Spinocerebellar Ataxia Type 20
Synonym: SCA20
Elsdon Storey, DPhil, FRACP and RJM Gardner, MB ChB, FCCMG.
Author Information and AffiliationsInitial Posting: February 27, 2007; Last Update: April 18, 2019.
Estimated reading time: 11 minutes
Summary
Clinical characteristics.
Spinocerebellar ataxia type 20 (SCA20) is characterized by a slowly progressive ataxia and dysarthria. Approximately two thirds of those affected also display palatal tremor ("myoclonus") and/or abnormal phonation clinically resembling spasmodic adductor dysphonia. Dysarthria, which may be abrupt in onset, precedes the onset of ataxia in about two thirds of affected individuals, sometimes by a number of years. Hypermetric horizontal saccades (without nystagmus or disturbance of vestibulo-ocular reflex gain) are seen in about half of affected persons. Although minor pyramidal signs (brisk knee jerks, crossed adductor spread) may be seen, spasticity and extensor plantar responses are not. Cognition is normal. Clinical information is based on the findings in 16 personally examined affected members of a single Australian family of Anglo-Celtic descent.
Diagnosis/testing.
The diagnosis of SCA20 is based on clinical findings and neuroimaging. Within five years of disease onset CT scan shows pronounced dentate calcification, typically without concomitant pallidal calcification. In addition to evidence of dentate calcification, MRI shows mild-to-moderate pan cerebellar atrophy and normal cerebrum and brain stem (except for increased inferior olivary T2 signal in those with palatal tremor). The locus for SCA20 lies within the pericentromeric region of chromosome 11; the gene is unknown. A 260-kb duplication of 11q12.2-11q12.3 has been proposed as the probable cause of SCA20 in the index family.
Management.
Treatment of manifestations: Physical and occupational therapy; guidance from a speech pathologist expert in the management of neurogenic dysphagia.
Prevention of secondary complications: Prevention of falls by using appropriate gait aids and home modifications; personal alarm system.
Diagnosis
Suggestive Findings
Spinocerebellar ataxia type 20 (SCA20) should be considered in individuals with a slowly progressive ataxia without sensory features who have the following findings:
Additional findings may include the following:
Hypermetric horizontal saccades (without nystagmus or disturbance of vestibuloocular reflex gain) in about half
Mild hyperreflexia (typically without spasticity or extensor plantar responses) in a minority
Postural tremor of arms with or without involvement of the head (seen in a minority; may be the first symptom)
Neuroimaging
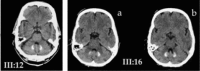
CT scan () shows pronounced dentate calcification, typically without concomitant pallidal calcification, at an early stage of the illness (≤3 years from onset).
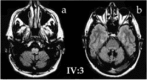
MRI () shows mild-to-moderate pan cerebellar atrophy, with low dentate signal on T1- and T2-weighted sequences (consistent with calcification) and normal cerebrum and brain stem (apart from increased inferior olivary T2 signal in some, as a correlate of palatal tremor).
III:12. CT scan showing heavy dentate calcification and pan cerebellar atrophy in an individual age 62 years with mild ataxia, three years after symptom onset III:16. CT scan slices at two levels of the dentate showing heavy dentate calcification and (more...)
MRI axial proton density images showing (a) inferior olivary hypertrophy and (b) low dentate signal consistent with dentate calcification in an individual age 54 years; 16 years after onset Note: Roman:arabic numeral combinations refer to pedigree numbers (more...)
Neurophysiology. Nerve conduction studies are normal.
Laboratory features. Indices of calcium metabolism (serum concentrations of calcium, phosphate, magnesium, alkaline phosphatase, parathyroid hormone, 25-hydroxy vitamin D) are normal.
Establishing the Diagnosis
The diagnosis of SCA20 is established in a proband with the above Suggestive Findings and the following characteristic radiographic features:
Brain CT. Pronounced dentate calcification, typically without concomitant pallidal calcification
Brain MRI. Mild-to-moderate pan cerebellar atrophy and normal cerebrum and brain stem (except for increased inferior olivary T2 signal in those with palatal tremor)
Note: The candidate region for SCA20 lies within the pericentromeric region of chromosome 11, encompassing chr11:44,045,910-69,634,192 (loci D11S903-FGF3). Although the candidate region includes SPTBN2 (pathogenic variants in which are responsible for SCA5), SPTBN2 pathogenic variants were excluded as the cause of SCA20 [Lorenzo et al 2006]. Knight et al [2008] reported a 260-kb heterozygous duplication within the candidate region, at 11q12.2-11q12.3 (chr11:61,453,940-61,746,019), cosegregating with SCA20 in the index family. It remains unknown whether this cosegregating duplication contains a locus or loci that are pathogenic in the duplicated state, or the duplication is merely in linkage disequilibrium with a closely linked locus that is the actual basis of the cerebellar phenotype (or whether there is another more complex genetic explanation).
Molecular genetic testing approaches can include a combination of chromosomal microarray analysis or exome array.
Chromosomal microarray analysis (CMA) uses oligonucleotide or SNP arrays to detect genome-wide large deletions/duplications that cannot be detected by sequence analysis.
Table 1.
Genomic Testing Used in Spinocerebellar Ataxia Type 20
View in own window
| Duplication 1 | Method | Proportion of Probands with a Pathogenic Variant Detectable by Method |
|---|
| Heterozygous duplication at 11q12.2-11q12.3 2 | CMA 3 | 100% |
| Targeted duplication analysis 4 | 100% |
- 1.
- 2.
For this GeneReview, the 11q12.2-11q12.3 recurrent duplication, observed in all reported individuals with SCA20, is defined as a 260-kb duplication at the 11q12 region of chromosome 11.
- 3.
Chromosomal microarray analysis (CMA) using oligonucleotide arrays or SNP arrays. CMA designs in current clinical use target the 11q12.2-11q12.3 region.
- 4.
Targeted duplication analysis methods can include FISH, quantitative PCR (qPCR), and multiplex ligation-dependent probe amplification (MLPA), as well as other targeted quantitative methods.
Clinical Characteristics
Clinical Description
Clinical information on spinocerebellar ataxia type 20 (SCA20) is based on the index pedigree, an Australian family of Anglo-Celtic descent that is the only family with SCA20 reported to date [Knight et al 2004, Storey et al 2005, Storey & Gardner 2012].
The 16 affected family members had onset between age 19 and 64 years (mean 47).
SCA20 presents with dysarthria without ataxia in a majority (10/16); the dysarthria may be of abrupt (2/16) or subacute (1/16) onset. It often combines the clinical appearance of spasmodic adductor dysphonia with cerebellar dysarthria. Other initial symptoms were dysarthria with simultaneous gait ataxia (2/16), gait ataxia alone (2/16), upper-limb kinetic and isometric tremor (1/16), and episodic vertigo (1/16).
Progression of SCA20, as judged by cross-sectional data, appears to be relatively slow; all affected members of this family were able to walk with or without gait aids except one, who became wheelchair dependent after 40 years of symptoms. Another required a feeding gastrostomy after 15 years of symptoms.
The clinical picture usually includes palatal tremor ("myoclonus") without ear click (10/16), although this finding can be subtle. Gaze-evoked nystagmus is unusual (3/16). In two it was impersistent; in another affected individual persistent downbeating nystagmus was evident. Saccades are typically hypermetric into downgaze (10/16) and horizontally (8/16). The vestibulo-ocular reflex gain, as judged by dynamic vs static visual acuity, is normal, correlating with absence of movement-induced oscillopsia. Minor pyramidal signs (brisk knee jerks, crossed adductor spread) are seen in a minority (5/16), but none have spastic tone or extensor plantar responses. Postural and kinetic tremor of the upper limbs, the presenting feature in one individual, was evident in only one other family member. Only one displayed intention tremor (as distinct from dysmetria and dyssynergia) on the finger/nose test.
Other extrapyramidal features (apart from slowing of repetitive movements without movement decay) are absent. None had a history of cognitive decline.
Penetrance
The penetrance is unknown, as the involved gene has not been identified.
Anticipation
Only four parent-child pairs could be documented by self-report regarding age of onset, which was younger by an average of 12 years in the offspring. This information is inadequate to confirm or refute anticipation. Large CAG/CTG and ATTCT/AGAAT repeats have been excluded in the region of interest [Knight et al 2004].
Prevalence
The prevalence of SCA20 is unknown; it is assumed to be very rare, given that no further individuals have been reported since the publication of the original family in 2008.
Differential Diagnosis
The differential diagnosis of spinocerebellar ataxia type 20 (SCA20) is essentially that of its component features, as the constellation of progressive, dominantly inherited ataxia, early dentate calcification, and (often) palatal tremor is distinctive.
Inherited ataxia. See Hereditary Ataxia Overview.
Dentate calcification appears early in SCA20; it was seen in five affected individuals who had been symptomatic for five years or less.
While dentate calcification is common in the general population with increasing age, affecting 0.7% of those older than age 65 years in one study [
Harrington et al 1981], it rarely occurs in the absence of basal ganglia calcification (as it did in 9/11 individuals in the family with SCA20).
Hyperparathyroidism and pseudohypoparathyroidism (see
Disorders of GNAS Inactivation) with basal ganglia (and dentate) calcification may be dominantly inherited, and can be excluded on biochemical testing.
Palatal tremor ("myoclonus") may be seen in the following situations, while dentate calcification is not:
Dysphonia (which is apparent in SCA20 rather than confirmed on formal voice analysis) may also be seen with ataxia and motor neuropathy (OMIM 606183) [Barbieri et al 2001]; this latter syndrome appears to be inherited in an autosomal recessive manner. The presence of motor neuropathy and the absence of dentate calcification and palatal tremor also serve to distinguish this syndrome from SCA20.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease in an individual diagnosed with spinocerebellar ataxia type 20 (SCA20), the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Treatment of Manifestations
Affected persons should be followed by a neurologist with consultation from physiatrists and physical and occupational therapists.
Although neither exercise nor physical therapy has been shown to stem the progression of incoordination or muscle weakness, individuals should maintain activity.
Canes and walkers help prevent falls. Modification of the home with such conveniences as grab bars, raised toilet seats, and ramps to accommodate motorized chairs may be necessary.
Speech therapy and communication devices such as writing pads and computer-based devices may benefit those with dysarthria or dysphonia.
Weighted eating utensils and dressing hooks help maintain a sense of independence.
Weight control is important because obesity can exacerbate difficulties with ambulation and mobility.
When dysphagia becomes troublesome, videofluoroscopic swallow evaluation can identify the consistency of food least likely to trigger aspiration.
Prevention of Secondary Complications
Secondary complications are unlikely in the early years of the disease.
Later, prevention of falls via appropriate gait aids and home modifications, and (if falls are frequent) a personal alarm system may be required. To limit the likelihood of fractures resulting from falls, bone density should be estimated and osteoporosis treated if present.
Vitamin supplements are recommended, particularly if caloric intake is reduced.
Weight control is important because obesity can exacerbate difficulties with ambulation and mobility.
Surveillance
The following are appropriate:
Agents/Circumstances to Avoid
Affected individuals should avoid alcohol as well as medications known to cause nerve damage (e.g., isoniazid).
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Other
Tremor-controlling drugs do not work well for cerebellar tremors.
Education for affected individuals and their families is the cornerstone of management.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Spinocerebellar ataxia type 20 (SCA20) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
If a parent of the
proband is affected, the risk to the sibs is 50%.
If the parents are clinically unaffected, the risk to the sibs of a
proband is likely to be low.
Offspring of a proband. The risk to offspring of an individual with SCA20 is 50%.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, the parent's family members are at risk.
Prenatal Testing
Because the gene in which pathogenic variants are responsible for SCA20 has not been identified, prenatal testing is not possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
NCBI Genes and Disease
Ataxia UK
United Kingdom
Phone: 0800 995 6037; +44 (0) 20 7582 1444 (from abroad)
Email: help@ataxia.org.uk
euro-ATAXIA (European Federation of Hereditary Ataxias)
United Kingdom
Email: lporter@ataxia.org.uk
Spanish Ataxia Federation (FEDAES)
Spain
Phone: 601 037 982
Email: info@fedaes.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Spinocerebellar Ataxia Type 20: Genes and Databases
View in own window
| Gene | Chromosome Locus | Protein | ClinVar |
|---|
| Unknown | 11p13-q11 | Unknown | |
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
Introduction. The 260-kb duplicated segment reported by Knight et al [2008] includes ten known and two unknown genes. It was not found in 1,129 control samples, suggesting that it is not merely a copy number variant.
Mechanism of disease causation is unknown, but suspected to be a dosage effect of one or more genes in the 11q12.2-12.3 region.
Genes of interest in this region. One of the known genes in the duplicated region, DAGLA, is potentially an attractive candidate as it is highly expressed in murine Purkinje cells.
Chapter Notes
Revision History
18 April 2019 (sw) Comprehensive update posted live
7 June 2012 (me) Comprehensive update posted live
6 January 2009 (es) Revision: 260-kb
duplication of 11q12.2-11q12.3 identified as suspected cause of SCA20
27 February 2007 (me) Review posted live
6 February 2007 (es) Original submission
References
Literature Cited
Barbieri F, Pellecchia MT, Esposito E, Di Stasio E, Castaldo I, Santorelli F, Perretti A, Santoro L, De Michele G. Adult-onset familial laryngeal abductor paralysis, cerebellar ataxia, and pure motor neuropathy.
Neurology. 2001;56:1412–4. [
PubMed: 11376202]
de Yebenes JG, Vazquez A, Rabano J, de Seijas EV, Urra DG, Obregon MC, Barquero MS, Arribas MA, Moreno JL, Alenda JR. Hereditary branchial myoclonus with spastic paraparesis and cerebellar ataxia: a new autosomal dominant disorder.
Neurology. 1988;38:569–72. [
PubMed: 3352913]
Harrington MG, Macpherson P, McIntosh WB, Allam BF, Bone I. The significance of the incidental finding of basal ganglia calcification on computed tomography.
J Neurol Neurosurg Psychiatry. 1981;44:1168–70. [
PMC free article: PMC491242] [
PubMed: 7334414]
Knight MA, Gardner RJ, Bahlo M, Matsuura T, Dixon JA, Forrest SM, Storey E. Dominantly inherited ataxia and dysphonia with dentate calcification: spinocerebellar ataxia type 20.
Brain. 2004;127:1172–81. [
PubMed: 14998916]
Knight MA, Hernandez D, Diede SJ, Dauwerse HG, Rafferty I, van de Leemput J, Forrest SM, Gardner RJM, Storey E, van Ommen GJ, Tapscott SJ, Fischbeck KH, Singleton AB. A duplication at chromosome 11q12.2-11q12.3 is associated with spinocerebellar ataxia type 20.
Hum Mol Genet. 2008;17:3847–53. [
PMC free article: PMC2588641] [
PubMed: 18801880]
Lorenzo DN, Forrest SM, Ikeda Y, Dick KA, Ranum LP, Knight MA. Spinocerebellar ataxia type 20 is genetically distinct from spinocerebellar ataxia type 5.
Neurology. 2006;67:2084–5. [
PubMed: 17159129]
Sperling MR, Herrmann C Jr. Syndrome of palatal myoclonus and progressive ataxia: two cases with magnetic resonance imaging.
Neurology. 1985;35:1212–4. [
PubMed: 4022358]
Storey E, Gardner RJM. Spinocerebellar ataxia type 20.
Handb Clin Neurol. 2012;103:567–73. [
PubMed: 21827916]
Storey E, Knight MA, Forrest SM, Gardner RJ. Spinocerebellar ataxia type 20.
Cerebellum. 2005;4:55–7. [
PubMed: 15895561]