Summary
Clinical characteristics.
Virtually all individuals with Woodhouse-Sakati syndrome (WSS) have the endocrine findings of hypogonadism (evident at puberty) and progressive childhood-onset hair thinning that often progresses to alopecia totalis in adulthood. More than half of individuals have the neurologic findings of progressive extrapyramidal movements (dystonic spasms with dystonic posturing with dysarthria and dysphagia), moderate bilateral postlingual sensorineural hearing loss, and mild intellectual disability. To date, more than 40 families (including 33 with a molecularly confirmed diagnosis) with a total of 88 affected individuals have been reported in the literature.
Diagnosis/testing.
The diagnosis of WSS is established in a proband with suggestive clinical, neuroimaging, and neurophysiologic findings by identification of biallelic pathogenic variants in DCAF17 on molecular genetic testing.
Management.
Treatment of manifestations: Treatment is symptomatic and should be managed by a multidisciplinary team. Hypogonadism requires hormone replacement therapy to induce secondary sex characteristics and promote bone health at the usual age of puberty. Alopecia is treated symptomatically for cosmetic reasons only. Treatment for dystonia is routine; oral medications are tried first and followed in some instances by botulinum toxin injection and/or deep-brain stimulation. Dysarthria often benefits from consultation with a speech therapist. Those with dysphagia often require measures to reduce oral secretions, use of thickened liquids and pureed foods to avoid aspiration, and eventually a gastrostomy to help maintain caloric intake. Standard treatment for diabetes mellitus, hypothyroidism, hearing loss, and intellectual disability.
Surveillance: Monitoring for endocrine abnormalities is recommended at the following ages: hypogonadism beginning at age 12-14 years; diabetes mellitus and hypothyroidism beginning at age 20 years; serum IGF-1 every three to five years following diagnosis; annual neurologic assessment for dystonia; speech and language assessment for dysarthria and dysphagia as needed; annual developmental assessment throughout childhood; annual audiology evaluation.
Agents/circumstances to avoid: Persons with dystonia should avoid situations in which the risk of falling is increased.
Evaluation of relatives at risk: Molecular genetic testing for the DCAF17 pathogenic variants identified in the proband is appropriate for evaluation of apparently asymptomatic older and younger sibs to identify as early as possible those who would benefit from early identification and treatment of potential complications.
Genetic counseling.
WSS is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the pathogenic DCAF17 variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Diagnosis
No consensus clinical diagnostic criteria for Woodhouse-Sakati syndrome (WSS) have been published.
Suggestive Findings
WSS should be suspected in individuals with any combination of the following clinical, family history, neuroimaging, and neurophysiology findings.
Endocrine
- Hypogonadism (100% of individuals), hypogonadotropic in males and hypergonadotropic in females
- Primary amenorrhea in females
- Lack of development of secondary sexual characteristics in males and females
- Low insulin-like growth factor 1 (IGF-1) (100%)
- Adolescent- to young adult-onset diabetes mellitus (66%)
- Hypothyroidism (30%)
Alopecia. Hair loss beginning in childhood or adolescence, resulting in partial-to-complete loss of scalp hair and eyelashes (100%) (Figure 1A, 1H)

Figure 1.
Affected individuals from different families with WSS who have variable degrees of alopecia or hair loss and neurologic involvement A. Male age 23 with flat occiput; temporal and frontal alopecia
Neurologic
- Adolescent to young-adult onset of extrapyramidal findings including focal (later generalized) dystonia (65%), chorea, dysarthria, and dysphagia
- Sensorineural hearing loss (SNHL) with onset in childhood (62%)
- Intellectual disability (58%)
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Supportive Findings
Neuroimaging findings on brain MRI
- Partially empty sella and a small pituitary gland (Figure 2A) [Abusrair et al 2018]
- Progressive frontoparietal/periventricular white matter lesions (Figure 2B, 2C) [Abusrair et al 2018, Lehéricy et al 2020]
- Iron deposition in the globus pallidus (Figure 2D), and to a lesser extent, in the substantia nigra and red nucleus [Abusrair et al 2018, Lee et al 2020]
- Rarely, prominent perivascular spaces and diffusion restriction involving the splenium of the corpus callosum (Figure 2E, 2F) [Abusrair et al 2018]
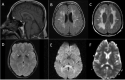
Figure 2.
Brain MRI of individuals with WSS. Arrows indicate the following findings: A. Sagittal T1-weighted image, showing small pituitary gland and partially empty sella.
Neurophysiology findings on evoked-potential (EP) analysis [Abusrair et al 2020]
- Prolonged P100 latencies on pattern reversal visual EPs
- Prolonged cortical N19 response on median somatosensory EPs
- Absent or prolonged P37 cortical response on tibial somatosensory EPs
Establishing the Diagnosis
The diagnosis of WSS is established in a proband with suggestive clinical, neuroimaging, and neurophysiologic findings by identification of biallelic pathogenic variants in DCAF17 on molecular genetic testing (see Table 1).
Molecular testing approaches can include gene-targeted testing (single-gene testing or multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of WSS has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of DCAF17 identifies biallelic pathogenic variants in all individuals with typical findings of WSS. DCAF17 exon and whole-gene deletion/duplication variants have not been reported. However, gene-targeted deletion/duplication analysis may be useful to confirm homozygosity of a pathogenic variant detected by sequence analysis when parental DNA is not available (see Molecular Genetics).
Note: Targeted analysis for individuals who are Saudi Arabian or Qatari may be performed first. To date, all affected individuals in these populations have been homozygous for the same founder pathogenic variant, c.436delC [Alazami et al 2008, Ben-Omran et al 2011].
A multigene panel that includes DCAF17 and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Woodhouse-Sakati Syndrome
Clinical Characteristics
Clinical Description
Woodhouse-Sakati syndrome (WSS) is characterized by the endocrine findings of hypogonadism, diabetes mellitus, and hypothyroidism and progressive childhood-onset alopecia along with neurologic findings of progressive extrapyramidal movements, sensorineural hearing loss, and intellectual disability. To date, 88 individuals from more than 40 families have been reported [Steindl et al 2010, Nanda et al 2014, Abdulla et al 2015, Bohlega et al 2019].
Two clinical types of WSS have been described [Bohlega et al 2019] with variable prognosis. However, intrafamilial variability is common and both types can occur within a family:
- Type 1. Severe and progressive neurologic disability at a younger age (range 9-17 years) causing significant impairment of the quality of life and activities of daily living. Manifestations include severe intellectual disability and dystonia.
- Type 2. Absent or mild neurologic involvements that do not affect activities of daily living
Endocrine
Hypogonadism, present in all affected individuals, manifests as delayed puberty with lack of secondary sexual characteristics. The nature of hypogonadism has been difficult to characterize as both hypergonadotropic and hypogonadotropic hypogonadism have been described [Agopiantz et al 2014]; in about 30% of affected individuals the hormonal profile does not neatly fit either group. Sense of smell is normal.
Women typically have primary amenorrhea. Detailed endocrine investigation in more than 50 of the women described in the literature typically revealed severely reduced or absent estradiol and high FSH and LH, consistent with hypergonadotropic hypogonadism. There appears to be decreased hypothalamic-pituitary responsiveness, as the FSH and LH are not as high as expected for the degree of ovarian failure [Woodhouse & Sakati 1983, Rachmiel et al 2011, Agopiantz et al 2014].
The ovaries are streak or underdeveloped, and not visualized by laparotomy, laparoscopy, or autopsy [Al-Semari & Bohlega 2007, Ben-Omran et al 2011, Rachmiel et al 2011]. Ovarian biopsy showed fibrous tissue with no identifiable oocysts [Woodhouse & Sakati 1983, Agopiantz et al 2014].
Men have moderately low testosterone and – in contrast to women – inappropriately low gonadotropins, consistent with hypogonadotropic hypogonadism, which may be of central or central and peripheral etiology. Semen analysis may show azoospermia [Agopiantz et al 2014, Ali et al 2016]. One male had cryptorchidism [Rachmiel et al 2011].
Although the testes are of normal size, testicular biopsy reveals reduced spermatogenesis with predominance of Sertoli cells and few Leydig cells [Agopiantz et al 2014].
Low insulin-like growth factor 1 (IGF-1) is present in all individuals [Ali et al 2016]. Reduction of IGF-1 is more pronounced in females [Al-Semari & Bohlega 2007, Ben-Omran et al 2011]. The low IGF-1 levels may reflect low sex steroids resulting from hypogonadism.
The growth pattern is normal and growth hormone levels are usually normal; short stature is not a part of this syndrome [Agopiantz et al 2014].
Diabetes mellitus. Type 2 diabetes (either insulin dependent or non-insulin dependent) was reported in 66% of all individuals and 96% of those older than age 25 years [Al-Semari & Bohlega 2007, Agopiantz et al 2014].
Hypothyroidism of peripheral origin (primary but without evidence of autoimmunity) was found in 30% of individuals, typically around age 20 years [Al-Semari & Bohlega 2007].
Other. No abnormalities of the corticotropic axis or prolactin [Al-Semari & Bohlega 2007, Agopiantz et al 2014] have been reported.
Ectodermal
Alopecia. All affected individuals have predominantly frontotemporal alopecia with sparse, thin scalp hair. Hair loss begins in childhood and often progresses to alopecia totalis in the third or fourth decade or earlier. Eyelashes and eyebrows are absent or sparse. In men, facial hair is absent or underdeveloped.
Scanning electron microscopy of the hair shows longitudinal grooves with no specific abnormalities [Al-Semari & Bohlega 2007].
Facial skin is often wrinkled in advanced stages, conferring a progeroid appearance [Woodhouse & Sakati 1983, Al-Semari & Bohlega 2007, Agopiantz et al 2014].
Anodontia. Total loss of teeth is rare; when it occurs, it is usually seen at a later stage [Al-Semari & Bohlega 2007, Agopiantz et al 2014].
Nails appear to be normal.
Neurologic
Extrapyramidal abnormal movement was seen in more than 56% of reported individuals. In particular, dystonic spasms with dystonic posturing were seen in the majority, including segmental dystonia affecting the craniocervical region, oromandibular region, or one extremity. Often, the first neurologic manifestation is abnormal posturing movements that typically start insidiously in childhood or the early teens. Dysarthria (often with a high-pitched voice) and dysphagia are common.
In a majority of individuals, dystonia becomes generalized and disabling (Figure 1B, 1F, 1H). Progressive dystonia of the trunk may lead to severe dystonic scoliosis. As the dystonia progresses, gait difficulties ultimately lead to immobility.
Inter- and intrafamilial variability is common [Al-Semari & Bohlega 2007, Ben-Omran et al 2011, Ali et al 2016 , Bohlega et al 2019]. For example, families with the founder DCAF17 pathogenic variant (c.436delC) can have dystonia [Al-Semari & Bohlega 2007] or not [Bohlega et al 2019]. Of note, although extrapyramidal features were not mentioned in the original report of Woodhouse and Sakati [1983], it was found that up to 65% of affected individuals develop variable degrees of dystonia at some point in the disease course (Figure 1H) [Bohlega et al 2019 ].
Sensorineural hearing loss (SNHL). Moderate bilateral SNHL was noted in 62% of reports. When present, deafness is invariably postlingual, usually starting in adolescence [Ben-Omran et al 2011].
Intellectual disability is described in 58% of individuals. It is typically mild and usually overshadowed by accompanying severe and disabling dystonia, dysarthria, and SNHL. The authors have observed ten individuals who were able to complete a college education and hold permanent manual occupations [Author, personal observation].
Other. Seizures with onset in early childhood, tremors, and mild Parkinsonism features have rarely been reported [Al-Semari & Bohlega 2007, Schneider & Bhatia 2008, Bohlega et al 2019].
Polyneuropathy with stocking glove sensory loss and diminished deep tendon reflexes but normal strength has been reported [Schneider & Bhatia 2008].
Other Findings
Dysmorphic facial features include a long triangular face, prominent nasal bridge, widely spaced eyes, and sparse eyebrows, creating a characteristic facial appearance (Figure 1D, 1E, 1G).
Bilateral keratoconus was reported in four individuals [Al-Swailem et al 2006, Schneider & Bhatia 2008, Ben-Omran et al 2011].
Electrocardiographic (EKG) abnormalities (lengthening of the ST segments and T-wave flattening) were described in the original report [Woodhouse & Sakati 1983] but rarely reported subsequently [Koshy et al 2008, Schneider & Bhatia 2008]. Of note, these EKG abnormalities were asymptomatic and individuals with WSS have no major cardiac manifestations.
Genotype-Phenotype Correlations
There is no clear genotype-phenotype correlation. Even individuals with the same Saudi Arabian founder DCAF17 pathogenic variant (c.436delC) have displayed marked phenotypic variability.
Prevalence
To date, more than 40 families with an estimated 88 affected individuals have been reported. Of these, 51 individuals from 33 families have had the diagnosis confirmed molecularly.
The carrier frequency of the Saudi Arabian founder variant (c.436delC) is 0.00243309 [Abouelhoda et al 2016].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in DCAF17.
Differential Diagnosis
Table 2.
Disorders/Phenotypes to Consider in the Differential Diagnosis of Woodhouse-Sakati Syndrome
Other disorders to consider in the differential diagnosis of Woodhouse-Sakati syndrome (WSS):
- Hereditary dystonia. Hereditary dystonia is associated with extensive clinical and genetic heterogeneity (see Hereditary Dystonia Overview). Unlike WSS, hypogonadism and alopecia are not known to be features associated with hereditary dystonia.
- Hereditary hearing loss and deafness. Nonsyndromic hereditary hearing loss is characterized by extreme genetic heterogeneity: to date, more than 6,000 causative variants have been identified in more than 110 genes. Syndromic hearing impairment is known to be associated with more than 400 genetic syndromes. (See Hereditary Hearing Loss and Deafness Overview.)
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Woodhouse-Sakati syndrome (WSS), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Woodhouse-Sakati Syndrome
Treatment of Manifestations
No specific treatment exists for WSS. Treatment is aimed at relieving symptoms [Albanese et al 2015].
Table 4.
Treatment of Manifestations in Individuals with Woodhouse-Sakati Syndrome
Surveillance
Table 5.
Recommended Surveillance for Individuals with Woodhouse-Sakati Syndrome
Agents/Circumstances to Avoid
Persons with dystonia should avoid situations in which the risk of falling is increased.
Evaluation of Relatives at Risk
Molecular genetic testing for known familial DCAF17 pathogenic variants is appropriate for the evaluation of apparently asymptomatic older and younger sibs of a proband in order to identify as early as possible those who will benefit from early identification and treatment of potential complications.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Woodhouse-Sakati syndrome (WSS) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected individual are obligate heterozygotes (i.e., presumed to be carriers of one DCAF17 pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a DCAF17 pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for a DCAF17 pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting biallelic DCAF17 pathogenic variants and being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- The neurologic phenotype in a sib who inherits biallelic pathogenic variants cannot be predicted based on the phenotype in the proband; an affected sib may have a neurologic phenotype ranging from normal to severe regardless of the neurologic features observed in the proband [Bohlega et al 2019].
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Unless an individual with WSS has children with an affected individual or a carrier, his/her offspring will be obligate heterozygotes (carriers) for a pathogenic variant in DCAF17.
Other family members. Each sib of the proband’s parents is at a 50% risk of being a carrier of a DCAF17 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the DCAF17 pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the DCAF17 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for WSS are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- NBIA Disorders Association
- Dystonia Medical Research FoundationPhone: 312-755-0198; 800-377-DYST (3978)Email: dystonia@dystonia-foundation.org
- Dystonia UKUnited KingdomEmail: info@dystonia.org.uk
- NBIAcureCenter of Excellence for NBIA Clinical Care and ResearchInternational Registry for NBIA and Related DisordersOregon Health & Science UniversityEmail: info@nbiacure.org
- Treat Iron-Related Childhood Onset Neurodegeneration (TIRCON)GermanyEmail: TIRCON@med.uni-muenchen.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Woodhouse-Sakati Syndrome: Genes and Databases
Table B.
OMIM Entries for Woodhouse-Sakati Syndrome (View All in OMIM)
Molecular Pathogenesis
Alternate splicing of DCAF17 results in multiple transcripts of variable length. The longest transcript, NM_025000.4, has 14 exons. In comparison another major transcript, NM_001164821.1, lacks two exons in the coding region for a total of 12 exons. Transcript NM_025000.4 encodes DDB1- and CUL4-associated factor 17, a 520 amino-acid protein known as the α isoform (NP_079276.2). This is a nucleolar protein of unknown function expressed in various tissues including brain, skin, and liver – which correlates to some extent with the multiorgan involvement in individuals with Woodhouse-Sakati syndrome (WSS).
Reported DCAF17 pathogenic variants include small intragenic deletions, nonsense and splice site variants, and small indels [Alazami et al 2010, Habib et al 2011, Abdulla et al 2015, Ali et al 2016]. Pathogenic DCAF17 missense variants have not been described.
Of the 33 families with molecularly confirmed WSS, biallelic compound heterozygous DCAF17 pathogenic variants has been reported in only one [Ali et al 2016]. The remaining families were homozygous for DCAF17 pathogenic variants. Gene-targeted deletion/duplication analysis may be useful to confirm apparent homozygosity of a pathogenic variant detected by sequence analysis when parental DNA samples are not available.
Mechanism of disease causation. The types of pathogenic variants known to result in WSS predict premature translation termination, missplicing, or nonsense-mediated decay, suggesting that WSS results from loss of DCAF17 function.
Table 6.
Notable DCAF17 Pathogenic Variants
Chapter Notes
Author History
Ali Abusrair, MD (2021-present)
Fowzan S Alkuraya, MD (Hons), ABP, ABMGG; King Faisal Specialist Hospital and Research Center (2016-2021)
Saeed A Bohlega, MD, FRCPC, FAAN (2016-present)
Revision History
- 8 July 2021 (sw) Comprehensive update posted live
- 4 August 2016 (bp) Review posted live
- 2 February 2016 (sab) Original submission
References
Literature Cited
- Abdulla MC, Alazami AM, Alungal J, Koya JM, Musambil M. Novel compound heterozygous frameshift mutations of C2orf37 in a familial Indian case of Woodhouse-Sakati syndrome. J Genet. 2015;94:489–92. [PubMed: 26440089]
- Abouelhoda M, Sobahy T, El-Kalioby M, Patel N, Shamseldin H, Monies D, Al-Tassan N, Ramzan K, Imtiaz F, Shaheen R, Alkuraya FS. Clinical genomics can facilitate countrywide estimation of autosomal recessive disease burden. Genet Med. 2016;18:1244–9. [PubMed: 27124789]
- Abusrair A, AlHamoud I, Bohlega S. Multimodal evoked potential profiles in Woodhouse-Sakati syndrome. J Clin Neurophysiol. 2020. Epub ahead of print. [PubMed: 33417382]
- Abusrair AH, Bohlega S, Al-Semari A, Al-Ajlan FS, Al-Ahmadi K, Mohamed B, AlDakheel A. Brain MR imaging findings in Woodhouse-Sakati syndrome. AJNR Am J Neuroradiol. 2018;39:2256–62. [PMC free article: PMC7655421] [PubMed: 30409855]
- Agopiantz M, Corbonnois P, Sorlin A, Bonnet C, Klein M, Hubert N, Pascal-Vigneron V, Jonveaux P, Cuny T, Leheup B, Weryha G. Endocrine disorders in Woodhouse-Sakati syndrome: a systematic review of the literature. J Endocrinol Invest. 2014;37:1–7. [PubMed: 24464444]
- Alazami AM, Al-Saif A, Al-Semari A, Bohlega S, Zlitni S, Alzahrani F, Bavi P, Kaya N, Colak D, Khalak H, Baltus A, Peterlin B, Danda S, Bhatia KP, Schneider SA, Sakati N, Walsh CA, Al-Mohanna F, Meyer B, Alkuraya FS. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am J Hum Genet. 2008;83:684–91. [PMC free article: PMC2668059] [PubMed: 19026396]
- Alazami AM, Schneider SA, Bonneau D, Pasquier L, Carecchio M, Kojovic M, Steindl K, de Kerdanet M, Nezarati MM, Bhatia KP, Degos B, Goh E, Alkuraya FS. C2orf37 mutational spectrum in Woodhouse-Sakati syndrome patients. Clin Genet. 2010;78:585–90. [PubMed: 20507343]
- Albanese A, Romito LM, Calandrella D. Therapeutic advances in dystonia. Mov Disord. 2015;30:1547–56. [PubMed: 26301801]
- Ali RH, Shah K, Nasir A, Steyaert W, Coucke PJ, Ahmad W. Exome sequencing revealed a novel biallelic deletion in the DCAF17 gene underlying Woodhouse Sakati syndrome (WSS). Clin Genet. 2016;90:263–9. [PubMed: 26612766]
- Al-Semari A, Bohlega S. Autosomal-recessive syndrome with alopecia, hypogonadism, progressive extra-pyramidal disorder, white matter disease, sensory neural deafness, diabetes mellitus, and low IGF1. Am J Med Genet A. 2007;143A:149–60. [PubMed: 17167799]
- Al-Swailem SA, Al-Assiri AA, Al-Torbak AA. Woodhouse Sakati syndrome associated with bilateral keratoconus. Br J Ophthalmol. 2006;90:116–7. [PMC free article: PMC1856898] [PubMed: 16361682]
- Ben-Omran T, Ali R, Almureikhi M, Alameer S, Al-Saffar M, Walsh CA, Felie JM, Teebi A. Phenotypic heterogeneity in Woodhouse-Sakati syndrome: two new families with a mutation in the C2orf37 gene. Am J Med Genet A. 2011;155A:2647–53. [PMC free article: PMC6905109] [PubMed: 21964978]
- Bohlega S, Abusrair AH, Al-Ajlan FS, Alharbi N, Al-Semari A, Bohlega B, Abualsaud D, Alkuraya F. Patterns of neurological manifestations in Woodhouse-Sakati syndrome. Parkinsonism Relat Disord. 2019 Dec;69:99–103. [PubMed: 31726291]
- Crowell JL, Shah BB. Surgery for dystonia and tremor. Curr Neurol Neurosci Rep. 2016;16:22. [PubMed: 26838349]
- Habib R, Basit S, Khan S, Khan MN, Ahmad W. A novel splice site mutation in gene C2orf37 underlying Woodhouse-Sakati syndrome (WSS) in a consanguineous family of Pakistani origin. Gene. 2011;490:26–31. [PubMed: 21963443]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Koshy G, Danda S, Thomas N, Mathews V, Viswanathan V. Three siblings with Woodhouse-Sakati syndrome in an Indian family. Clin Dysmorphol. 2008;17:57–60. [PubMed: 18049083]
- Lee JH, Yun JY, Gregory A, Hogarth P, Hayflick SJ. Brain MRI pattern recognition in neurodegeneration with brain iron accumulation. Front Neurol. 2020;11:1024. [PMC free article: PMC7511538] [PubMed: 33013674]
- Lehéricy S, Roze E, Goizet C, Mochel F. MRI of neurodegeneration with brain iron accumulation. Curr Opin Neurol. 2020;33:462–73. [PubMed: 32657887]
- Nanda A, Pasternack SM, Mahmoudi H, Ishorst N, Grimalt R, Betz RC. Alopecia and hypotrichosis as characteristic findings in Woodhouse-Sakati syndrome: report of a family with mutation in the C2orf37 gene. Pediatr Dermatol. 2014;31:83–7. [PubMed: 24015686]
- Rachmiel M, Bistritzer T, Hershkoviz E, Khahil A, Epstein O, Parvari R. Woodhouse-Sakati syndrome in an Israeli-Arab family presenting with youth-onset diabetes mellitus and delayed puberty. Horm Res Paediatr. 2011;75:362–6. [PubMed: 21304230]
- Schneider SA, Bhatia KP. Dystonia in the Woodhouse Sakati syndrome: A new family and literature review. Mov Disord. 2008;23:592–6. [PubMed: 18175354]
- Steindl K, Alazami AM, Bhatia KP, Wuerfel JT, Petersen D, Cartolari R, Neri G, Klein C, Mongiardo B, Alkuraya FS, Schneider SA. A novel C2orf37 mutation causes the first Italian cases of Woodhouse Sakati syndrome. Clin Genet. 2010;78:594–7. [PubMed: 21044051]
- Woodhouse NJ, Sakati NA. A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. J Med Genet. 1983;20:216–9. [PMC free article: PMC1049050] [PubMed: 6876115]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: August 4, 2016; Last Update: July 8, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Bohlega SA, Abusrair A. Woodhouse-Sakati Syndrome. 2016 Aug 4 [Updated 2021 Jul 8]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

