-III and STGal-IV are expressed in most tissues and cells in adult mammals. These observations are consistent with the identification of α2–3-sialylated glycans on many different cell types, including different glycoproteins and glycolipids. In contrast, ST3Gal-I transcripts in humans and mice are abundant in the spleen, liver, bone marrow, thymus, and salivary glands and are less abundant in other tissues. Like ST3Gal-I, ST3Gal-II is also more restricted in its expression pattern, with transcripts most abundant in the brain and much lower levels of expression in some other tissues. The relatively robust expression of ST3Gal-II in the brain is consistent with the abundance of α2–3-sialylated glycolipids in this organ and with the part played by this enzyme in glycolipid (ganglioside) synthesis (Figure 16.23). The ST3Gal-V locus is also expressed in the brain, skeletal muscle, testes, and the liver.
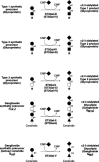
Figure 16.23
Synthesis of glycoproteins and glycolipids bearing terminal α2–3-linked sialic acids by the ST3Gal series of sialyltransferases. Enzymes in parentheses contribute at relatively low levels in vitro to the reactions indicated.
In vertebrates, α2–3 sialic acid linkages are found on terminal galactose moieties (Figure 16.23). Such structures are substrates for further modification by α1–3 fucosylation (see Figure 16.14 and Chapter 26), β1–4 GalNAc linkage formation in limited circumstances (see Figure 16.21), α2–6 sialylation (Figure 16.24), α2–8 sialylation (Figures 16.25 and 16.26), and sulfation (Figure 16.27). In contrast, the α2–3-sialylated structures represented in Figure 16.23 are generally not substrates for other enzymes, including α1–2 fucosyltransferases, α1–3 galactosyltransferases, GlcNAcTs, or GalNAcTs. The latter enzymes may compete for terminal α2–3 sialylation as a glycan chain-terminating modification.

Figure 16.24
Synthesis of α2–6-sialylated termini on O-glycans, and glycolipids (and see Chapters 8 and 9) by the ST6GalNAc series of sialyltransferases. Enzymes in parentheses contribute at relatively low levels in vitro to the reactions indicated. (more...)

Figure 16.25
Structure and synthesis of polysialic acid on N-glycans.

Figure 16.26
Structure and synthesis of polysialic acid on glycolipids. Enzymes in parentheses contribute at low levels in vitro to the reactions indicated.
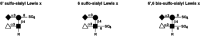
Figure 16.27
Sulfation of α2–3-sialylated, α1–3 fucosylated glycans represented by the sialyl Lex tetrasaccharide have been observed at the 6-hydroxyl positions of the galactose residue and the GlcNAc residue. Both may contribute to (more...)
Structures bearing α2–3 sialic acid linkages have been assigned a function in only a few circumstances as of this writing. As discussed in detail in Chapter 26, the glycan ligands of the selectin family of leukocyte adhesion molecules include α2–3-sialylated structures, modified by α1–3 fucosylation or by α1–3 fucosylation and sulfation. The α2–3-linked sialic acid component of such selectin ligands is likely essential for physiologic ligand formation involving E-selectin, P-selectin, and L-selectin. Analyses of the tertiary structures of the CRD of E-selectin derived from crystallographic studies and mutagenesis studies imply that specific positively charged amino acid residues interact with the negatively charged sialic acid moiety on the glycolipid and glycoprotein counterreceptors. Recognition is specific for sialic acid in α2–3-linkage, since adhesive interactions are not supported by variants of selectin ligands in which α2–3-linked sialic acid is substituted with α2–6-linked sialic acid.
Structures bearing α2–3 sialic acid linkages may contribute to the homeostatic maintenance of circulating half-life of plasma glycoproteins, by virtue of “masking” terminal galactose residues that would ordinarily contribute to the removal of such proteins from the circulation through the asialoglycoprotein receptor (see Chapter 25). The physiological role for this process in uncertain, however, at least with respect to the asialoglycoprotein receptor, since mice deficient in this receptor do not have increased plasma levels of desialylated glycoproteins or lipoproteins in their circulation. However, recent studies demonstrate that these mice accumulate endogenous, but as yet unidentified, ligands for this receptor. The converse also seems to be true, in that α2–3 sialic-acid-terminated glycans bearing a β1–4 GalNAc moiety (see Figure 16.21) undergo enhanced asialoglycoprotein receptor-dependent clearance, at least in the case of one plasma protein.
In other studies, the role of ST3Gal-I in the production of the Siaα2–3Galβ1–3GalNAcα-Ser/Thr glycan is important for the viability of peripheral CD8+ T cells. Mice lacking the ST3Gal-I enzyme exhibit a decreased cytotoxic-T-cell response with an increase in the apoptotic death of naive CD8+ T cells (see Chapter 33). The timing, specificity, and location of this enhanced apoptotic death indicate that it is unlikely to be due to the liver asialoglycoprotein receptor and may result from an endogenous lectin yet to be recognized that binds to Galβ1–3GalNAcα-Ser/Thr glycans.
The α2–3 sialic acid linkages that decorate some glycoproteins and glycolipids also contribute to microbial pathogenesis. The most extensively studied system in this context concerns the role of such structures as receptors for the influenza virus. Sialic acid in α2–3-linkage is recognized by the influenza virus hemagglutinin commonly found in birds and pigs and prior to its recombination, yielding binding to α2–6 sialic acid linkages as found frequently in isolates from the human population. The viral hemagglutinin mediates attachment of the virus to cells that express α2–3 sialic acid linkages, promoting viral neuraminidase-dependent release of sialic acids with delivery of the viral genome to the interior of the cell (see Chapter 28).
Structures bearing α2–3 sialic acid linkages have also been implicated in contributing to bacterial pathogenesis. For example, α2-3 sialic acid structures support adhesion of H. pylori, the spirochete implicated in the pathogenesis of gastritis, gastric ulcers, and lymphoma of the gastrointestinal tract mucosa. However, it remains to be determined if these in vitro observations have a physiological correlate. In contrast, there is strong evidence for a physiological role for the ganglioside GM1 (GM1a; Galβ1–3GalNAcβ1–4[Siaα2–3]Galβ1–4Glcβ1-Cer) as a receptor for cholera toxin, produced by Vibrio cholerae, and heat-labile enterotoxin (LT-1), produced by enterotoxigenic E. coli (see Chapter 28). Cholera toxin is responsible for the severe enteropathogenicity that accompanies infection with V. cholerae. Heat-labile enterotoxin is the causative agent of traveler's diarrhea and contributes to a substantial amount of childhood mortality in the developing countries. Both toxins are heterohexamers composed of a single toxic subunit with ADP-ribosylating activity (the A subunit), complexed with a pentamer composed of five B subunits that bind GM1. The cytotoxic A subunit is delivered to enterocytes by binding of the heterohexamer to the intestinal epithelial cells via the branched pentasaccharide of the ganglioside GM1a. Binding of these toxins to GM1a is clearly of pathophysiological relevance; glycan-based inhibitors of these interactions are currently under evaluation in humans for their ability to diminish the symptoms and progression of cholera and traveler's diarrhea.
α2–6-sialylated Structures (139,140,149,155–157)
Sialic acid in α2–6 linkage is expressed by various vertebrate cells and tissue types and displayed on a wide variety of glycoproteins and glycolipids. Synthesis of α2–6-linked sialic moieties is directed by members of a family of at least five different α2–6 sialyltransferases (see Figure 16.24). The genes of these have been defined by molecular cloning approaches (ST6Gal-I, ST6GalNAc-I, ST6GalNAc-II, ST6GalNAc-III, and ST6GalNAc-IV). A gene corresponding to a sixth activity, ST6GlcNAc-I, has not yet been genetically isolated. Surveys of tissues and cells in a variety of mammals, and some lower vertebrates, indicate that α2–6-sialylated glycans may be found in various (but not all) cell types, as they appear less ubiquitous than the α2–3-linked sialic-acid-bearing glycans.
In vertebrates, α2–6 sialic acid linkages are found on terminal galactose moieties, on terminal or subterminal moieties, or on an internal GalNAc moiety. Glycans modified by terminal α2–6 sialic acid are generally not modified further, except possibly by some members of the poly α2–8 sialyltransferase family, as discussed below. The products of ST6Gal-I are typically found on N-glycans, although the in vitro acceptor substrate specificity of this enzyme indicates that the β1–4-linked galactose of a terminal lactosamine repeat unit on O-glycans can be modified by this enzyme. ST6Gal-I is expressed at a relatively high level in hepatocytes and lymphocytes and is responsible for α2–6 sialylation of serum glycoproteins and glycoproteins of the antigen receptor complex. In contrast, the α2–6-sialylated products and the ST6GalNAc-I and ST6GalNAc-II enzymes are restricted to structures displayed on O-glycans. The ST6GalNAc-III enzyme is responsible for substitution of core N-acetylgalactosamine moiety on O-lycans, whereas the ST6GalNAc-IV enzyme appears to use glycosphingolipid precursors as the preferred acceptor.
Few definitive functions have been assigned to α2–6-linked sialic acid modifications. As noted above (and see Chapter 28), α2–6-linked sialic acid serves as a receptor for human infectious strains of the influenza virus. The common occurrence of α2–6-linked terminal sialic acid moieties on plasma glycoproteins may mask terminal galactose and N-acetylgalactosamine moieties on the glycoproteins that would ordinarily lead to their clearance by the hepatic asialoglycoprotein receptor. As also discussed previously, experiments using mice with an induced mutation in the asialoglycoprotein receptor indicate that this notion is not as straightforward as once thought, since such mice do not have obvious quantitative alterations in plasma glycoprotein levels.
The clearest information concerning the function of some α2–6-linked sialic acids comes from a characterization of a role for such structures by a genetically induced absence of ST6Gal-I and in the context as ligands for CD22, a member of the I-type lectin family (see also Chapters 24 and 33). CD22 is restricted in its expression to the surfaces of B lymphocytes. The extracellular domain of CD22 specifically recognizes Siaα2–6Galβ1–4GlcNAc-, where the sialylation is the product of the ST6Gal-I enzyme. This glycan has been found on a variety of leukocytic glycoproteins, including the protein tyrosine phosphatase CD45, with attendant possibilities for CD22-dependent signal transduction processes mediated by interactions between CD22 on B lymphocytes and α2–6-sialylated glycans on CD45. This structure is also displayed by CD22 itself, implying that CD22 may participate in homophilic interactions, both between cells and on the same cells. Since CD22 has been associated with the antigen receptor on B-lymphocyte proteins, and since its cytosolic domain is tyrosine-phosphorylated in some circumstances, it had been proposed that homophilic and heterophilic interactions involving CD22 might also effect biologically important signal transduction events in the immune system.
Mice lacking ST6Gal-I manifest an immunodeficiency characterized by a diminished antibody response to T-lymphocyte-dependent and -independent antigens, by reduced B-lymphocyte proliferation in response to cross-linking of the B-cell surface glycoproteins CD40 and surface IgM, by reductions in the expression of B-cell surface IgM and CD22, and by an approximately 65% reduction in serum IgM levels. B-lymphocyte antigen receptor-dependent signal transduction processes are also attenuated in these mice. These observations disclose an important role for the ST6Gal-I in the immune system. Considered together, inconsistency exists among the different reports on the phenotypes observed in mice with induced mutations in the CD22 locus (discussed in Chapter 24), and these phenotypes are not necessarily completely consistent with, or the biological converse of, the phenotype obtained with the CD22 ligand-deficient (STGal-I null) mice. Some studies have implied that other lectins may exist that bind to the ST6Gal-I-generated glycans. Additional experiments will be required to resolve these issues.
α2–8-sialylated Structures (139,140,158–168)
Glycans modified by sialic acid in α2–8 linkage have been identified as developmentally regulated antigens in the vertebrate CNS (Chapter 34); the biology of α2–8-sialylated glycoconjugates has been studied most extensively in the context of neural development. These α2–8-sialylated glycans are expressed in other, nonneuronal tissues during embryogenesis, however, and in the adult vertebrate, and these glycans have been observed on transformed cells. At least five different α2–8 sialyltransferases direct the synthesis of α2–8 sialylated structures (see Figures 16.25 and 16.26). Two of these enzymes, termed ST8Sia-II (also termed STX) and ST8Sia-IV (also termed PST-1 and PST), catalyze the synthesis of linear polymers of α2–8 sialic acid, termed polysialic acid, or PSA (see Figure 16.25). These polymers may be composed of 100 or more sialic acid residues. N-glycans with terminal sialic acid moieties in α2–3 linkage serve as the substrate for attachment of the initial α2–8-linked sialic acid residue, although there is some evidence that α2–6-linked sialic acids may also support attachment of the first α2–8-linked sialic acid residue. This so-called “initiase” reaction is followed by an “elongation” reaction in which the α2–8 sialic acid moiety added in the preceding step serves as the attachment site for the next α2–8-linked sialic acid. These two “activities” may be subsumed by ST8Sia-II and ST8Sia-IV themselves or are the province of distinct sialyltransferases.
PSA is most extensively studied as a component of the posttranslational modifications of N-CAM, a member of the immunoglobulin superfamily (and see Chapter 34). The α-subunit of the voltage-gated sodium channel may also be modified with PSA. The polysialyltransferases ST8Sia-II and ST8Sia-IV are each subject to autocatalytic polysialylation which can occur on N-glycans that decorate each enzyme, although polysialylation is not a prerequisite for polysialyltransferase activity. This process can yield membrane-associated, poly-α2–8-sialylated forms of the enzymes, which may account for the observation that some cultured cell lines which do not express N-CAM or the sodium channel can be induced to express surface-localized PSA when transfected with ST8Sia-II or with ST8Sia-IV.
Modification of N-CAM by PSA is apparently directed by specific peptide segments composed of NCAM's immunoglobulin domain 5 and its adjacent Ig4 and fibronectin type III repeats, along with a membrane attachment segment. Regulated expression of PSA during development is directed in part by tissue-specific regulation of expression of ST8Sia-II and ST8Sia-IV (Chapter 34). ST8Sia-II is prominently expressed in fetal brain, but it declines markedly during the postnatal period. In contrast, ST8Sia-IV continues to be expressed postnatally in the brain and in nonneuronal tissues such as the heart, lung, and spleen. Posttranscriptional mechanisms can also regulate PSA expression.
PSA negatively modulates the homotypic adhesive properties of N-CAM on opposing cells. This property correlates with the observation that the embryonic form of N-CAM is extensively modified by PSA and is less able to participate in homotypic adhesive interactions than is the adult form of PSA, which is not highly modified by PSA. PSA can also diminish interactions promoted by other adhesion molecules, including L1-dependent attachment to laminin or collagen. Since PSA is highly negatively charged, is highly hydrated, and contributes up to one third of the molecular mass of N-CAM, it is believed that PSA negatively modulates cell adhesion processes by physical interference with apposition of the plasma membranes of adjacent cells. PSA can also positively modulate N-CAM-dependent adhesive interactions with other adhesion counterreceptors, however, and can modulate the adhesive activity of glycoproteins such as L1 on the same cell. Mechanisms to account for these observations are the subject of ongoing study. In general, however, PSA-dependent modulation of cell-cell and cell-matrix interactions are believed to be important to targeting of growing axons to sites of innervation, to the migration of neuronal cells during neural development, and to physiological plasticity in the CNS (see Chapter 34). A confirmation of these hypotheses, and discovery of other potential functions for PSA, will await the generation and analysis of mice that are specifically deficient in PSA expression.
The second general type of α2–8 sialic acid occurs in certain members of the ganglioside family. These are constructed by three α2–8 sialyltransferases that are distinct from the two responsible for PSA synthesis. These are termed ST8Sia-I (also known as GD3 synthase), ST8Sia-III, and ST8Sia-V (see Figure 16.26). They can construct single or oligomeric α2–8 sialic acid linkages but not the polysialylated structures elaborated by ST8Sia-II or ST8Sia-IV. These three enzymes are generally thought to act primarily on glycolipid substrates, but in vitro studies suggest that ST8Sia-III can also utilize N-glycans. The precise nature of the products formed by these enzymes, in vivo, is not clear at present. An overview of some of their in vitro substrate specificities is presented in Figure 16.26. These enzymes are prominently expressed in the brain, where each exhibits a distinct developmentally regulated expression pattern. ST8Sia-I is also expressed in the kidney and thymus, where it presumably contributes to the synthesis of α2–8-sialylated gangliosides. In vitro experiments imply that certain α2–8-sialylated gangliosides may participate in signal transduction processes in neuronal cell types, yielding differentiative phenotypes. In vivo correlates for these observations are not yet available, and other clear functions for the α2–8-sialylated gangliosides remain to be discovered.
Sulfated Glycans: L-Selectin Ligands, HNK-1, and Keratan Sulfate (169–182)
In principle, any free hydroxyl group on a monosaccharide component of a glycoconjugate is a potential position for modification by sulfation. However, in vertebrates, glycan sulfation is generally restricted to Gal, GlcNAc, GlcA, IdA, and GalNAc moieties. Sulfation can occur at internal or terminal positions. Chapter 11 discusses the internally sulfated glycans represented by heparin, heparan sulfate, and chondroitin sulfate proteoglycans that contribute to the ECM of vertebrate organisms. Only sulfated terminal glycan structures are discussed in this chapter. The most common and well-understood members of this group of glycans include the sulfated structures that contribute to selectin counterreceptor activity, the HNK-1 epitope, and keratan sulfate. This group also includes the sulfated structures found on some pituitary glycoprotein hormones. This latter topic is discussed in some detail above and is not dealt with further here.
Certain sulfated glycans contribute to the process used by lymphocytes to home to peripheral lymph nodes. These glycans have been identified as components of the glycocalyx on the specialized endothelium that lines the postcapillary venules in peripheral lymph nodes and other secondary lymphoid organs. These so-called high endothelial venules support adhesion of lymphocytes via the C-type lectin L-selectin. L-selectin-dependent adhesion occurs through recognition of mucin-type L-selectin counterreceptors elaborated by high endothelial venules. Glycan structural analyses disclose that sulfated forms of α2–3-sialylated, α1–3-fucosylated glycans represented by the sialyl Lex tetrasaccharide (Figures 16.14 and 16.27) provide an essential contribution to the L-selectin counterreceptor activity of these glycoproteins. Sulfation has been observed at the 6-hydroxyl positions of the galactose residue and the GlcNAc residue, both of which may contribute to L-selectin ligand activity. The biosynthesis of these structures, the enzymes that participate in this process, and their functional attributes are discussed further in Chapter 26.
The HNK-1 antigen is a terminally sulfated glycan that was first described on human natural killer cells and has the cluster designation CD57. The HNK-1 epitope is also expressed by a variety of cell types in the vertebrate nervous system, where its cell-type-specific expression patterns change during neural development (and see Chapter 34). The HNK-1 glycan is composed of a 3-O-sulfated GlcA moiety, in which GlcA is attached in β1–3 linkage to a terminal galactose (Figure 16.28). This structure is synthesized by specific glucuronosyltransferases that act on terminal (poly)lactosamine units of N-linked glycans. Glucuronylation is followed by 3-O-sulfation of the GlcA by one or more specific sulfotransferases. The HNK-1 epitope has also been described on O-glycans of glycoproteins, on proteoglycans, and on glycolipids, where similar enzymatic processes lead to its synthesis. There is evidence that different glucuronosyltransferases may be involved in the synthesis of HNK-1 epitopes on glycoproteins and glycolipids.
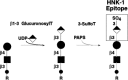
Figure 16.28
Synthesis and structure of the HNK-1 epitope.
The HNK-1 structure has been described as a posttranslational modification of a variety of neuronal cell proteins, including N-CAM-1, contactin, myelin-associated glycoprotein, telencephalin, L1, contactin, and P0, the major glycoprotein of peripheral nerve myelin. This sulfated glycan can function as a ligand for laminin, for L-selectin and P-selectin, and for a cerebellar adhesion protein termed amphoterin. HNK-1 can also mediate homotypic adhesive interactions involving P0. HNK-1-dependent adhesive interactions have been implicated in cell migration processes involving cell-cell and cell-matrix interactions and may participate in reinnervation of muscles by motor neurons.
Keratan sulfate represents a prominent posttranslational modification of several ECM proteins, whose structures and functions are discussed in detail in Chapter 11. Keratan sulfate is a polylactosamine attached to proteins via typical N-glycans (KS type I) or by a typical O-glycan moiety (KS type II). It is believed that keratan sulfate is synthesized in a stepwise manner by enzymes involved in polylactosamine synthesis, in coordination with a pair of 6-O-sulfotransferase activities (Figure 16.29). A keratan-specific N-acetylglucosamine 6-O-sulfotransferase activity utilizes unsubstituted (terminal) N-acetylglucosamine moieties but will not effectively modify internal N-acetylglucosamine residues. This enzyme will not utilize the GalNAc moieties in chondroitin-sulfate-type glycans, whose component is the GalNAc-GlcA disaccharide repeat. In contrast, a second molecularly distinct 6-O-sulfotransferase activity has been described that can apparently catalyze 6-O-sulfation of GlcNAc moieties in keratan sulfate and GalNAc moieties in chondroitin sulfate. The relative quantitative contributions made by each GlcNAc 6-O-sulfotransferase to keratan sulfate synthesis remain to be determined.
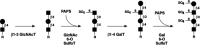
Figure 16.29
Synthesis of keratan sulfate by enzymes involved in polylactosamine synthesis and with a pair of 6-O-sulfotransferase activities. Keratan sulfate is attached (R) to proteins via an N-linked glycan (KS type I) or an O-linked glycan (KS type II).
The galactose 6-O-sulfotransferase efficiently sulfates galactose moieties within keratan sulfate chains but is apparently more efficient in this sulfation reaction when the galactose to be modified is adjacent to a 6-O-sulfated GlcNAc moiety. These observations imply that sulfation of GlcNAc moieties can only occur when free GlcNAc moieties exist during synthesis and elongation of polylactosamine chains, whereas sulfation of galactose moieties may occur at any time during polylactosamine chain synthesis.
There are as yet few defined functions for keratan sulfate. Corneal keratan sulfate (KS I) apparently maintains the proper spatial organization of type I collagen fibrils in the cornea that promotes transparency of this structure. Alterations in the degree of sulfation of keratan sulfate are associated with corneal opacity in a disease known as macular corneal dystrophy. Alterations in the amount of sulfation of brain keratan sulfate have been found in the brains of Alzheimer's disease patients. The pathophysiological relevance of the latter observation has not yet been defined.
Future Directions
The presently large number of terminal glycan chain modifications continues to expand with ongoing experimentation involving glycan isolation and increasingly sensitive structural analyses. Where it has been explored, the careful regulation of terminal glycan structural diversity in whole animals implies that these structures may function to transfer biological signals between cells and their neighbors or between cells and the molecules in their environment. There are also examples where specific terminal glycan moieties mediate receptor-dependent interactions leading to glycoprotein clearance or to cell adhesion. Nevertheless, the significant structural diversity displayed by glycans is not yet accompanied by a corresponding functional diversity. As noted in Chapter 33, genetic modification of the glycosylation phenotype in intact organisms represents a powerful approach to exploring and confirming postulated roles for terminal glycan structures, as well as for discovering new functions for them. The large number of glycosyltransferase loci that contribute to the terminal glycan structural diversity, the overlap existing in some of their biosynthetic pathways, and the multiplicity of organs and tissues where these structures may be found together imply that a correspondingly large number of such experiments will be needed before a more complete understanding can be expected of the functions generated by glycan diversification.
References
- 1.
- Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem. 1985;54:631–664. [PubMed: 3896128]
- 2.
- Sadler J.E. 1984. Biosynthesis of glycoproteins: Formation of O-linked oligosaccharides. In Biology of carbohydrates (ed. V. Ginsburg and P. Robbins), vol. 2, pp. 199–288. Wiley, New York.
- 3.
- van Echten G, Sandhoff K. Ganglioside metabolism. Enzymology, topology, and regulation. J. Biol. Chem. 1993;268:5341–5344. [PubMed: 8449895]
- 4.
- Feizi T. Demonstration by monoclonal antibodies that carbohydrate structures of glycoproteins and glycolipids are onco-developmental antigens. Nature. 1985;314:53–57. [PubMed: 2579340]
- 5.
- Hakomori S -I. Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res. 1996;56:5309–5318. [PubMed: 8968075]
- 6.
- Varki A. Biological roles of oligosaccharides: All of the theories are correct. Glycobiology. 1993;3:97–130. [PMC free article: PMC7108619] [PubMed: 8490246]
- 7.
- Joziasse D H. Mammalian glycosyltransferases: Genomic organization and protein structure. Glycobiology. 1992;2:271–277. [PubMed: 1421748]
- 8.
- Baenziger J U. Protein-specific glycosyltransferases: How and why they do it! FASEB J. 1994;8:1019–1025. [PubMed: 7926366]
- 9.
- Perillo N L, Marcus M E, Baum L G. Galectins: Versatile modulators of cell adhesion, cell proliferation, and cell death. J. Mol. Med. 1998;76:402–412. [PubMed: 9625297]
- 10.
- Almeida R, Amado M, David L, Levery S B, Holmes E H, Merkx G, van Kessel A G, Rygaard E, Hassan H, Bennett E, Clausen H. A family of human β4-galactosyltransferases. Cloning and expression of two novel UDP-galactose:β-n-acetylglucosamine β1,4-galactosyltransferases, β4Gal-T2 and β4Gal-T3. J. Biol. Chem. 1997;272:31979–31991. [PubMed: 9405390]
- 11.
- Nomura T, Takizawa M, Aoki J, Arai H, Inoue K, Wakisaka E, Yoshizuka N, Imokawa G, Dohmae N, Takio K, Hattori M, Matsuo N. Purification, cDNA cloning, and expression of UDP-Gal: Glucosylceramide β-1,4-galactosyltransferase from rat brain. J. Biol. Chem. 1998;273:13570–13577. [PubMed: 9593693]
- 12.
- Schwientek T, Almeida R, Levery S B, Holmes E H, Bennett E, Clausen H. Cloning of a novel member of the UDP-galactose: β-N-acetylglucosamine β1,4-galactosyltransferase family, β4Gal-T4, involved in glycosphingolipid biosynthesis. J. Biol. Chem. 1998;273:29331–29340. [PubMed: 9792633]
- 13.
- Kolbinger F, Streiff M B, Katopodis A G. Cloning of a human UDP-galactose:2-acetamido-2-deoxy-d-glucose 3β-galactosyltransferase catalyzing the formation of type 1 chains. J. Biol. Chem. 1998;273:433–440. [PubMed: 9417100]
- 14.
- Amado M, Almeida R, Carneiro F, Levery S B, Holmes E H, Nomoto M, Hollingsworth M A, Hassan H, Schwientek T, Nielsen P A, Bennett E P, Clausen H. A family of human β3-galactosyltransferases: Characterization of four members of a UDP-galactose: β-N-acetyl-glucosamine/β-nacetyl-galactosamine β-1,3-galactosyltransferase family. J. Biol. Chem. 1998;273:12770–12778. [PubMed: 9582303]
- 15.
- Ujita M, McAuliffe J, Schwientek T, Almeida R, Hindsgaul O, Clausen H, Fukuda M. Synthesis of poly-N-acetyllactosamine in Core 2 branched O-glycans. The requirement of novel β-1,4-galactosyltransferase IV and β-1,3-N-acetylglucosaminyltransferase. J. Biol. Chem. 1998;273:34843–34849. [PubMed: 9857011]
- 16.
- Sasaki K, Kurata-Miura K, Ujita M, Angata K, Nakagawa S, Sekine S, Nishi T, Fukuda M. Expression cloning of cDNA encoding a human β-1,3-N-acetylglucosaminyltransferase that is essential for poly-N-acetyllactosamine synthesis. Proc. Natl. Acad. Sci. 1997;94:14294–14299. [PMC free article: PMC24948] [PubMed: 9405606]
- 17.
- Zhou D, Dinter A, Gutiérrez Gallego R, Kamerling J P, Vliegenthart J F G, Berger E G, Hennet T. A β-1,3-N-acetylglucosaminyltransferase with poly-N-acetyllactosamine synthase activity is structurally related to β-1,3-galactosyltransferases. Proc. Natl. Acad. Sci. 1999;96:406–411. [PMC free article: PMC15149] [PubMed: 9892646]
- 18.
- Wang W C, Lee N, Aoki D, Fukuda M N, Fukuda M. The poly-N-acetyllactosamines attached to lysosomal membrane glycoproteins are increased by the prolonged association with the Golgi complex. J. Biol. Chem. 1991;266:23185–23190. [PubMed: 1744116]
- 19.
- Muthing J, Spanbroek R, Peter-Katalinic J, Hanisch F G, Hanski C, Hasegawa A, Unland F, Lehmann J, Tschesche H, Egge H. Isolation and structural characterization of fucosylated gangliosides with linear poly-N-acetyllactosaminyl chains from human granulocytes. Glycobiology. 1996;6:147–156. [PubMed: 8727787]
- 20.
- Do K Y, Smith D F, Cummings R D. Lamp-1 in Cho Cells is a Primary Carrier of Poly-n-acetyllactosamine Chains and is Bound Preferentially by a Mammalian S-type Lectin. Biochem. Biophys. Res. Commun. 1990;173:1123–1128. [PubMed: 2268316]
- 21.
- Merkle R K, Cummings R D. Relationship of the terminal sequences to the length of poly-N-acetyllactosamine chains in asparagine-linked oligosaccharides from the mouse lymphoma cell line BW5147. Immobilized tomato lectin interacts with high affinity with glycopeptides containing long poly-N-acetyllactosamine chains. J. Biol. Chem. 1987;262:8179–8189. [PubMed: 3597368]
- 22.
- Elices M J, Goldstein I J. Initiation of poly-N-acetyllactosamine chain biosynthesis occurs preferentially on complex multi-antennary asparagine-linked oligosaccharides. Carbohydr. Res. 1990;203:109–118. [PubMed: 2146013]
- 23.
- Yousefi S, Higgins E, Daoling Z, Pollex-Kruger A, Hindsgaul O, Dennis J W. Increased UDP-GlcNAc:Gal β1-3GalNAc-R (GlcNAc to GalNAc) β-1, 6-N-acetylglucosaminyltransferase activity in metastatic murine tumor cell lines. Control of polylactosamine synthesis. J. Biol. Chem. 1991;266:1772–1782. [PubMed: 1824844]
- 24.
- Wilkins P P, McEver R P, Cummings R D. Structures of the O-glycans on P-selectin glycoprotein ligand-1 from HL-60 cells. J. Biol. Chem. 1996;271:18732–18742. [PubMed: 8702529]
- 25.
- Prieto P A, Larsen R D, Cho M, Rivera H N, Shilatifard A, Lowe J B, Cummings R D, Smith D F. Expression of human H-type α1,2-fucosyltransferase encoding for blood group H(O) antigen in Chinese hamster ovary cells. Evidence for preferential fucosylation and truncation of polylactosamine sequences. J. Biol. Chem. 1997;272:2089–2097. [PubMed: 8999907]
- 26.
- Fukuda M. 1994. Cell surface carbohydrates: Cell-type specific expression. In Molecular glycobiology (ed. M. Fukuda), pp. 1–52. Oxford University Press, Oxford, United Kingdom.
- 27.
- Renouf D V, Hounsell E F. Conformational studies of the backbone (poly-N-acetyllactosamine) and the core region sequences of O-linked carbohydrate chains. Int. J. Biol. Macromol. 1993;15:37–42. [PubMed: 8443131]
- 28.
- Merkle R K, Cummings R D. Asparagine-linked oligosaccharides containing poly-N-acetyllactosamine chains are preferentially bound by immobilized calf heart agglutinin. J. Biol. Chem. 1998;263:16143–16149. [PubMed: 3182789]
- 29.
- Bierhuizen M F, Mattei M G, Fukuda M. Expression of the developmental I antigen by a cloned human cDNA encoding a member of a β-1,6-N-acetylglucosaminyltransferase gene family. Genes Dev. 1993;7:468–478. [PubMed: 8449405]
- 30.
- Mattila P, Salminen H, Hirvas L, Niittymaki J, Salo H, Niemela R, Fukuda M, Renkonen O, Renkonen R. The centrally acting β1,6N-acetylglucosaminyltransferase (GlcNAc to Gal). Functional expression, purification, and acceptor specificity of a human enzyme involved in midchain branching of linear poly-N-acetyllactosamines. J. Biol. Chem. 1998;273:27633–27639. [PubMed: 9765298]
- 31.
- Feizi T. The blood group Ii system: A carbohydrate antigen system defined by naturally monoclonal or oligoclonal autoantibodies of man. Immunol. Commun. 1981;10:127–156. [PubMed: 6169630]
- 32.
- Hakomori S. Blood group ABH and Ii antigens of human erythrocytes: Chemistry, polymorphism, and their developmental change. Semin. Hematol. 1981;18:39–62. [PubMed: 6782678]
- 33.
- Childs R A, Kapadia A, Feizi T. Expression of blood group I and i active carbohydrate sequences on cultured human and animal cell lines assessed by radioimmunoassays with monoclonal cold agglutinins. Eur. J. Immunol. 1980;10:379–384. [PubMed: 6157539]
- 34.
- Yeh J -C, Ong E, Fukuda M. Molecular cloning and expression of a novel β-1,6-N-acetylglucosaminyltransferase that forms Core 2, Core 4, and I branches. J. Biol. Chem. 1999;274:3215–3221. [PubMed: 9915862]
- 35.
- Landsteiner K. Individual differences in human blood. Science. 1931;73:405–411. [PubMed: 17755773]
- 36.
- Daniels G L, Anstee D J, Cartron J P, Dahr W, Issitt P D, Jorgensen J, Kornstad L, Levene C, Lomas-Francis C, Lubenko A. et al. Blood Group Terminology 1995. Isbt Working Party on Terminology for Red Cell Surface Antigens. Vox Sang. 1995;69:265–279. [PubMed: 8578746]
- 37.
- Mollison P.L., Engelfriet C.P., and Contreras M. 1997. Blood transfusion in clinical medicine, 10th edition. Blackwell, Oxford, United Kingdom.
- 38.
- Garratty G. Problems associated with passively transfused blood group alloantibodies. Am. J. Clin. Pathol. 1998;109:769–777. [PubMed: 9620038]
- 39.
- Capon S M, Goldfinger D. Acute hemolytic transfusion reaction, a paradigm of the systemic inflammatory response: New insights into pathophysiology and treatment. Transfusion. 1995;35:513–520. [PubMed: 7770905]
- 40.
- Watkins W M. Biochemistry and genetics of the ABO, Lewis, and P blood group systems. Adv. Hum. Genet. 1980;10:1–136. [PubMed: 6156588]
- 41.
- Lowe J.B. 1999. Red cell membrane antigens. In The molecular basis of blood diseases (ed. G. Stamatoyannopoulos et al.). W.B. Saunders, Orlando, Florida. (In press.)
- 42.
- Clausen H, Hakomori S. ABH and related histo-blood group antigens: Immunochemical differences in carrier isotypes and their distribution. Vox Sang. 1989;56:1–20. [PubMed: 2464874]
- 43.
- Clausen H, Levery S B, Nudelman E, Tsuchiya S, Hakomori S. Repetitive A epitope (type 3 chain A) defined by blood group A1-specific monoclonal antibody TH-1: Chemical basis of qualitiative A1 and A2 distinction. Proc. Natl. Acad. Sci. 1985;82:1199–1203. [PMC free article: PMC397222] [PubMed: 2579390]
- 44.
- Le Pendu J, Lambert F, Samuelsson B E, Breimer M E, Seitz R C, Urdaniz M P, Suesa N, Ratcliffe M, Francoise A, Poschmann A, Vinas J, Oriol R. Monoclonal antibodies specific for type 3 and type 4 chain-based blood group determinants: Relationship to the A1 and A2 subgroups. Glycoconj. J. 1986;3:255–271.
- 45.
- Clausen H, Levery S B, Nudelman E, Baldwin M, Hakomori S. Further characterization of type 2 and type 3 chain blood group A glycosphingolipids from human erythrocyte membranes. Biochemistry. 1986;25:7075–7085. [PubMed: 3801409]
- 46.
- Betteridge A, Watkins W M. Acceptor substrate specificities of human α-2-l-fucosyltransferases from different tissues. Biochem. Soc. Trans. 1986;13:1126–1127.
- 47.
- Laine R A, Rush J S. Chemistry of human erythrocyte polylactosamine glycopeptides (erythroglycans) as related to ABH blood group antigenic determinants. Adv. Exp. Med. Biol. 1988;228:331–347. [PubMed: 2459929]
- 48.
- Koscielak J, Miller-Podraza H, Krauze R, Piasek A. Isolation and characterization of poly(glycosyl)ceramides (megaloglycolipids) with A, H, and I blood-group activities. Eur. J. Biochem. 1976;71:9–18. [PubMed: 827447]
- 49.
- Eastlund T. The histo-blood group ABO system and tissue transplantation. Transfusion. 1988;38:975–988. [PubMed: 9767749]
- 50.
- Kisailus E C, Kabat E A. Immunochemical Studies on Blood Groups. Lxvi. Competitive Binding Assays of A1 and A2 Blood Group Substances with Insolubilized Anti-a Serum and Insolubilized a Agglutinine from Dolichos Biflorus. J. Exp. Med. 1978;147:830–843. [PMC free article: PMC2184207] [PubMed: 75942]
- 51.
- Moreno C, Lundblad A, Kabat E A. Immunochemical studies on blood groups. LI. A comparative study of the reaction of A1 and A2 blood group glycoproteins with human anti-A. J. Exp. Med. 1971;134:439–457. [PMC free article: PMC2139049] [PubMed: 4104425]
- 52.
- Yamamoto F -I, Marken J, Tsuji T, White T, Clausen H, Hakomori S. Cloning and characterization of DNA complementary to human UDP-GalNAc:Fucα1→2Gal α1→3GalNAc transferase (histo-blood group A transferase) mRNA. J. Biol. Chem. 1990;264:1146–1151. [PubMed: 2104828]
- 53.
- Yamamoto F -I, Clausen H, White T, Marken J, Hakomori S -I. Molecular genetic basis of the histo-blood group ABO system. Nature. 1990;345:229–233. [PubMed: 2333095]
- 54.
- Mourant A.E. 1978. Blood groups and diseases. A study of associations of diseases with blood groups and other polymorphisms. Oxford University Press, Oxford, United Kingdom.
- 55.
- Garratty G. Blood group antigens as tumor markers, parasitic/bacterial/viral receptors, and their association with immunologically important proteins. Immunol. Invest. 1995;24:213–232. [PubMed: 7713584]
- 56.
- Greenwell P. Blood group antigens: Molecules seeking a function? Glycoconj. J. 1997;14:159–173. [PubMed: 9111133]
- 57.
- Phillips M D, Santhouse A. von Willebrand disease: Recent advances in pathophysiology and treatment. Am. J. Med. Sci. 1998;316:77–86. [PubMed: 9704661]
- 58.
- Mohlke K L, Purkayastha A A, Westrick R J, Smith P L, Petryniak B, Lowe J B, Ginsburg D. Mvwf, a dominant modifier of murine von Willebrand factor, results from altered lineage-specific expression of a glycosyltransferase. Cell. 1998;96:111–120. [PubMed: 9989502]
- 59.
- Boren T, Normark S, Falk P. Helicobacter pylori: Molecular basis for host recognition and bacterial adherence. Trends Microbiol. 1994;2:221–228. [PubMed: 8081648]
- 60.
- Aird I, Bentall H H, Mehigan J A, Roberts F J A. The blood groups in relation to peptic ulceration and carcinoma of colon, rectum, breast, and bronchus: An association between the ABO groups and peptic ulceration. Brit. Med. J. 1954;ii:315–321. [PMC free article: PMC2078797] [PubMed: 13182205]
- 61.
- Milne R W, Dawes C. The relative contributions of different salivary glands to the blood group activity of whole saliva in humans. Vox Sang. 1973;25:298–307. [PubMed: 4796232]
- 62.
- Oriol R, Danilovs J, Hawkins B R. A new genetic model proposing that the Se gene is a structural gene closely linked to the H gene. Am. J. Hum. Genet. 1981;33:421–431. [PMC free article: PMC1685044] [PubMed: 7246545]
- 63.
- Clarke C A, Edwards J, Wyn Haddock D R W, Howel-Evans A W, McConnell R B, Sheppard P M. ABO blood groups and secretor character in duodenal ulcer. Brit. Med. J. 1956; ii:725–731. [PMC free article: PMC2035396] [PubMed: 13364310]
- 64.
- Boren T, Falk P, Roth K A, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993; 262:1892–1895. [PubMed: 8018146]
- 65.
- Le Pendu J, Cartron J P, Lemieux R U, Oriol. R. The presence of at least two different H-blood-group-related βDGal α-2-L-fucosyltransferases in human serum and the genetics of blood group H substances. Am. J. Hum. Genet. 1985;37:749–760. [PMC free article: PMC1684624] [PubMed: 9556663]
- 66.
- Kelly R J, Ernst L K, Larsen R D, Bryant J G, Robinson J S, Lowe J B. Molecular basis for H blood group deficiency in Bombay (Oh) and para-Bombay individuals. Proc. Natl. Acad. Sci. 1994;91:5843–5847. [PMC free article: PMC44093] [PubMed: 7912436]
- 67.
- Rouquier S, Lowe J B, Kelly R J, Fertitta A L, Lennon G G, Giorgi D. Molecular cloning of a human genomic region containing the H blood group α(1,2) fucosyltransferase gene and two H restriction fragments. Isolation of a candidate for the human Secretor blood locus-related DNA group locus. J. Biol. Chem. 1995;270:4632–4639. [PubMed: 7876234]
- 68.
- Kelly R J, Rouquier S, Giorgi D, Lennon G G, Lowe J B. Sequence and expression of a candidate for the human Secretor blood group α(1,2) fucosyltransferase gene (FUT2). Homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the non-secretor phenotype. J. Biol. Chem. 1995;270:4640–4649. [PubMed: 7876235]
- 69.
- Costache M, Cailleau A, Fernandez-Mateos P, Oriol R, Mollicone R. Advances in molecular genetics of α-2- and α-3/4-fucosyltransferases. Transfus. Clin. Biol. 1997;4:367–382. [PubMed: 9269717]
- 70.
- Clausen H, Holmes E, Hakomori S. Novel blood group H glycolipid antigens exclusively expressed in blood group A and AB erythrocytes (type 3 chain H). II. Differential conversion of different H substrates by A1 and A2 enzymes, and type 3 chain H expression in relation to secretor status. J. Biol. Chem. 1986;261:1388–1392. [PubMed: 3944092]
- 71.
- Dejter-Juszynski M, Harpaz N, Flowers H M, Sharon N. Blood-group ABH-specific macroglycolipids of human erythrocytes: Isolation in high yield from a crude membrane glycoprotein fraction. Eur. J. Biochem. 1978;83:363–373. [PubMed: 631124]
- 72.
- Mourant A E. A “new” human blood group antigen of frequent occurrence. Nature. 1946;158:237–238. [PubMed: 20995492]
- 73.
- Rege V P, Painter T J, Watkins W M, Morgan W T J. Isolation of a serologically active fucose containing trisaccharide from human blood group Lea substrate. Nature. 1964;240:740–742. [PubMed: 14235664]
- 74.
- Hanfland P, Graham H. Immunochemistry of the Lewis-blood-group system: Partial characterization of Le(a), Le(b) and H-type 1 (LedH)-blood-group active glycosphingolipids from human plasma. Arch. Biochem. Biophys. 1981;210:383–395. [PubMed: 7294833]
- 75.
- Hanfland P, Kardowicz M, Peter-Katalinic J, Pfannschmidt G, Crawford R J, Graham H A, Egge H. Immunochemistry of the Lewis blood group system: Isolation and structures of the Lewis-c active and related glycosphingolipids from the plasma of blood-group O Le(a-b-) nonsecretors. Arch. Biochem. Biophys. 1986;246:655–672. [PubMed: 2423032]
- 76.
- Marcus D M, Cass L E. Glycosphingolipids with Lewis blood group activity: Uptake by human erythrocytes. Science. 1969;164:553–555. [PubMed: 5778002]
- 77.
- Johnson P H, Yates A D, Watkins W M. Human salivary fucosyltransferases: Evidence for two distinct α-3-l-fucosyltransferase activities one of which is associated with the Lewis blood group Le gene. Biochem. Biophys. Res. Commun. 1981;100:1611–1618. [PubMed: 7295318]
- 78.
- Kukowska-Latallo J F, Larsen R D, Nair R P, Lowe J B. A cloned human cDNA determines expression of a mouse stage-specific embryonic antigen and the Lewis blood group α(1,3/1,4)fucosyltransferase. Genes Dev. 1990;4:1288–1303. [PubMed: 1977660]
- 79.
- Weston B W, Nair R P, Larsen R D, Lowe J B. Isolation of a novel human α(1,3)fucosyltransferase gene and molecular comparison to the human Lewis blood group α(1,3/1,4)fucosyltransferase gene. J. Biol. Chem. 1992;267:4152–4160. [PubMed: 1740457]
- 80.
- Weston B W, Smith P L, Kelly R J, Lowe J B. Molecular cloning of a fourth member of a human α(1,3)fucosyltransferase gene family: Multiple homologous sequences that determine expression of the Lewis x, sialyl Lewis x, and difucosyl sialyl Lewis x epitopes. J. Biol. Chem. 1992;267:24575–24584. [PubMed: 1339443]
- 81.
- Lowe J B, Kukowska-Latallo J F, Nair R P, Larsen R D, Marks R M, Macher B A, Kelly R J, Ernst L K. Molecular Cloning of a Human Fucosyltransferase Gene that Determines Expression of the Lewis X and Vim-2 Epitopes But Not Elam-1-dependent Cell Adhesion. J. Biol. Chem. 1991;266:17467–17477. [PubMed: 1716630]
- 82.
- Goelz S E, Hession C, Goff D, Griffiths B, Tizard R, Newman B, Chi-Rosso G, Lobb R. Elft: a Gene that Directs the Expression of An Elam-1 Ligand. Cell. 1990;63:1349–1356. [PubMed: 1702034]
- 83.
- Kumar R, Potvin B, Muller W A, Stanley P. Cloning of a human α(1,3)fucosyltransferase gene that encodes ELFT but does not confer ELAM-1 recognition on CHO transfections. J. Biol. Chem. 1991;266:21777–21783. [PubMed: 1718983]
- 84.
- Sasaki K, Kurata K, Funayama K, Nagata M, Watanabe E, Ohta S, Hanai N, Nishi T. Expression cloning of a novel α1,3-fucosyltransferase that is involved in biosynthesis of the sialyl Lewis x carbohydrate determinants in leukocytes. J. Biol. Chem. 1994;269:14730–14737. [PubMed: 8182079]
- 85.
- Natsuka S, Gersten K M, Zenita K, Kannagi R, Lowe J B. Molecular cloning of a cDNA encoding a novel human leukocyte α(1,3)fucosyltransferase capable of synthesizing the sialyl Lewis x determinant. J. Biol. Chem. 1994;269:16789–16794. [PubMed: 8207002]
- 86.
- Kudo T, Ikehara Y, Togayachi A, Kaneko M, Hiraga T, Sasaki K, Narimatsu H. Expression cloning and characterization of a novel murine α1,3-fucosyltransferase, mFuc-TIX, that synthesizes the Lewis x (CD15) epitope in brain and kidney. J. Biol. Chem. 1998;273:26729–26738. [PubMed: 9756916]
- 87.
- Mollicone R, Reguigne I, Kelly R J, Fletcher A, Watt J, Chatfield S, Aziz A, Cameron H S, Weston B W, Lowe J B, Oriol R. Molecular basis for Lewis α(1,3/1,4)-fucosyltransferase gene deficiency (FUT3) found in Lewis-negative Indonesian pedigrees. J. Biol. Chem. 1994;269:20987–20994. [PubMed: 8063716]
- 88.
- Elmgren A, Mollicone R, Costache M, Borjeson C, Oriol R, Harrington J, Larson G. Significance of individual point mutations, T202C and C314T, in the human Lewis (FUT3) gene for expression of Lewis antigens by the human α(1,3/1,4)-fucosyltransferase, Fuc-TIII. J. Biol. Chem. 1997;272:21994–21998. [PubMed: 9268337]
- 89.
- Kansas G S. Selectins and their ligands: Current concepts and controversies. Blood. 1996;88:3259–3287. [PubMed: 8896391]
- 90.
- Forman D. Helicobacter pylori infection and cancer. Br. Med. Bull. 1998;54:71–78. [PubMed: 9604432]
- 91.
- Atherton J C. H. pylori virulence factors. Br. Med. Bull. 1998;54:105–120. [PubMed: 9604436]
- 92.
- Oberhuber G, Kranz A, Dejaco C, Dragosics B, Mosberger I, Mayr W, Radaszkiewicz T. Blood groups Lewis(b) and ABH expression in gastric mucosa: Lack of inter-relation with Helicobacter pylori colonisation and occurrence of gastric MALT lymphoma. Gut. 1997;41:37–42. [PMC free article: PMC1027225] [PubMed: 9274469]
- 93.
- Heneghan M A, Moran A P, Feeley K M, Egan E L, Goulding J, Connolly C E, McCarthy C F. Effect of host Lewis and ABO blood group antigen expression on Helicobacter pylori colonisation density and the consequent inflammatory response. FEMS Immunol. Med. Microbiol. 1998;20:257–266. [PubMed: 9626930]
- 94.
- Monteiro M A, Chan K H, Rasko D A, Taylor D E, Zheng P Y, Appelmelk B J, Wirth H P, Yang M, Blaser M J, Hynes S O, Moran A P, Perry M B. Simultaneous expression of type 1 and type 2 Lewis blood group antigens by Helicobacter pylori lipopolysaccharides. Molecular mimicry between H. pylori lipopolysaccharides and human gastric epithelial cell surface glycoforms. J. Biol. Chem. 1998;273:11533–11543. [PubMed: 9565568]
- 95.
- Marcus D M, Kundu S K, Suzuki A. The P blood group system: Recent progress in immunochemistry and genetics. Semin. Hematol. 1981;18:63–71. [PubMed: 7010611]
- 96.
- Yang Z, Bergstrom J, Karlsson K A. Glycoproteins with Gal alpha 4Gal are absent from human erythrocyte membranes, indicating that glycolipids are the sole carriers of blood group P activities. J. Biol. Chem. 1994;269:14620–14624. [PubMed: 8182069]
- 97.
- Lomberg H, Eden C S. Influence of P blood group phenotype on susceptibility to urinary tract infection. FEMS Microbiol. Immunol. 1989;1:363–370. [PubMed: 2631876]
- 98.
- Wold A E, Thorssen M, Hull S, Svanborg E C. Attachment of Escherichia coli to mannose- or Galα1–4Galβ-containing receptors in human colonic epithelial cells. Infect. Immun. 1988;56:2531–2537. [PMC free article: PMC259607] [PubMed: 2901402]
- 99.
- Lomberg H, Hanson L A, Jacobsson B, Jodal U, Leffler H, Svanborg E C. Correlation of P blood group, vesicoureteral reflux, and bacterial attachment in patients with recurrent pyelonephritis. N. Engl. J. Med. 1983;308:1189–1192. [PubMed: 6341837]
- 100.
- Stapleton A E, Stroud M R, Hakomori S I, Stamm W E. The globoseries glycosphingolipid sialosyl galactosyl globoside is found in urinary tract tissues and is a preferred binding receptor in vitro for uropathogenic Escherichia coli expressing pap-encoded adhesins. Infect. Immun. 1998;66:3856–3861. [PMC free article: PMC108435] [PubMed: 9673272]
- 101.
- Striker R, Nilsson U, Stonecipher A, Magnusson G, Hultgren S J. Structural requirements for the glycolipid receptor of human uropathogenic Escherichia coli. Mol. Microbiol. 1995;16:1021–1029. [PubMed: 7476178]
- 102.
- Tomisawa S, Kogure T, Kuroume T, Leffler H, Lomberg H, Shimabukoro N, Terao K, Svanborg E C. P blood group and proneness to urinary tract infection in Japanese children. Scand. J. Infect. Dis. 1989;21:403–408. [PubMed: 2587941]
- 103.
- Roberts J A, Marklund B I, Ilver D, Haslam D, Kaack M B, Baskin G, Louis M, Mollby R, Winberg J, Normark S. The Gal(alpha 1–4)Gal-specific tip adhesin of Escherichia coli P-fimbriae is needed for pyelonephritis to occur in the normal urinary tract. Proc. Natl. Acad. Sci. 1994;91:11889–11893. [PMC free article: PMC45341] [PubMed: 7991552]
- 104.
- Brown K E, Young N S. Parvovirus B19 in human disease. Annu. Rev. Med. 1997;48:59–67. [PubMed: 9046945]
- 105.
- Brown K E, Anderson S M, Young N S. Erythrocyte P antigen: Cellular receptor for B19 parvovirus. Science. 1993;262:114–117. [PubMed: 8211117]
- 106.
- Brown K E, Hibbs J R, Gallinella G, Anderson S M, Lehman E D, McCarthy P, Young N S. Resistance to parvovirus B19 infection due to lack of virus receptor (erythrocyte P antigen). N. Engl. J. Med. 1994;330:1192–1196. [PubMed: 8139629]
- 107.
- Galili U. Evolution and pathophysiology of the human natural anti-alpha-galactosyl IgG (anti-Gal) antibody. Springer Semin. Immunopathol. 1993;15:155–171. [PubMed: 7504839]
- 108.
- Galili U, Kobrin E, Macher B A, Shohet S B. Anti-Gal and human red cell aging. Prog. Clin. Biol. Res. 1989;319:225–241. [PubMed: 2482975]
- 109.
- Bleil J D, Wassarman P M. Galactose at the nonreducing terminus of O-linked oligosaccharides of mouse egg zona pellucida glycoprotein ZP3 is essential for the glycoprotein's sperm receptor activity. Proc. Natl. Acad. Sci. 1988;85:6778–6782. [PMC free article: PMC282061] [PubMed: 2842789]
- 110.
- Thall A D, Maly P, Lowe J B. Oocyte Gal alpha 1,3Gal epitopes implicated in sperm adhesion to the zona pellucida glycoprotein ZP3 are not required for fertilization in the mouse. J. Biol. Chem. 1995;270:21437–21440. [PubMed: 7545161]
- 111.
- Winand R J, Devigne J W, Meurisse M, Galili U. Specific stimulation of Graves' disease thyrocytes by the natural anti-Gal antibody from normal and autologous serum. J. Immunol. 1994;153:1386–1395. [PubMed: 8027563]
- 112.
- Galili U, LaTemple D C. Natural anti-Gal antibody as a universal augmenter of autologous tumor vaccine immunogenicity. Immunol. Today. 1997;18:281–285. [PubMed: 9190114]
- 113.
- Cooper D K. Xenoantigens and xenoantibodies. Xenotransplantation. 1998;5:6–17. [PubMed: 9507728]
- 114.
- Lambrigts D, Sachs D H, Cooper D K. Discordant organ xenotransplantation in primates: World experience and current status. Transplantation. 1998;66:547–561. [PubMed: 9753331]
- 115.
- Osman N, McKenzie I F, Ostenried K, Ioannou Y A, Desnick R J, Sandrin M S. Combined transgenic expression of alpha-galactosidase and alpha1,2-fucosyltransferase leads to optimal reduction in the major xenoepitope Galalpha(1,3)Gal. Proc. Natl. Acad. Sci. 1997;94:14677–14682. [PMC free article: PMC25090] [PubMed: 9405672]
- 116.
- Takeuchi Y, Porter C D, Strahan K M, Preece A F, Gustafsson K, Cosset F L, Weiss R A, Collins M K. Sensitization of cells and retroviruses to human serum by (alpha 1–3) galactosyltransferase. Nature. 1996;379:85–88. [PubMed: 8538747]
- 117.
- Siddiqui B, Hakomori S -I. A revised structure for the Forssman glycolipid hapten. J. Biol. Chem. 1971;246:5766–5769. [PubMed: 4328838]
- 118.
- Fredman P. Glycosphingolipid tumor antigens. Adv. Lipid Res. 1993;25:213–234. [PubMed: 8368149]
- 119.
- Mori E, Mori T, Sanai Y, Nagai Y. Radioimmuno-thin-layer chromatographic detection of Forssman antigen in human carcinoma cell lines. Biochem. Biophys. Res. Commun. 1982;108:926–932. [PubMed: 6983878]
- 120.
- Haslam D B, Baenziger J U. Expression cloning of Forssman glycolipid synthetase: A novel member of the histo-blood group ABO gene family. Proc. Natl. Acad. Sci. 1996;93:10697–10702. [PMC free article: PMC38217] [PubMed: 8855242]
- 121.
- Koski C L, Chou D K, Jungalwala F B. Anti-peripheral nerve myelin antibodies in Guillain-Barre syndrome bind a neutral glycolipid of peripheral myelin and cross-react with Forssman antigen. J. Clin. Invest. 1989;84:280–287. [PMC free article: PMC303980] [PubMed: 2738153]
- 122.
- Strokan V, Rydberg L, Hallberg E C, Molne J, Breimer M E. Characterisation of human natural anti-sheep xenoantibodies. Xenotransplantation. 1998;5:111–121. [PubMed: 9584825]
- 123.
- Zinkl G M, Zuk A, van der Bijl P, van Meer G, Matlin K S. An antiglycolipid antibody inhibits Madin-Darby canine kidney cell adhesion to laminin and interferes with basolateral polarization and tight junction formation. J. Cell Biol. 1996;133:695–708. [PMC free article: PMC2120812] [PubMed: 8636242]
- 124.
- Manzella S M, Hooper L V, Baenziger J U. Oligosaccharides containing β 1,4-linked N-acetylgalactosamine, a paradigm for protein-specific glycosylation. J. Biol. Chem. 1996;271:12117–12120. [PubMed: 8647799]
- 125.
- Baenziger J U, Kumar S, Brodbeck R M, Smith P L, Beranek M C. Circulatory half-life but not interaction with the lutropin/chorionic gonadotropin receptor is modulated by sulfation of bovine lutropin oligosaccharides. Proc. Natl. Acad. Sci. 1992;89:334–338. [PMC free article: PMC48231] [PubMed: 1729704]
- 126.
- Smith P L, Baenziger J U. Molecular basis of recognition by the glycoprotein hormone-specific N-acetylgalactosamine-transferase. Proc. Natl. Acad. Sci. 1992;89:329–333. [PMC free article: PMC48230] [PubMed: 1370352]
- 127.
- Fiete D J, Beranek M C, Baenziger J U. A cysteine-rich domain of the “mannose” receptor mediates GalNAc-4-SO4 binding. Proc. Natl. Acad. Sci. 1998;95:2089–2093. [PMC free article: PMC19259] [PubMed: 9482843]
- 128.
- Lefrancois L, Bevan M J. Functional modifications of cytotoxic T-lymphocyte T200 glycoprotein recognized by monoclonal antibodies. Nature. 1985;314:449–452. [PubMed: 2580241]
- 129.
- Lefrancois L, Puddington L, Machamer C E, Bevan M J. Acquisition of cytotoxic T lymphocyte-specific carbohydrate differentiation antigens. J. Exp. Med. 1985;162:1275–1293. [PMC free article: PMC2187862] [PubMed: 2413157]
- 130.
- Lefrancois L. Carbohydrate differentiation antigens of murine T cells: Expression on intestinal lymphocytes and intestinal epithelium. J. Immunol. 1987;138:3375–3384. [PubMed: 2437191]
- 131.
- Conzelmann A, Kornfeld S. A murine cytotoxic T lymphocyte cell line resistant to Vicia villosa lectin is deficient in UDP-GalNAc:β-galactose β1,4-N-acetylgalactosaminyltransferase. J. Biol. Chem. 1984;259:12528–12535. [PubMed: 6436236]
- 132.
- Conzelmann A, Kornfeld S. Beta-linked N-acetylgalactosamine residues present at the nonreducing termini of O-linked oligosaccharides of a cloned murine cytotoxic T lymphocyte line are absent in a Vicia villosa lectin-resistant mutant cell line. J. Biol. Chem. 1984;259:12536–12542. [PubMed: 6333421]
- 133.
- Donald A S R, Yates A D, Soh C P C, Morgan W T J, Watkins W M. A blood group Sda-active pentasaccharide isolated from Tamm-Horsfall urinary glycoprotein. Biochem. Biophys. Res. Commun. 1983;115:625–631. [PubMed: 6414473]
- 134.
- Conzelmann A, Lefrancois L. Monoclonal antibodies specific for T cell-associated carbohydrate determinants react with human blood group antigens CAD and SDA. J. Exp. Med. 1988;167:119–131. [PMC free article: PMC2188809] [PubMed: 2447219]
- 135.
- Smith P L, Lowe J B. Molecular cloning of a murine N-acetylgalactosaminyltransferase cDNA that determines expression of the T-lymphocyte-specific CT oligosaccharide differentiation antigen. J. Biol. Chem. 1994;269:15162–15171. [PubMed: 7515051]
- 136.
- Nagata Y, Yamashiro S, Yodoi J, Lloyd K O, Shiku H, Furukawa K. Expression cloning of β1,4 N-acetylgalactosaminyltransferase cDNAs that determine the expression of GM2 and GD2 gangliosides. J. Biol. Chem. 1992;267:12082–12089. [PubMed: 1601877]
- 137.
- Takamiya K, Yamamoto A, Furukawa K, Yamashiro S, Shin M, Okada M, Fukumoto S, Haraguchi M, Takeda N, Fujimura K, Sakae M, Kishikawa M, Shiku H, Furukawa K, Aizawa S. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc. Natl. Acad. Sci. 1996;93:10662–10667. [PMC free article: PMC38211] [PubMed: 8855236]
- 138.
- Takamiya K, Yamamoto A, Furukawa K, Zhao J, Fukumoto S, Yamashiro S, Okada M, Haraguchi M, Shin M, Kishikawa M, Shiku H, Aizawa S, Furukawa K. Complex gangliosides are essential in spermatogenesis of mice: Possible roles in the transport of testosterone. Proc. Natl. Acad. Sci. 1998;95:12147–12152. [PMC free article: PMC22799] [PubMed: 9770454]
- 139.
- Tsuji S. Molecular cloning and functional analysis of sialyltransferases. J. Biochem. (Tokyo). 1996;120:1–13. [PubMed: 8864835]
- 140.
- Harduin-Lepers A, Recchi M -A, Delannoy P. 1994. The year of the sialyltransferases. Glycobiology. 1995;5:741–758. [PubMed: 8720072]
- 141.
- Kono M, Ohyama Y, Lee Y C, Hammamoto T, Kojima N, Tsuji S. Mouse β-galactoside α2,3sialyltransferases: Comparison of in vitro substrate specificities and tissue specific expression. Glycobiology. 1997;7:469–479. [PubMed: 9184827]
- 142.
- Ishii A, Ohta M, Watanabe Y, Matsuda K, Ishiyama K, Sakoe K, Nakamura M, Inokuchi J, Sanai Y, Saito M. Expression cloning and functional characterization of human cDNA for ganglioside GM3 synthase. J. Biol. Chem. 1998;273:31652–31655. [PubMed: 9822625]
- 143.
- Kono M, Takashima S, Liu H, Inoue M, Kojima N, Lee Y C, Hamamoto T, Tsuji S. Molecular cloning and functional expression of a fifth-type alpha 2,3-sialyltransferase (mST3Gal V: GM3 synthase). Biochem. Biophys. Res. Commun. 1998;253:170–175. [PubMed: 9875239]
- 144.
- Ashwell G, Harford J. Carbohydrate-specific receptors of the liver. Annu. Rev. Biochem. 1982;51:531–554. [PubMed: 6287920]
- 145.
- Weigel P H. Galactosyl and N-acetylgalactosaminyl homeostasis: A function for mammalian asialoglycoprotein receptors. Bioessays. 1994;16:519–524. [PubMed: 7945281]
- 146.
- Braun J R, Willnow T E, Ishibashi S, Ashwell G, Herz J. The major subunit of the asialoglycoprotein receptor is expressed on the hepatocellular surface in mice lacking the minor receptor subunit. J. Biol. Chem. 1996;271:21160–21166. [PubMed: 8702886]
- 147.
- Ishibashi S, Hammer R E, Herz J. Asialoglycoprotein receptor deficiency in mice lacking the minor receptor subunit. J. Biol. Chem. 1994;269:27803–27806. [PubMed: 7961705]
- 148.
- Karlsson K -A. Animal glycosphingolipids as membrane attachment sites for bacteria. Annu. Rev. Biochem. 1989;58:309–350. [PubMed: 2673013]
- 149.
- Karlsson K A. Microbial recognition of target-cell glycoconjugates. Curr. Opin. Struct. Biol. 1995;5:622–635. [PubMed: 8574698]
- 150.
- Weis W, Brown J H, Cusak S, Paulson J C, Skehel J J, Wiley D C. Structure of the influenza virus hemagglutinin complexed with its receptor, sialic acid. Nature. 1988;333:426–431. [PubMed: 3374584]
- 151.
- Hirmo S, Kelm S, Schauer R, Nilsson B, Waldstrom T. Adhesion of Helicobacter pylori strains to α-2,3-linked sialic acids. Glycoconj. J. 1996;13:1005–1011. [PubMed: 8981092]
- 152.
- Simon P M, Goode P L, Mobasseri A, Zopf D. Inhibition of Helicobacter pylori binding to gastrointestinal epithelial cells by sialic acid-containing oligosaccharides. Infect. Immun. 1997;65:750–757. [PMC free article: PMC176121] [PubMed: 9009338]
- 153.
- Merritt E A, Kuhn P, Sarfaty S, Erbe J L, Holmes R K, Hol W G. The 1.25 Å resolution refinement of the cholera toxin B-pentamer: Evidence of peptide backbone strain at the receptor-binding site. J. Mol. Biol. 1998;282:1043–1059. [PubMed: 9753553]
- 154.
- Merritt E A, Sarfaty S, Feil I K, Hol W. Structural foundation for the design of receptor antagonists targeting Escherichia coli heat-labile enterotoxin. Structure. 1997;5:1485–499. [PubMed: 9384564]
- 155.
- Weinstein J, de Souza-e-Silva U, Paulson J C. Sialylation of glycoprotein oligosaccharides N-linked to asparagine. Enzymatic characteristization of a Galβ1–3(4)GlcNAc α2–3sialyltransferase and a Galβ1–4GlcNAc α2–6sialyltransferase from rat liver. J. Biol. Chem. 1982;257:13845–13853. [PubMed: 7142180]
- 156.
- Weinstein J, Lee E U, McEntee K, Lai P, Paulson J C. Primary structure of β-galactoside α2,6sialyltransferase. J. Biol. Chem. 1987;262:17735–17743. [PubMed: 3121604]
- 157.
- Hennet T, Chui D, Paulson J C, Marth J D. Immune regulation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. 1998;95:4504–4509. [PMC free article: PMC22519] [PubMed: 9539767]
- 158.
- Eckhardt M, Muhlenhoff M, Bethe A, Koopman J, Frosch M, Gerardy-Schahn R. Molecular characterization of eukaryotic polysialyltransferase-1. Nature. 1995;373:715–718. [PubMed: 7854457]
- 159.
- Livingston B D, Paulson J C. Polymerase chain reaction cloning of a developmentally regulated member of the sialyltransferase gene family. J. Biol. Chem. 1993;268:11504–11507. [PubMed: 7685014]
- 160.
- Scheidegger E P, Sternberg L R, Roth J, Lowe J B. A human STX cDNA confers polysialic acid expression in mammalian cells. J. Biol. Chem. 1995;270:22685–22688. [PubMed: 7559389]
- 161.
- Sasaki K, Kurata K, Kojima N, Kurosawa N, Ohta S, Hanai N, Tsuji S, Nishi T. Expression cloning of a GM3-specific alpha-2,8-sialyltransferase (GD3 synthase). J. Biol. Chem. 1994;269:15950–15056. [PubMed: 8195250]
- 162.
- Nakayama J, Fukuda M. A human polysialyltransferase directs in vitro synthesis of polysialic acid. J. Biol. Chem. 1996;271:1829–1832. [PubMed: 8567623]
- 163.
- Nara K, Watanabe Y, Maruyama K, Kasahara K, Nagai Y, Sanai Y. Expression cloning of a CMP-NeuAc:NeuAc α2,8-sialyltransferase (GD3 synthase) from human melanoma cells. Proc. Natl. Acad. Sci. 1994;91:7952–7956. [PMC free article: PMC44522] [PubMed: 8058740]
- 164.
- Haraguchi M, Yamashiro S, Yamamoto A, Furukawa K, Takamiya K, Lloyd K O, Shiku H, Furukawa K. Isolation of GD3 synthase gene by expression cloning of GM3 alpha-2,8-sialyltransferase cDNA using anti-GD2 monoclonal antibody. Proc. Natl. Acad. Sci. 1994;91:10455–10459. [PMC free article: PMC45039] [PubMed: 7937974]
- 165.
- Nakayama J, Fukuda M N, Hirabayashi Y, Kanamori A, Sasaki K, Nishi T, Fukuda M. Expression cloning of a human GT3 synthase. GD3 and GT3 are synthesized by a single enzyme. J. Biol. Chem. 1996;271:3684–3891. [PubMed: 8631981]
- 166.
- Yoshida Y, Kojima N, Kurosawa N, Hamamoto T, Tsuji S. Molecular cloning of Sia α2,3Gal β1,4GlcNAc α2,8-sialyltransferase from mouse brain. J. Biol. Chem. 1996;270:14628–14633. [PubMed: 7782326]
- 167.
- Kono M, Yoshida Y, Kojima N, Tsuji S. Molecular cloning and expression of a fifth type of α2,8-sialyltransferse (ST8Sia V). Its substrate specificity is similar to that of SAT-V/III, which synthesize GD1c, GD1a, GQ1b and GT3. J. Biol. Chem. 1996;271:29366–29371. [PubMed: 8910600]
- 168.
- Rutishauser U. Polysialic acid and the regulation of cell interactions. Curr. Opin. Cell Biol. 1996;8:679–684. [PubMed: 8939657]
- 169.
- Bowman K G, Hemmerich S, Bhakta S, Singer M S, Bistrup A, Rosen S D, Bertozzi C R. Identification of an N-acetylglucosamine-6-O-sulfotransferase activity specific to lymphoid tissue: An enzyme with a possible role in lymphocyte homing. Chem. Biol. 1998;5:447–460. [PubMed: 9710564]
- 170.
- Rosen S D, Bertozzi C R. Leukocyte adhesion: Two selectins converge on sulphate. Curr. Biol. 1996;6:261–264. [PubMed: 8805242]
- 171.
- Uchimura K, Muramatsu H, Kaname T, Ogawa H, Yamakawa T, Fan Q W, Mitsuoka C, Kannagi R, Habuchi O, Yokoyama I, Yamamura K, Ozaki T, Nakagawara A, Kadomatsu K, Muramatsu T. Human N-acetylglucosamine-6-O-sulfotransferase involved in the biosynthesis of 6-sulfo sialyl Lewis X: Molecular cloning, chromosomal mapping, and expression in various organs and tumor cells. J. Biochem. 1998;124:670–678. [PubMed: 9722682]
- 172.
- Schachner M, Martini R. Glycans and the modulation of neural-recognition molecule function. Trends Neurosci. 1995;18:183–191. [PubMed: 7539963]
- 173.
- Voshol H, van Zuylen C W E M, Orberger G, Vliegenthart J F G, Schachner M. Structure of the HNK-1 carbohydrate epitope on bovine peripheral myelin glycoprotein P0. J. Biol. Chem. 1996;271:22957–22960. [PubMed: 8798480]
- 174.
- Bakker H, Friedmann I, Oka S, Kawasaki T, Nifant'ev N, Schachner M, Mantei N. Expression cloning of a cDNA encoding a sulfotransferase involved in the biosynthesis of the HNK-1 carbohydrate epitope. J. Biol. Chem. 1997;272:29942–29946. [PubMed: 9368071]
- 175.
- Yuen C -T, Chai W, Loveless R W, Lawson A M, Margolis R U, Feizi T. Brain contains HNK-1 immunoreactive O-glycans of the sulfoglucuronyl lactosamine series that terminate in 2-linked or 2,6-linked hexose (mannose). J. Biol. Chem. 1997;272:8924–8931. [PubMed: 9083013]
- 176.
- Terayama K, Oka S, Seiki T, Miki Y, Nakamura A, Kozutsumi Y, Takio K, Kawasaki T. Cloning and functional expression of a novel glucuronyltransferase involved in the biosynthesis of the carbohydrate epitope HNK-1. Proc. Natl. Acad. Sci. 1997;94:6093–6098. [PMC free article: PMC21007] [PubMed: 9177175]
- 177.
- Ong E, Yeh J -C, Ding Y, Hindsgaul O, Fukuda M. Expression cloning of a human sulfotransferase that directs the synthesis of the HNK-1 glycan on the neural cell adhesion molecule and glycolipids. J. Biol. Chem. 1998;273:5190–5195. [PubMed: 9478973]
- 178.
- Fukuta M, Inazawa J, Torii T, Tsuzuki K, Shimada E, Habuchi O. Molecular cloning and characterization of human keratan sulfate Gal-6-sulfotransferase. J. Biol. Chem. 1997;272:32321–32328. [PubMed: 9405439]
- 179.
- Sugumaran G, Katsman M, Drake R R. Purification, photoaffinity labeling, and characterization of a single enzyme for 6-sulfation of both chondroitin sulfate and keratan sulfate. J. Biol. Chem. 1995;270:22483–22487. [PubMed: 7673238]
- 180.
- Habuchi O, Hirahara Y, Uchimura K, Fukuta M. Enzymatic sulfation of galactose residue of keratan sulfate by chondroitin 6-sulfotransferase. Glycobiology. 1996;6:51–57. [PubMed: 8991509]
- 181.
- Chakravarti S, Magnuson T, Lass J H, Jepsen K J, LaMantia C, Carroll H. Lumican regulates collagen fibril assembly: Skin fragility and corneal opacity in the absence of lumican. J. Cell Biol. 1998;141:1277–1286. [PMC free article: PMC2137175] [PubMed: 9606218]
- 182.
- Lindahl B, Eriksson L, Spillmann D, Caterson B, Lindahl U. Selective loss of cerebral keratan sulfate in Alzheimer's disease. J. Biol. Chem. 1996;271:16991–16994. [PubMed: 8663590]
- Background
- Regulated Glycosylation of Constitutively Expressed Precursors to Terminal Chain Structures
- Type-2 Chains
- Type-1 Chains
- Polylactosamines
- β1–6GlcNAc Branching Structures
- The A, B, and H Blood Group Structures
- Lewis Blood Group Structures
- P Blood Group Structures
- The α1–3Gal Structure
- The Forssman Antigen
- Sulfated Terminal β-linked GalNAc Structures on Pituitary Glycoproteins
- Sialylated Terminal β-linked GalNAc Structures
- α2–3-sialylated Structures
- α2–6-sialylated Structures
- α2–8-sialylated Structures
- Sulfated Glycans: L-Selectin Ligands, HNK-1, and Keratan Sulfate
- Future Directions
- References
- Structures Common to Different Types of Glycans - Essentials of GlycobiologyStructures Common to Different Types of Glycans - Essentials of Glycobiology
Your browsing activity is empty.
Activity recording is turned off.
See more...