TUBB4A-Related Leukodystrophy
Norah Nahhas, MD, Alex Conant, BS, Eline Hamilton, MD, Julian Curiel, BS, Cas Simons, PhD, Marjo van der Knaap, MD, PhD, and Adeline Vanderver, MD.
Author Information and AffiliationsInitial Posting: November 3, 2016.
Estimated reading time: 17 minutes
Summary
Clinical characteristics.
TUBB4A-related leukodystrophy comprises a phenotypic spectrum in which the MRI findings range from hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) at the severe end to isolated hypomyelination at the mild end. Progressive neurologic findings reflect involvement of the pyramidal tracts (spasticity, brisk deep tendon reflexes, and Babinski sign), extrapyramidal system (rigidity, dystonia, choreoathetosis, oculogyric crisis, and perioral dyskinesia), cerebellum (ataxia, intention tremor, dysmetria), and bulbar function (dysarthria, dysphonia, and swallowing). Cognition is variably affected, usually less severely than motor function. Typically, those with H-ABC present in early childhood (ages 1-3 years) and those with isolated hypomyelination in later childhood or adulthood. The rate of progression varies with disease severity.
Management.
Treatment of manifestations: Functionally disabling spasticity requires medical management and physical therapy; dystonia requires medical management and – when refractory to medical management – possibly surgical intervention. Swallowing dysfunction may require use of a gastrostomy tube for feeding. Seizures, constipation, and gastroesophageal reflux disease are treated in the routine manner.
Prevention of secondary complications: Calcium and vitamin D supplementation as required to prevent osteoporosis; attention to skin care and frequent repositioning to help prevent pressure sores in individuals with decreased mobility; annual flu vaccination; use of routine fall prevention strategies, adaptive equipment (e.g., wheelchairs and walkers), and physical therapy to help prevent secondary injury.
Surveillance: Routine evaluations of (1) swallowing and feeding to reduce the risk of aspiration; (2) nutrition to prevent malnutrition; (3) orthopedic and joint integrity to detect joint dislocation and scoliosis. At least yearly: (1) medical evaluations to assess weight and medications; (2) evaluations by specialists in occupational therapy, physical therapy, speech therapy, rehabilitation medicine. Annual neurologic assessment to detect emerging complications.
Genetic counseling.
TUBB4A-related leukodystrophy is inherited in an autosomal dominant manner. Most affected individuals have the disorder as the result of a de novo pathogenic variant. The risk to sibs of a proband with clinically unaffected parents is low but greater than that of the general population because of the possibility of germline mosaicism or somatic and germline mosaicism in a parent. Individuals with TUBB4A-related leukodystrophy are not known to reproduce.
Diagnosis
Suggestive Findings
TUBB4A-related leukodystrophy should be suspected in individuals with the following clinical and brain MRI findings that define the two hypomyelination phenotypes.
Clinical findings
Onset during infancy or childhood
Motor developmental delay
Presence of pyramidal and extrapyramidal signs
Gait ataxia and cerebellar dysfunction
Dysarthria, aphonia, or "whispering" dysphonia
Brain MRI findings
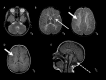
Progressive atrophy of the basal ganglia involving the neostriatum (i.e., the putamen and caudate nucleus) predominantly, often with a significant decrease in size of the putamen (which can disappear over time) and to a lesser degree the head of the caudate. The thalamus and globus pallidus are typically spared. Note that although changes in the putamen are evident in many children with the H-ABC
phenotype by age two years, in some children the changes may not be evident until later childhood.
Diffuse cerebral hypomyelination manifest as mild T2-weighted hyperintensity involving the supratentorial white matter, corpus callosum, and internal capsule, and typically isointense or mildly hyperintense T1-weighted signal
Cerebellar findings of white matter T1-weighted signal that is isointense or mildly hyper- or hypointense relative to gray matter structures. Cerebellar atrophy prominently affecting the vermis is a common but not obligatory feature of H-ABC.
MRI findings A. Cerebellar white matter T1-weighted signal that is isointense or mildly hyper- or hypointense relative to gray matter structures; cerebellar atrophy prominently affects the vermis. There are no specific signal changes to the brain stem (more...)
Establishing the Diagnosis
The diagnosis of a TUBB4A-related leukodystrophy is established in a proband with characteristic clinical and MRI findings and a heterozygous TUBB4A pathogenic variant identified by molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel or single-gene testing) and genomic testing (comprehensive genomic sequencing) depending on the phenotype.
Gene-targeted testing requires the clinician to determine which gene(s) are likely involved, whereas genomic testing may not. Because the phenotype of TUBB4A-related leukodystrophy is broad, children with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a mild phenotype indistinguishable from many other inherited hypomyelinating leukodystrophies are more likely to be diagnosed using genomic testing (see Option 2).
Option 1. Gene-Targeted Testing
When the phenotypic findings, such as hypomyelination with basal ganglia atrophy, suggest the diagnosis of TUBB4A-related leukodystrophy, molecular genetic testing approaches can include the following:
To consider: multigene panel. A multigene leukodystrophy panel that includes
TUBB4A and other genes of interest (see
Differential Diagnosis) may be considered; however, the diagnostic
sensitivity of a multigene panel may be low in this instance because primary neuronal disorders with MRI findings that resemble a classic leukodystrophy (like
TUBB4A-related leukodystrophy) are often not included in leukodystrophy panels.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Option 2. Genomic Testing
When the phenotype is indistinguishable from many other inherited disorders with leukodystrophy or with atypical white matter changes on MRI, comprehensive genomic testing, which does not require the clinician to determine which gene(s) are likely involved, may be considered. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in TUBB4A-Related Leukodystrophy
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
TUBB4A
| Sequence analysis 3 | 71/71 affected persons |
| Gene-targeted duplication/deletion analysis 4 | None reported |
- 1.
- 2.
- 3.
- 4.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
Clinical Characteristics
Clinical Description
TUBB4A-related leukodystrophy typically presents between ages one and three years; onset range is a few months of age in more severe forms [van der Knaap et al 2002] to later childhood or adulthood in some instances of isolated hypomyelination [Hamilton et al 2014, Pizzino et al 2014, Shimojima et al 2015].
The disorder is progressive; rate of progression varies with disease severity. Males and females are similarly affected.
Manifestations can include the following.
Delayed motor development. Some children have a period of normal motor development with subsequent deterioration [van der Knaap et al 2002].
Cognitive dysfunction. Cognition is variably affected but usually less severely than motor function. Learning difficulty is common; social awareness is usually preserved.
Pyramidal involvement. Bilateral or unilateral upper-motor neuron dysfunction (spasticity, brisk deep tendon reflexes, and Babinski signs) typically manifests in early childhood [Mercimek-Mahmutoglu et al 2005, Wakusawa et al 2006].
Of note, some individuals with a heterozygous TUBB4A pathogenic variant and a spastic paraplegia have been described as having mild white matter changes on brain MRI [Kancheva et al 2015], thus expanding the spectrum of TUBB4A-related leukodystrophy. To date, however, molecular genetic testing of individuals with as-yet unclassified hereditary spastic paraplegia has not commonly identified a causative heterozygous TUBB4A pathogenic variant [Kumar et al 2015].
Extrapyramidal involvement due to neostriatal involvement includes rigidity, dystonia, choreoathetosis, oculogyric crisis, and perioral dyskinesia. Extrapyramidal features – in particular hemidystonia – can be the first manifestation of this condition. Extrapyramidal features can be exacerbated by changes in body position or by visual and acoustic stimuli.
Gait dysfunction. Although some individuals achieve independent ambulation, many never do. Gait instability and falls are common.
Cerebellar signs can include ataxia, intention tremor, dysmetria, and nystagmus.
Dysarthria, dysphonia, and swallowing dysfunction. Communication and feeding difficulties emerge over time, necessitating a gastrostomy tube for feeding in many individuals.
Other less common findings (seen in severe cases) can include the following [van der Knaap et al 2002, Sasaki et al 2009, Simons et al 2013, Ferreira et al 2014, Hamilton et al 2014]:
Individuals mosaic for a TUBB4A pathogenic variant. Of note, a few parents of individuals with TUBB4A-related hypomyelination have been asymptomatic and mosaic for a TUBB4A pathogenic variant [Simons et al 2013].
Neurophysiologic studies
Electroencephalogram is usually normal or demonstrates slow background activity.
Electromyogram and nerve conduction studies are normal.
Brain stem evoked potentials are usually delayed.
Visual evoked potentials are usually normal.
Laboratory findings. Cerebrospinal fluid (CSF) analysis is typically normal [van der Knaap et al 2002]. Of note, in some individuals a low level of CSF 5-methyltetrahydrofolic acid was observed with normal plasma folate levels and normal CSF 5-MTHF reductase or decreased CSF homovanillic acid [Tomás-Vila et al 2014, Tonduti et al 2016]; this has not been broadly described [Mercimek-Mahmutoglu & Stockler-Ipsiroglu 2007] and thus is not thought to be a primary metabolic defect.
Genotype-Phenotype Correlations
Four TUBB4A pathogenic variants are consistently associated with specific phenotypes:
Prevalence
The exact prevalence is unknown; 71 affected individuals have been reported to date.
Differential Diagnosis
Hypomyelinating leukodystrophies with early childhood onset and/or extrapyramidal signs should be considered in the differential diagnosis.
Pelizaeus-Merzbacher disease
(PMD) is an X-linked disorder caused by a PLP1 intragenic pathogenic variant or a large PLP1 deletion/duplication. It typically presents during infancy or early childhood with a combination of nystagmus, upper motor neuron dysfunction, gait ataxia, and extrapyramidal signs. Brain MRI shows diffuse hypomyelination but lacks the classic atrophy of the cerebellum and basal ganglia of a TUBB4A-related leukodystrophy, hypomyelination with atrophy of basal ganglia and cerebellum (H-ABC).
Pelizaeus-Merzbacher-like disease 1
(PMLD1) is an autosomal recessive disorder caused by biallelic GJC2 pathogenic variants. PMLD1 usually presents during early childhood with manifestations similar to those of the H-ABC phenotype including: developmental delay, speech delay, pyramidal and extrapyramidal involvement, cerebellar signs, and preservation of mental functions. Much like those with PMD, affected children manifest nystagmus early in the disease course [Uhlenberg et al 2004], which – although it is described in TUBB4A-related leukodystrophy – is not typical.
Pol III-related leukodystrophies are autosomal recessive disorders caused by biallelic pathogenic variants in one of three genes (POLR3A, POLR3B, or POLR1C). Manifestations include spasticity, gait ataxia, extrapyramidal movement disorders, and cerebellar signs, similar to those of TUBB4A-related leukodystrophy. Other manifestations of Pol III-related leukodystrophies include abnormal dentition and hypogonadotropic hypogonadism, which are not commonly associated with TUBB4A-related leukodystrophy.
SOX 10-associated leukodystrophy/peripheral and central demyelination, Waardenburg syndrome, and Hirschsprung disease (PCWH) (OMIM 609136) is an autosomal dominant disorder caused by heterozygous pathogenic variants in SOX10. Manifestations overlap with TUBB4A-related leukodystrophy, including developmental delay, spasticity, gait ataxia, and extrapyramidal movement disorders. Additional manifestations of PCWH include:
Free sialic acid storage disorders are autosomal recessive neurodegenerative disorders caused by biallelic pathogenic variants in SLC17A5 that result in defective sialic acid storage and transport. Whereas the most severe form (infantile free sialic acid storage disease) includes coarse facial features and non-neurologic manifestations such as hepatosplenomegaly and cardiomegaly, the milder form (Salla disease) is similar to H-ABC and includes progressive neurologic deterioration with spasticity, extrapyramidal movement disorders, and seizures.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with a TUBB4A-related leukodystrophy, the following evaluations are recommended [Van Haren et al 2015]:
Evaluation by a pediatric neurologist for evidence of developmental delay, spasticity, and extrapyramidal movement disorders
Assessment of developmental milestones and cognitive function
Assessment of functional disability and equipment needs by a physiotherapist
Assessment of speech (communication) and feeding (swallowing)
Audiologic assessment
Orthopedic evaluation for evidence of scoliosis and/or joint deformity, particularly in individuals with significant dystonia
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Although there is no curative treatment for TUBB4A-related leukodystrophies, quality of life can be improved in the following ways.
Spasticity that is functionally disabling can lead to joint contractures and scoliosis; both require physical therapy (stretching and positioning) and medical management. Oral GABA agonists such as baclofen and diazepam can be used. In some instances intrathecal baclofen pumps can be considered. For focal spasticity, intramuscular botulinum toxin may be helpful.
Dystonia can be managed with:
When dystonia is refractory to medical management, a baclofen pump may be considered. Of note, to date deep brain stimulation has not been studied in TUBB4A-related leukodystrophy.
Swallowing dysfunction may result in use of a gastrostomy tube for feeding to reduce the risk of aspiration.
Dysarthria may warrant augmentative communication tools.
Anticonvulsant medications should be used when seizures are present.
Constipation, commonly due to neurologic dysfunction and poor intestinal motility, can be treated with diet, laxatives, and stool softeners.
Gastroesophageal reflux disease is common and should be considered in the evaluation of pain.
Functional ability can be improved by use of walkers or wheeled mobility devices and other necessary equipment.
Accommodations in school such as an individual educational plan are often needed. With such accommodations many children with the classic H-ABC
phenotype perform at or near grade level for many years, although cognitive decline may be seen later.
Family support and advocacy groups can provide needed psychosocial support for affected individuals.
Prevention of Secondary Complications
The following recommendations – based on consensus – have been developed for all leukodystrophies [Van Haren et al 2015].
Calcium and vitamin D supplementation as required to prevent osteoporosis
Skin care and frequent repositioning to help prevent pressure sores in individuals with decreased mobility
Annual flu vaccination
Fall prevention strategies, adaptive equipment (e.g., wheelchairs and walkers), and physical therapy (to increase strength) to help prevent secondary injury
Surveillance
The following are appropriate:
Routine evaluations of swallowing and feeding to reduce the risk of aspiration, and nutrition to prevent malnutrition
At least yearly:
Medical evaluation including physical examination to assess weight and medications
Evaluations by specialists in occupational therapy, physical therapy, speech therapy, and rehabilitation medicine
Evaluation by orthopedists to assess for scoliosis and joint dislocation
Annual neurologic evaluation to assess symptoms and any emerging complications
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
TUBB4A-related leukodystrophy is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Sibs of a proband
The risk to the sibs of the
proband depends on the genetic status of the proband's parents.
Because
TUBB4A-related leukodystrophy has occurred as the result of a
de novo pathogenic variant in most cases reported to date, the risk to the sibs of a
proband is presumed to be low but greater than that of the general population because of the possibility of
germline mosaicism or somatic and germline mosaicism in a parent.
Offspring of a proband. Each child of an individual with TUBB4A-related leukodystrophy would have a 50% chance of inheriting the TUBB4A pathogenic variant; however, individuals with TUBB4A-related leukodystrophy are not known to reproduce.
Other family members. Given that most probands with TUBB4A-related leukodystrophy reported to date have the disorder as a result of a de novo TUBB4A pathogenic variant, the risk to other family members is presumed to be low.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
European Leukodystrophy Association (ELA)
Phone: 03 83 30 93 34
Leukodystrophy Australia
Australia
Phone: 1800-141-400
Email: info@leuko.org.au
Medical Home Portal
United Leukodystrophy Foundation
Phone: 800-SAV-LIVE; 815-748-3211
Email: office@ulf.org
Myelin Disorders Bioregistry Project
Phone: 215-590-1719
Email: sherbinio@chop.edu
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
TUBB4A-Related Leukodystrophy: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Gene structure. Alternate splicing results in multiple transcript variants that encode different isoforms (see Table A, Gene for details). The transcript variant NM_006087.3 is 2583 bp and comprises four exons [Lee et al 1984].
Pathogenic variants. See Table 2 and Table 3 (pdf) for a listing of pathogenic variants.
Variant c.745G>A is the most common TUBB4A pathogenic variant; it is consistently associated with the classic phenotype of hypomyelination with atrophy of basal ganglia and cerebellum (H-ABC).
Variant c.4C>G is not associated with a leukodystrophy but with DYT4 ("hereditary whispering dysphonia") [Hersheson et al 2013].
Table 2.
TUBB4A Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.4C>G 1 | p.Arg2Gly |
NM_006087.3
NP_006078.2
|
| c.730G>A 2, 3 | p.Gly244Ser |
| c.745G>A 2, 3, 4, 5 | p.Asp249Asn |
| c.785G>A 2, 3, 4 | p.Arg262His |
| c.1228G>A 2, 4 | p.Glu410Lys |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
- 2.
- 3.
- 4.
- 5.
To view information on additional variants, see Table 3 (pdf).
Normal gene product. The transcript variant NM_006087.3 encodes a β-tubulin protein isoform of 444 amino acids. β-tubulin proteins can bind to α-tubulins to form heterodimers, which form copolymers that assemble into microtubules. Microtubules are an essential component of the cytoskeleton and play important roles in many cellular processes such as mitosis, motility, and transport.
TUBB4A is predominantly expressed in the CNS, especially in the cerebellum and putamen and white matter [Nogales 2001, Hersheson et al 2013].
Abnormal gene product. Pathogenic variants in TUBB4A result in changes in β-tubulin structure that are thought to affect microtubule polymerization or stability [Savage et al 1994].
References
Literature Cited
Blumkin L, Halevy A, Ben-Ami-Raichman D, Dahari D, Haviv A, Sarit C, Lev D, van der Knaap MS, Lerman-Sagie T, Leshinsky-Silver E. Expansion of the spectrum of TUBB4A-related disorders: a new phenotype associated with a novel mutation in the TUBB4A gene.
Neurogenetics. 2014;15:107–13. [
PubMed: 24526230]
Bondurand N, Dastot-Le Moal F, Stanchina L, Collot N, Baral V, Marlin S, Attie-Bitach T, Giurgea I, Skopinski L, Reardon W, Toutain A, Sarda P, Echaieb A, Lackmy-Port-Lis M, Touraine R, Amiel J, Goossens M, Pingault V. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4.
Am J Hum Genet. 2007;81:1169–85. [
PMC free article: PMC2276340] [
PubMed: 17999358]
Carvalho D, Santos S, Martins B, Marques FP. TUBB4A novel mutation reinforces the genotype-phenotype correlation of hypomyelination with atrophy of the basal ganglia and cerebellum.
Brain. 2015;138:e327. [
PubMed: 25168210]
Erro R, Hersheson J, Ganos C, Mencacci NE, Stamelou M, Batla A, Thust SC, Bras JM, Guerreiro RJ, Hardy J, Quinn NP, Houlden H, Bhatia KP. H-ABC syndrome and DYT4: Variable expressivity or pleiotropy of TUBB4 mutations?
Mov Disord. 2015a;30:828–33. [
PubMed: 25545912]
Erro R, Hersheson J, Houlden H, Bhatia KP. A novel TUBB4A mutation suggests that genotype-phenotype correlation of H-ABC syndrome needs to be revisited.
Brain. 2015b;138:e370. [
PubMed: 25614026]
Ferreira C, Poretti A, Cohen J, Hamosh A, Naidu S. Novel TUBB4A mutations and expansion of the neuroimaging phenotype of hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC).
Am J Med Genet A. 2014;164A:1802–7. [
PMC free article: PMC10506160] [
PubMed: 24706558]
Hamilton EM, Polder E, Vanderver A, Naidu S, Schiffmann R, Fisher K, Raguž AB, Blumkin L. H-ABC Research Group, van Berkel CG, Waisfisz Q, Simons C, Taft RJ, Abbink TE, Wolf NI, van der Knaap MS. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation.
Brain. 2014;137:1921–30. [
PMC free article: PMC4345790] [
PubMed: 24785942]
Hamilton EM, Wolf NI, van der Knaap MS. Reply: A novel TUBB4A mutation suggests that genotype-phenotype correlation of H-ABC syndrome needs to be revisited.
Brain. 2015;138:e371. [
PubMed: 25619510]
Hersheson J, Mencacci NE, Davis M, MacDonald N, Trabzuni D, Ryten M, Pittman A, Paudel R, Kara E, Fawcett K, Plagnol V, Bhatia KP, Medlar AJ, Stanescu HC, Hardy J, Kleta R, Wood NW, Houlden H. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia.
Ann Neurol. 2013;73:546–53. [
PMC free article: PMC3698699] [
PubMed: 23424103]
Inoue K, Shilo K, Boerkoel CF, Crowe C, Sawady J, Lupski JR, Agamanolis DP. Congenital hypomyelinating neuropathy, central dysmyelination, and Waardenburg-Hirschsprung disease: phenotypes linked by SOX10 mutation.
Ann Neurol. 2002;52:836–42. [
PubMed: 12447940]
Kancheva D, Chamova T, Guergueltcheva V, Mitev V, Azmanov DN, Kalaydjieva L, Tournev I, Jordanova A. Mosaic dominant TUBB4A mutation in an inbred family with complicated hereditary spastic paraplegia.
Mov Disord. 2015;30:854–8. [
PubMed: 25772097]
Kumar KR, Vulinovic F, Lohmann K, Park JS, Schaake S, Sue CM, Klein C. Mutations in TUBB4A and spastic paraplegia.
Mov Disord. 2015;30:1857–8. [
PubMed: 26477786]
Mercimek-Mahmutoglu S, Stockler-Ipsiroglu S. Cerebral folate deficiency and folinic acid treatment in hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) syndrome.
Tohoku J Exp Med. 2007;211:95–6. [
PubMed: 17202777]
Mercimek-Mahmutoglu S, van der Knaap MS, Baric I, Prayer D, Stoeckler-Ipsiroglu S. Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC). Report of a new case.
Neuropediatrics. 2005;36:223–6. [
PubMed: 15944912]
Miyatake S, Osaka H, Shiina M, Sasaki M, Takanashi J, Haginoya K, Wada T, Morimoto M, Ando N, Ikuta Y, Nakashima M, Tsurusaki Y, Miyake N, Ogata K, Matsumoto N, Saitsu H. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies.
Neurology. 2014;82:2230–7. [
PubMed: 24850488]
Nogales E. Structural insight into microtubule function.
Annu Rev Biophys Biomol Struct. 2001;30:397–420. [
PubMed: 11441808]
Pizzino A, Pierson TM, Guo Y, Helman G, Fortini S, Guerrero K, Saitta S, Murphy JL, Padiath Q, Xie Y, Hakonarson H, Xu X, Funari T, Fox M, Taft RJ, van der Knaap MS, Bernard G, Schiffmann R, Simons C, Vanderver A. TUBB4A de novo mutations cause isolated hypomyelination.
Neurology. 2014;83:898–902. [
PMC free article: PMC4153852] [
PubMed: 25085639]
Purnell SM, Bleyl SB, Bonkowsky JL. Clinical exome sequencing identifies a novel TUBB4A mutation in a child with static hypomyelinating leukodystrophy.
Pediatr Neurol. 2014;50:608–11. [
PMC free article: PMC4029864] [
PubMed: 24742798]
Sagnelli A, Magri S, Farina L, Chiapparini L, Marotta G, Tonduti D, Consonni M, Scigliuolo GM, Benti R, Pareyson D, Taroni F, Salsano E, Di Bella D. Early-onset progressive spastic paraplegia caused by a novel TUBB4A mutation: brain MRI and FDG-PET findings.
J Neurol. 2016;263:591–3. [
PubMed: 26810722]
Sasaki M, Takanashi J, Tada H, Sakuma H, Furushima W, Sato N. Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum.
Brain Dev. 2009;31:582–7. [
PubMed: 18851904]
Savage C, Xue Y, Mitani S, Hall D, Zakhary R, Chalfie M. Mutations in the Caenorhabditis elegans beta-tubulin gene mec-7: effects on microtubule assembly and stability and on tubulin autoregulation.
J Cell Sci. 1994;107:2165–75. [
PubMed: 7983175]
Shimojima K, Okumura A, Ikeno M, Nishimura A, Saito A, Saitsu H, Matsumoto N, Yamamoto T. A de novo TUBB4A mutation in a patient with hypomyelination mimicking Pelizaeus-Merzbacher disease.
Brain Dev. 2015;37:281–5. [
PubMed: 24974158]
Simons C, Wolf NI, McNeil N, Caldovic L, Devaney JM, Takanohashi A, Crawford J, Ru K, Grimmond SM, Miller D, Tonduti D, Schmidt JL, Chudnow RS, van Coster R, Lagae L, Kisler J, Sperner J, van der Knaap MS, Schiffmann R, Taft RJ, Vanderver A. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum.
Am J Hum Genet. 2013;92:767–73. [
PMC free article: PMC3644625] [
PubMed: 23582646]
Tomás-Vila M, Menor F, Ley-Martos M, Jumillas-Luján MJ, Marco-Hernández AV, Barbero P. Hypomyelination with atrophy of the basal ganglia and cerebellum. Contribution of two new cases to a recently reported entity.
Rev Neurol. 2014;58:161–5. [
PubMed: 24504878]
Tonduti D, Aiello C, Renaldo F, Dorboz I, Saaman S, Rodriguez D, Fettah H, Elmaleh M, Biancheri R, Barresi S, Boccone L, Orcesi S, Pichiecchio A, Zangaglia R, Maurey H, Rossi A, Boespflug-Tanguy O, Bertini E. TUBB4A-related hypomyelinating leukodystrophy: New insights from a series of 12 patients.
Eur J Paediatr Neurol. 2016;20:323–30. [
PubMed: 26643067]
Uhlenberg B, Schuelke M, Rüschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, Stoltenburg-Didinger G, Aksu F, Topaloğlu H, Nürnberg P, Hübner C, Weschke B, Gärtner J. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease.
Am J Hum Genet. 2004;75:251–60. [
PMC free article: PMC1216059] [
PubMed: 15192806]
van der Knaap MS, Naidu S, Pouwels PJ, Bonavita S, van Coster R, Lagae L, Sperner J, Surtees R, Schiffmann R, Valk J. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum.
AJNR Am J Neuroradiol. 2002;23:1466–74. [
PMC free article: PMC7976795] [
PubMed: 12372733]
Van Haren K, Bonkowsky JL, Bernard G, Murphy JL, Pizzino A, Helman G, Suhr D, Waggoner J, Hobson D, Vanderver A, Patterson MC. GLIA Consortium. Consensus statement on preventive and symptomatic care of leukodystrophy patients.
Mol Genet Metab. 2015;114:516–26. [
PubMed: 25577286]
Wakusawa K, Haginoya K, Kitamura T, Togashi N, Ishitobi M, Yokoyama H, Higano S, Onuma A, Nara T, Iinuma K. Effective treatment with levodopa and carbidopa for hypomyelination with atrophy of the basal ganglia and cerebellum.
Tohoku J Exp Med. 2006;209:163–7. [
PubMed: 16707859]