Summary
Clinical description.
In adults, X-linked spondyloepiphyseal dysplasia tarda (X-linked SEDT) is characterized by disproportionately short stature with short trunk and arm span significantly greater than height. At birth, affected males are normal in length and have normal body proportions. Affected males exhibit linear growth deficiency beginning around age six to eight years. Final adult height is typically 137-163 cm. Progressive joint and back pain with osteoarthritis ensues; hip, knee, and shoulder joints are commonly involved but to a variable degree. Hip replacement is often required as early as age 40 years. Interphalangeal joints are typically spared. Motor and cognitive milestones are normal.
Diagnosis/testing.
The clinical diagnosis of X-linked SEDT can be established in a male proband with characteristic radiographic findings (which typically appear prior to puberty) including: multiple epiphyseal abnormalities, platyspondyly with characteristic superior and inferior "humping" seen on lateral view, scoliosis, hypoplastic odontoid process, short femoral necks, and coxa vara; evidence of premature osteoarthritis appears in young adulthood. The molecular diagnosis of X-linked SEDT can be established in a male proband with suggestive findings and a hemizygous pathogenic variant in TRAPPC2 identified by molecular genetic testing. The molecular diagnosis of X-linked SEDT can be established in a female proband with osteoarthritis and a heterozygous pathogenic variant in TRAPPC2 identified by molecular genetic testing.
Management.
Treatment of manifestations: Treatment for scoliosis and kyphoscoliosis per orthopedic surgeon; surgical intervention may include spine surgery (correction of scoliosis or kyphosis). Pain management as needed for osteoarthritis; joint replacement (hip, knee, shoulder) as needed.
Surveillance: Cervical spine films prior to school age and before any surgical procedure involving general anesthesia to assess for clinically significant odontoid hypoplasia. Annual follow up for assessment scoliosis and joint pain.
Agents/circumstances to avoid: Extreme neck flexion and extension in individuals with odontoid hypoplasia. Activities and occupations that place undue stress on the spine and weight-bearing joints.
Evaluation of relatives at risk: Presymptomatic testing in males at risk may obviate unnecessary diagnostic testing for other causes of short stature and/or osteoarthritis.
Genetic counseling.
By definition, X-linked SEDT is inherited in an X-linked manner. When performed, molecular genetic testing of all mothers of affected sons determined that regardless of family history all were carriers of a pathogenic variant in TRAPPC2. Carrier females are at a 50% risk of transmitting the TRAPPC2 pathogenic variant in each pregnancy: males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be carriers and will not be affected. None of the sons of an affected male will be affected; all daughters will be carriers of the TRAPPC2 pathogenic variant. Carrier testing of at-risk female relatives and prenatal testing for pregnancies at increased risk are possible if the pathogenic variant in the family has been identified.
Diagnosis
No consensus clinical diagnostic criteria for X-linked spondyloepiphyseal dysplasia tarda have been published.
Suggestive Findings
X-linked spondyloepiphyseal dysplasia tarda (X-linked SEDT) should be suspected in males with the following findings:
- Disproportionate short stature in adolescence or adulthood and a relatively short trunk and barrel-shaped chest. Upper- to lower-body segment ratio is usually about 0.8. Arm span typically exceeds height by 10-20 cm. Short neck, dorsal kyphosis, and lumbar hyperlordosis may be evident by puberty.
- Early-onset osteoarthritis, especially in the hip joints
- A family history consistent with X-linked recessive inheritance. A positive family history is contributory but not necessary.
- Absence of cleft palate and retinal detachment (frequently present in SED congenita; see Differential Diagnosis)
Establishing the Diagnosis
The clinical diagnosis of X-linked SEDT can be established in a male proband with characteristic radiographic findings, or the molecular diagnosis can be established in a male proband with suggestive findings and a hemizygous pathogenic (or likely pathogenic) variant in TRAPPC2 identified by molecular genetic testing (see Table 1) if radiographic findings are inconclusive.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a hemizygous TRAPPC2 variant of uncertain significance does not establish or rule out the diagnosis.
Radiographic Findings
The following radiographic findings may not be manifest in an affected male in early childhood and typically appear prior to puberty (Figure 1):
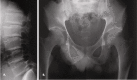
Figure 1.
Radiographs of a male age 31 years with SEDT A. Platyspondyly with superior and inferior humping of vertebral bodies
- Multiple epiphyseal abnormalities
- Platyspondyly (flattened vertebral bodies) with characteristic superior and inferior "humping" seen on lateral view; narrow disc spaces in adulthood
- Scoliosis / kyphoscoliosis
- Hypoplastic odontoid process
- Short femoral necks
- Coxa vara
- Evidence of premature osteoarthritis beginning in young adulthood
Radiographs of symptomatic males should be reviewed by a radiologist experienced with bone dysplasias.
Molecular Genetic Testing
Testing approaches can include single-gene testing and a multigene panel:
- Single-gene testing. Sequence analysis of TRAPPC2 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
- A multigene panel that includes TRAPPC2 and other genes of interest (see Differential Diagnosis) may also be considered. This method may be especially useful if expert radiographic interpretation is not available. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Table 1.
Molecular Genetic Testing Used in X-linked Spondyloepiphyseal Dysplasia Tarda
Clinical Characteristics
Clinical Description
Males. At birth, affected males are normal in length and have normal body proportions. Affected males exhibit linear growth deficiency beginning around grade school (age 6-8 years). Adults with X-linked spondyloepiphyseal dysplasia tarda (X-linked SEDT) have disproportionately short stature with short trunk and arm span significantly greater than height. Final adult height is typically 137-163 cm [Whyte et al 1999, Jones et al 2013, Rimoin et al 2013].
Scoliosis/kyphoscoliosis and odontoid hypoplasia are known radiographic features. Data on the incidence, onset, and severity of these features have not been published.
Osteoarthritis. Progressive joint and back pain with osteoarthritis ensues; hip, knee, and shoulder joints are commonly involved to variable degrees. Hip replacement is often required as early as age 40 years. Interphalangeal joints are typically spared.
Affected males achieve normal motor and cognitive milestones. Life span and intelligence appear normal.
Heterozygous females. Carrier females typically show no phenotypic changes, but mild symptoms of osteoarthritis have been reported [Whyte et al 1999].
Genotype-Phenotype Correlations
Data are inadequate to reliably correlate clinical severity to a specific TRAPPC2 pathogenic variant. All pathogenic variants identified thus far, irrespective of their molecular basis, result in an almost identical phenotype, including the true null variants.
Nomenclature
Spondyloepiphyseal dysplasia is a general term that describes the radiographic abnormalities seen in several skeletal dysplasias, including pseudoachondroplasia. The "congenita" form is evident at birth, whereas the "tarda" form is usually evident by school age.
SED tarda commonly refers to the X-linked recessive form of the disorder, although rare autosomal dominant and autosomal recessive "tarda" forms have been described.
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], X-linked spondyloepiphyseal dysplasia tarda is referred to as TRAPPC2-related X-linked spondyloepiphyseal dysplasia tarda and included in the spondyloepi(meta)physeal dysplasias group.
Prevalence
The prevalence is 1:150,000-1:200,000 [Wynne-Davies & Gormley 1985].
Pathogenic variants in TRAPPC2 have been found in several populations including European [Gedeon et al 2001], Japanese [Matsui et al 2001], and Chinese [Shu et al 2002], an observation suggesting that no specific population is at increased risk.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in TRAPPC2.
Differential Diagnosis
X-linked spondyloepiphyseal dysplasia tarda (X-linked SEDT) is distinguished from other forms of spondyloepiphyseal dysplasia (SED) by its later onset and X-linked inheritance (see Table 2).
Table 2.
Forms of Spondyloepiphyseal Dysplasia of Interest in the Differential Diagnosis of X-Linked Spondyloepiphyseal Dysplasia Tarda
Other
- SED tarda, autosomal forms (rare). A dominant form (OMIM 184100) may be caused by pathogenic variants in COL2A1; a recessive form has been described clinically but not molecularly defined.
- Scheuermann disease (OMIM 181440) is a term applied to premature osteoarthritis of the spine regardless of etiology.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with X-linked spondyloepiphyseal dysplasia tarda (X-linked SEDT), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with X-linked Spondyloepiphyseal Dysplasia Tarda
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with X-linked Spondyloepiphyseal Dysplasia Tarda
Surveillance
Table 5.
Recommended Surveillance for Individuals with X-linked Spondyloepiphyseal Dysplasia Tarda
Agents/Circumstances to Avoid
The following should be avoided:
- In individuals with odontoid hypoplasia, extreme neck flexion and extension
- Activities and occupations that place undue stress on the spine and weight-bearing joints
Evaluation of Relatives at Risk
If the TRAPPC2 pathogenic variant in the family is known, presymptomatic genetic testing of at-risk males allows early diagnosis and may obviate unnecessary diagnostic testing for other causes of short stature and/or osteoarthritis.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
By definition, X-linked spondyloepiphyseal dysplasia tarda (X-linked SEDT) is inherited in an X-linked manner.
Risk to Family Members
Parents of a proband
- The father of a male proband with X-linked SEDT will not have the disorder nor will he be hemizygous for the causative pathogenic variant; therefore, he does not require further evaluation/testing.
- In a family with more than one affected individual, the mother of an affected male is an obligate carrier. Note: If a woman has more than one affected child and no other affected relatives and if the familial pathogenic variant cannot be detected in her DNA, she most likely has germline mosaicism. Although no instances of maternal germline mosaicism have been reported, it remains a possibility.
- If a male is the only affected family member (i.e., a simplex case), the mother may be a carrier or the affected male may have a de novo pathogenic variant, in which case the mother is not a carrier.In reported individuals for whom molecular genetic testing was available in a research laboratory, all mothers of affected sons were carriers of a TRAPPC2 pathogenic variant regardless of family history [Gedeon et al 2001]. Mothers of affected sons who are not carriers have not been reported to date.
Sibs of a proband. The risk to sibs of a male proband with X-linked SEDT depends on the genetic status of the mother:
- If the mother of the proband has a TRAPPC2 pathogenic variant, the chance of transmitting the pathogenic variant in each pregnancy is 50%.
- Males who inherit the pathogenic variant will be affected;
- Females who inherit the pathogenic variant will be carriers and will usually not be affected (see Clinical Description, Heterozygous females).
- If the proband represents a simplex case and if the TRAPPC2 pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is presumed to be low but greater than that of the general population because of the theoretic possibility of maternal germline mosaicism.
Offspring of a male proband. Affected males transmit the TRAPPC2 pathogenic variant to all of their daughters and none of their sons.
Other family members. The maternal aunts and maternal female cousins of a male proband may be at risk of being carriers and the aunts' offspring, depending on their sex, may be at risk of being carriers or of being affected.
Carrier Detection
Identification of female heterozygotes requires either prior identification of the TRAPPC2 pathogenic variant in the family or, if an affected male is not available for testing, molecular genetic testing first by sequence analysis, and if no pathogenic variant is identified, by gene-targeted deletion/duplication analysis.
Note: Females who are heterozygotes (carriers) for X-linked SEDT may develop minimal clinical findings related to the disorder [Whyte et al 1999].
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of genetic status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the X-linked SEDT-causing pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Human Growth Foundation
- Little People of AmericaPhone: 888-LPA-2001; 714-368-3689Fax: 707-721-1896Email: info@lpaonline.org
- MAGIC FoundationPhone: 800-362-4423Email: contactus@magicfoundation.org
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
X-Linked Spondyloepiphyseal Dysplasia Tarda: Genes and Databases
Table B.
OMIM Entries for X-Linked Spondyloepiphyseal Dysplasia Tarda (View All in OMIM)
Molecular Pathogenesis
TRAPPC2 (previously SEDL) encodes the 140-amino acid protein "sedlin," which appears to be ubiquitously expressed [Gedeon et al 1999, Gécz et al 2000]. Sedlin is an essential component of the TRAPP (trafficking protein particle) complex that is required for the export of procollagen trimers (e.g., type II collagen) from the endoplasmic reticulum to the Golgi, which ultimately permits incorporation of these proteins into the extracellular matrix [Venditti et al 2012].
Mechanism of disease causation. Loss of function
TRAPPC2-specific laboratory technical considerations. TRAPPC2 contains six exons with the translation start site in exon 3. The exon and multiexon deletions (see also HGMD in Table A) would not be detected in heterozygous females by sequence analysis (see Table 1). Sequence analysis should include flanking intronic sequences, which is customary to evaluate splice junctions. This is particularly important with TRAPPC2, as there is an expressed pseudogene which is devoid of introns [Gécz et al 2000].
Notable TRAPPC2 variants. X-linked SEDT-causing pathogenic variants in TRAPPC2 include splice site, nonsense, and missense variants and deletions.
Table 6.
Notable Recurrent TRAPPC2 Pathogenic Variants
Chapter Notes
Author History
George E Tiller, MD, PhD (2001-present)
Vickie L Hannig, MS; Vanderbilt University Medical Center (2001-2020)
Revision History
- 6 April 2023 (sw) Revision: "TRAPPC2-Related X-Linked Spondyloepiphyseal Dysplasia Tarda" added as a synonym; Nosology of Genetic Skeletal Disorders: 2023 Revision [Unger et al 2023] added to Nomenclature
- 5 November 2020 (sw) Comprehensive update posted live
- 11 June 2015 (me) Comprehensive update posted live
- 15 February 2011 (me) Comprehensive update posted live
- 5 April 2006 (me) Comprehensive update posted live
- 10 February 2004 (me) Comprehensive update posted live
- 30 December 2003 (cd) Revision: change in test availability
- 1 November 2001 (me) Review posted live
- 16 May 2001 (gt) Original submission
References
Literature Cited
- Fiedler J, Le Merrer M, Mortier G, Heuertz S, Faivre L, Brenner RE. X-linked spondyloepiphyseal dysplasia tarda: Novel and recurrent mutations in 13 European families. Hum Mutat. 2004;24:103. [PubMed: 15221797]
- Gécz J, Hillman MA, Gedeon AK, Cox TC, Baker E, Mulley JC. Gene structure and expression study of the SEDL gene for spondyloepiphyseal dysplasia tarda. Genomics. 2000;69:242–51. [PubMed: 11031107]
- Gedeon AK, Colley A, Jamieson R, Thompson EM, Rogers J, Sillence D, Tiller GE, Mulley JC, Gecz J. Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda. Nat Genet. 1999;22:400–4. [PubMed: 10431248]
- Gedeon AK, Tiller GE, Le Merrer M, Heuertz S, Tranebjaerg L, Chitayat D, Robertson S, Glass IA, Savarirayan R, Cole WG, Rimoin DL, Kousseff BG, Ohashi H, Zabel B, Munnich A, Gecz J, Mulley JC. The molecular basis of X-linked spondyloepiphyseal dysplasia tarda. Am J Hum Genet. 2001;68:1386–97. [PMC free article: PMC1226125] [PubMed: 11349230]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jones KL, Jones MC, del Campo M. Smith's Recognizable Patterns of Human Malformation. 7 ed. Philadelphia, PA: WB Saunders; 2013.
- Matsui Y, Yasui N, Ozono K, Yamagata M, Kawabata H, Yoshikawa H. Loss of the SEDL gene product (Sedlin) causes X-linked spondyloepiphyseal dysplasia tarda: Identification of a molecular defect in a Japanese family. Am J Med Genet. 2001;99:328–30. [PubMed: 11252002]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rimoin DL, Lachman RS, Unger S. Chondrodysplasias. In: Rimoin DL, Pyeritz RE, Korf BR, eds. Emery & Rimoin’s Principles and Practice of Medical Genetics. 6 ed. New York, NY: Academic Press; 2013.
- Shaw MA, Brunetti-Pierri N, Kadasi L, Kovacova V, Van Maldergem L, De Brasi D, Salerno M, Gecz J. Identification of three novel SEDL mutations, including mutation in the rare, non-canonical splice site of exon 4. Clin Genet. 2003;64:235–42. [PubMed: 12919139]
- Shu SG, Tsai CR, Chi CS. Spondyloepiphyseal dysplasia tarda: report of one case. Acta Paediatr Taiwan. 2002;43:106–8. [PubMed: 12041616]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Tiller GE, Hannig VL, Dozier D, Carrel L, Trevarthen KC, Wilcox WR, Mundlos S, Haines JL, Gedeon AK, Gecz J. A recurrent RNA-splicing mutation in the SEDL gene causes X-linked spondyloepiphyseal dysplasia tarda. Am J Hum Genet. 2001;68:1398–407. [PMC free article: PMC1226126] [PubMed: 11326333]
- Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, Cohn DH, Cormier-Daire V, Girisha KM, Hall C, Krakow D, Makitie O, Mundlos S, Nishimura G, Robertson SP, Savarirayan R, Sillence D, Simon M, Sutton VR, Warman ML, Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023. Epub ahead of print. [PMC free article: PMC10081954] [PubMed: 36779427]
- Venditti R, Scanu T, Santoro M, Di Tullio G, Spaar A, Gaibisso R, Beznoussenko GV, Mironov AA, Mironov A Jr, Zelante L, Piemontese MR, Notarangelo A, Malhotra V, Vertel BM, Wilson C, De Matteis MA. Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science. 2012;337:1668–72. [PMC free article: PMC3471527] [PubMed: 23019651]
- Whyte MP, Gottesman GS, Eddy MC, McAlister WH. X-linked recessive spondyloepiphyseal dysplasia tarda. Clinical and radiographic evolution in a 6-generation kindred and review of the literature. Medicine (Baltimore). 1999;78:9–25. [PubMed: 9990351]
- Wynne-Davies R, Gormley J. The prevalence of skeletal dysplasias. An estimate of their minimum frequency and the number of patients requiring orthopaedic care. J Bone Joint Surg Br. 1985;67:133–7. [PubMed: 3155744]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: November 1, 2001; Last Revision: April 6, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Tiller GE. X-Linked Spondyloepiphyseal Dysplasia Tarda. 2001 Nov 1 [Updated 2023 Apr 6]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
