Clinical Description
RRM2B mitochondrial DNA maintenance defects (RRM2B-MDMD) are an important cause of both childhood- and adult-onset mitochondrial disease. To date, 78 individuals from 52 families with molecularly confirmed RRM2B-MDMD have been reported. In general, the clinical manifestations are similar in males and females.
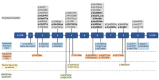
RRM2B-MDMD can, for the most part, be broadly categorized into two main groups: mtDNA depletion and multiple mtDNA deletions; and further characterized by phenotype, mode of inheritance, and disease pathogenesis ().
The two common forms of RRM2B-MDMD are RRM2B encephalomyopathic MDMD (the most severe form) and RRM2B autosomal dominant progressive external ophthalmoplegia (adPEO) (typically adult onset).
The two rare phenotypes – RRM2B mitochondrial neurogastrointestinal encephalopathy (MNGIE)-like and autosomal recessive progressive external ophthalmoplegia (arPEO) – are described briefly in this section. See Rare Forms.
Table 2.
Phenotypic Spectrum of RRM2B Mitochondrial DNA Maintenance Defects
View in own window
| Relative Prevalence | Phenotype | MOI | Onset | Comments | Disease Pathogenesis |
|---|
| Common | RRM2B encephalo-
myopathic MDMD | AR | Infancy | Severe multisystem disease often fatal in early life | mtDNA depletion |
| adPEO 1 | AD | Adulthood | Milder & often tissue-specific | Clonally expanded (multiple) mtDNA deletions 2 |
| Rare | MNGIE 3 | AR | Childhood or adulthood | GI dysmotility, PEO, ptosis, peripheral neuropathy & leukoencephalopathy | mtDNA depletion |
| arPEO 1 | AR | Early childhood or adulthood | Multisystem involvement & Kearns-Sayre syndrome-like 4 | Clonally expanded (multiple) mtDNA deletions 2 |
- 1.
- 2.
Accumulation of clonally expanded (multiple) mtDNA deletions causes tissue-specific cytochrome c oxidase (COX) deficiency.
- 3.
- 4.
RRM2B Encephalomyopathic MDMD
RRM2B encephalomyopathic MDMD, reported in 31 children, is the most severe form of RRM2B-MDMD, manifesting shortly after birth as hypotonia, poor feeding, and faltering growth (previously known as failure to thrive) requiring hospitalization. Subsequent assessments are likely to reveal multisystem involvement including sensorineural hearing loss, renal tubulopathy, and respiratory failure (see ) [Bourdon et al 2007, Bornstein et al 2008, Acham-Roschitz et al 2009, Kollberg et al 2009, Shaibani et al 2009, Spinazzola et al 2009, Stojanovic et al 2013, Pronicka et al 2016, Iwanicka-Pronicka et al 2019, Penque et al 2019, Keshavan et al 2020].
Table 3.
Select Clinical Features of RRM2B Encephalomyopathic MDMD
View in own window
| Feature | # of Children 1
w/Feature (%) | Comment |
|---|
|
Neuromuscular
| 31 (100%) | Truncal hypotonia (almost universally present) gross motor delay, feeding difficulties, poor head control, generalized weakness, areflexia, ptosis, PEO, discoordinated swallow, exercise intolerance |
|
Nervous system
|
Central
| 1-15 (3%-48%) | Encephalopathy: gross motor delay, seizures, focal &/or generalized UMN signs, microcephaly, neurologic regression, dystonia. MRI shows central hypomyelination, cerebral atrophy |
|
Peripheral
| 2 (6%) | Demyelinating peripheral neuropathy |
|
Respiratory
| 18 (58%) | Respiratory distress, respiratory failure. In some cases artificial ventilation may prolong life span. |
|
Renal
| 17 (55%) | Tubulopathy (proximal), nephrocalcinosis, aminoaciduria, glycosuria, lactic acidemia, hypocalcemia |
|
Faltering growth (previously known as failure to thrive)
| 16 (52%) | Likely due to multisystem involvement |
|
Hearing loss
| 11 (35%) | Sensorineural hearing loss may only be identified on formal assessment. |
|
Gastrointestinal
| 10 (32%) | Recurrent vomiting, feed intolerance, chronic diarrhea, cachexia |
|
Eye
| 4 (13%) | Ophthalmoparesis, pigmentary retinopathy (rod-cone dystrophy), cataracts, megalocornea, blindness, nystagmus |
|
Cardiovascular
| 4 (13%) | Left ventricular hypertrophy, cardiomyopathy, ventricular septal defect |
PEO = progressive external ophthalmoplegia; UMN = upper motor neuron
- 1.
Onset is typically in the first few months of life; affected children succumb in early childhood. Disease characteristics include myopathy manifesting as hypotonia, weakness associated with respiratory insufficiency, faltering growth (failure to thrive), and proximal renal tubulopathy.
Central nervous system (CNS) features can include encephalopathy (15 affected individuals), gross motor delay (15), feeding difficulties (14), seizures (12), and sensorineural hearing loss (11). Other less frequently reported CNS manifestations include cerebral atrophy, and generalized central hypomyelination.
Given the multisystem involvement, most infants with the encephalomyopathic phenotype have faltering growth and a poor prognosis.
RRM2B Autosomal Dominant Progressive External Ophthalmoplegia
RRM2B autosomal dominant progressive external ophthalmoplegia (adPEO) is characterized by a slowly progressive loss of function of the muscles that move the eye and retract the eyelid. The phenotype is a mild myopathy with proximal muscle weakness, bulbar dysfunction, and fatigue (see ) [Shaibani et al 2009, Fratter et al 2011, Pitceathly et al 2012, Sommerville et al 2014]. Mean age of disease onset is 46 years.
Table 4.
Select Features of RRM2B Autosomal Dominant Progressive External Ophthalmoplegia
View in own window
| Feature | # of Persons 1 w/Feature (%) |
|---|
|
Ophthalmologic
|
PEO
| 42 (100%) |
|
Ptosis
| 40 (95%) |
|
Neuromuscular
|
Myopathy
| 37 (88%) |
|
Exercise intolerance
| 33 (79%) |
|
Central nervous system
|
Cerebellar ataxia
| 35 (83%) |
|
Sensorineural hearing loss
| 26 (62%) |
|
Cognitive dysfunction
| 24 (57%) |
|
Bulbar dysfunction
| 9 (21%) |
|
Mood disturbance
| 20 (48%) |
|
Low body mass index
| 9 (21%) |
|
Gastrointestinal dysmotility
| 6 (14%) |
PEO = progressive external ophthalmoplegia
- 1.
The first individuals reported were from two large, unrelated families with autosomal dominant PEO [Tyynismaa et al 2009]. From published cases, the cardinal features of RRM2B adPEO are PEO and ptosis. Other commonly reported findings include myopathy, exercise intolerance, cerebellar ataxia, sensorineural hearing loss, cognitive dysfunction, mood disturbance, bulbar dysfunction and low body mass index. These features may have variable age of onset and severity.