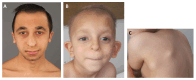
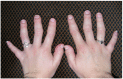
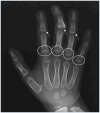
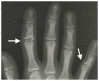
Clinical Description
Trichorhinophalangeal syndrome (TRPS) comprises TRPS I (caused by a heterozygous pathogenic variant in TRPS1) and TRPS II (caused by contiguous gene deletion of TRPS1, RAD21, and EXT1). Both types of TRPS are characterized by distinctive facial features, ectodermal features (fine, sparse, depigmented, and slow-growing hair, dystrophic nails, and small breasts), and skeletal findings (short stature, short feet, brachydactyly with ulnar or radial deviation of the fingers, and early, marked hip dysplasia). TRPS II is additionally characterized by multiple osteochondromas (typically first observed clinically on the scapulae and around the elbows and knees between ages one month and six years) and an increased risk of mild-to-moderate intellectual disability (see Table 2).
The largest cohort of individuals with TRPS reported to date includes 103 affected individuals from a large European collaborative study [Maas et al 2015]. This study and other studies [Giedion et al 1973, Lüdecke et al 2001, Güneş et al 2023] demonstrate that the phenotype of TRPS I within a family can vary markedly, and variability can be seen in all clinical and radiographic features.
Table 2.
Trichorhinophalangeal Syndrome: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature 1 |
|---|
TRPS I
(n=133) | TRPS II
(n=22) | Both TRPS I & II
(n=155) |
|---|
|
Characteristic facial features
| ~100% | ~100% | ~100% |
|
Ectodermal features
|
Sparse hair
| >85% | 70%-80% | >85% |
|
Abnormal nails
| 40%-50% | ~50% | 40%-50% |
|
Skeletal features
|
Short stature
| 40%-50% | ~80% | 50%-60% |
|
Cone-shaped epiphyses
| >95% | ~90% | >95% |
|
Brachydactyly
| 70%-80% | ~60% | 70%-80% |
|
Short metacarpals
| >65% | ~70% | >65% |
|
Hip dysplasia
| ~25% | ~60% | ~30% |
|
Osteopenia
| ~20% | 25%-30% | ~20% |
|
Osteochondromas
| ~0% | >85% 2 | ~10% |
|
Other
|
Intellectual disability
| ~10% | 50%-60% | ~15% 3 |
|
Microcephaly
| <10% | 50%-60% | 10%-20% 3 |
|
Cardiac anomalies
| ~10% | ~20% | ~10% |
- 1.
- 2.
Only occurs in TRPS II; due to EXT1 deletion in those with TRPS II
- 3.
Significantly more common in those with TRPS II
Facial Features
Characteristic facial features that are similar in both TRPS I and TRPS II include a large nose, broad nasal ridge and tip, underdeveloped alae, long philtrum, thin vermilion of the upper lip, and large, prominent ears. Other craniofacial features include a high forehead, a broad nasal septum, a horizontal smile, narrow, high-arched palate, micrognathia, and a horizontal chin furrow. Clear differences can be observed in the parts of the eyebrow: the medial eyebrow is almost always denser and wider than the lateral eyebrow [Lüdecke et al 2001, Vaccaro et al 2005, Maas et al 2015, Güneş et al 2023].
Ectodermal Features
Scalp hair. Almost all affected individuals have fine and sparse hair from a young age, particularly marked in the frontotemporal region [Jeon et al 2014, Wang et al 2020]. Scalp hair is typically slow growing and brittle. Hair color is frequently light, although it is not known if this is an associated finding.
One third of males lose their hair completely or almost completely within a few years of puberty. Women typically have more hair, but a high anterior hairline is common.
Variability exists and some individuals have near-normal scalp hair, and in some individuals the thickness and quality of hair improves with time.
Nails are thin and dystrophic in about half of individuals with TRPS; nail changes are more notable in the feet than the hands.
Teeth. Supernumerary teeth and malocclusion may be present. In addition, delayed eruption of the primary dentition and microdontia have been reported in a few individuals [Maas et al 2015].
Small breasts. Small breasts were found in four of 15 females (36%) [Maas et al 2015].
Skeletal Features
Short stature. Reduced linear growth is common in individuals with TRPS and is progressive [Lüdecke et al 2001, Maas et al 2015]. Lüdecke et al [2001] reported that birth length in 16 individuals was within the normal range, and the height z score of 28 adults with TRPS I was lower (-1.29) than that of 34 children (-0.96). Short stature was reported in 40%-50% of individuals with TRPS I and in 80% of individuals with TRPS II [Maas et al 2015, Güneş et al 2023, Öztürk et al 2023]. In individuals with a TRPS1 pathogenic variant reported between 1999 and 2023, 45.3% had short stature. The height z score for those with short stature was -3.27, compared to -0.87 for individuals without short stature.
There are a few reports of growth hormone deficiency in individuals with TRPS [Levy-Shraga et al 2020, Huang et al 2022a].
Joint manifestations. Short hands and feet are common in both TRPS I and TRPS II, typically with uneven shortening of one or more metacarpals or metatarsals and swelling of the proximal interphalangeal joints resulting in characteristic clinobrachydactyly [Lüdecke et al 2001]. Deviation of the phalanges and limited mobility of the small joints, which can be confused with rheumatoid arthritis, is very common [Maas et al 2015, Solc et al 2017, Öztürk et al 2023].
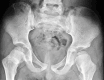
Hip dysplasia occurs especially in individuals with TRPS II, typically in pre-adolescence and early adulthood [Maas et al 2015, Grace & Ashby 2023]. By age 30 years, implantation of a prosthetic hip may be necessary. Degenerative skeletal changes can also be present in the cervical spine, knees, and ankles [Izumi et al 2010].
Delayed skeletal maturation has also been described [de Barros & Kakehasi 2016]. Note: Even in individuals with marked epiphyseal involvement, bone age can usually be determined reliably using the large number of unaffected epiphyses to assess skeletal maturation.
Osteopenia may be present in individuals with TRPS I and TRPS II but is likely more common in those with TRPS II. Reduced bone mass was described in two individuals with TRPS I [Stagi et al 2008]. Severe osteoporosis is reported in some affected individuals [Shao et al 2011, Macchiaiolo et al 2014].
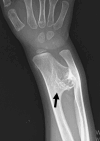
Multiple osteochondromas occur only in individuals with TRPS II due to the deletion of EXT1. Osteochondromas usually occur between age one month and six years. They first appear clinically on the shoulder blades and around the elbows and knees. The reported incidence of osteochondromas (85% in those with TRPS II) may be biased by the inclusion of very young individuals [Maas et al 2015]. The natural history is the same as seen in hereditary multiple osteochondromas [Hennekam 1991].
Psychomotor Development
The proportion of individuals with TRPS I with mild intellectual disability is reported to be slightly higher than in the general population. However, more than half of individuals with TRPS II have mild-to-moderate intellectual disability [Maas et al 2015, Güneş et al 2023]. Delays in motor development are usually associated with hip dysplasia and are therefore likely to be secondary [Maas et al 2015].
Other
Body weight is usually normal in relation to height.
Head circumference is typically normal throughout life in individuals with TRPS I. In those with TRPS II, 50%-60% of individuals have a head circumference below the 3rd centile [Lüdecke et al 2001, Maas et al 2015, Güneş et al 2023, Öztürk et al 2023].
Cardiac abnormalities vary from minor anomalies (persistent ductus arteriosus, persistent foramen ovale, bicuspid aortic valves, mitral valve regurgitation) to significant problems (aortic stenosis, anomalous venous return). Cardiac rhythm disturbances are rare.