Pelizaeus-Merzbacher-Like Disease 1
Synonyms: Hypomyelinating Leukodystrophy 2 (HLD2), PMLD1
Norah Nahhas, MD, Alex Conant, BS, Jennifer Orthmann-Murphy, MD, PhD, Adeline Vanderver, MD, and Grace Hobson, PhD.
Author Information and AffiliationsInitial Posting: December 21, 2017; Last Revision: January 17, 2019.
Estimated reading time: 17 minutes
Summary
Clinical characteristics.
Pelizaeus-Merzbacher-like disease 1 (PMLD1) is a slowly progressive leukodystrophy that typically presents during the neonatal or early-infantile period with nystagmus, commonly associated with hypotonia, delayed acquisition of motor milestones, speech delay, and dysarthria. Over time the hypotonia typically evolves into spasticity that affects the ability to walk and communicate. Cerebellar signs (gait ataxia, dysmetria, intention tremor, head titubation, and dysdiadochokinesia) frequently manifest during childhood. Some individuals develop extrapyramidal movement abnormalities (choreoathetosis and dystonia). Hearing loss and optic atrophy are observed in rare cases. Motor impairments can lead to swallowing difficulty and orthopedic complications, including hip dislocation and scoliosis. Most individuals have normal cognitive skills or mild intellectual disability – which, however, can be difficult to evaluate in the context of profound motor impairment.
Diagnosis/testing.
The diagnosis of PMLD1 is established in a proband with suggestive clinical and neuroimaging findings and identification of biallelic pathogenic variants in GJC2 on molecular genetic testing.
Management.
Treatment of manifestations: To date no definite treatment is available; treatment is mainly supportive and includes assuring adequate nutrition and providing standard treatment for developmental delay/cognitive impairment, neurologic complications (spasticity, ataxia, epilepsy, extrapyramidal movement disorders), communication difficulties, hearing loss, and visual impairment.
Surveillance: Routine assessment of growth, weight gain, vision, and hearing. Routine monitoring of disease progression, spine for evidence of scoliosis and hips for evidence of dislocation, and needs related to physical therapy, communication, and swallowing/feeding.
Genetic counseling.
PMLD1 is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier (heterozygote), and a 25% chance of being unaffected and not a carrier. Once the GJC2 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
Pelizaeus-Merzbacher-like disease 1 (PMLD1) should be suspected in individuals with the following classic clinical and neuroimaging findings:
Clinical findings
Nystagmus that typically presents during the neonatal period or early infancy
Mainly motor developmental delay and central hypotonia during infancy
Signs of upper motor neuron dysfunction (including spasticity, brisk deep tendon reflexes, and Babinski sign) usually affecting the lower limbs more than the upper limbs
Gait ataxia and other cerebellar signs
Mild choreiform movements and dystonia of the extremities that can become severe and disabling
Dysarthria and swallowing dysfunction
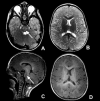
Neuroimaging findings. Findings on brain MRI include the following [Bugiani et al 2006, Steenweg et al 2010, Parikh et al 2015] (see ):
MRI in a male age 36 months with molecularly confirmed PMLD1. Note the diffuse T2-weighted hyperintensity (A and B) and diffuseT1 mild hyperintensity (C and D) consistent with hypomyelination. Involvement of pontine structures is seen in A (axial view) (more...)
Diffuse homogeneous hyperintense T2-weighted signal that affects the white matter of the cerebrum and cerebellum
Involvement of the corticospinal tracts with abnormal T
2-weighted signal extending into the brain stem resulting in extensive brain stem involvement not typically seen in Pelizaeus-Merzbacher disease (see
Differential Diagnosis)
Thin corpus callosum in older children
Brain atrophy and ventricular dilatation as a consequence of white matter loss without specific cerebellar atrophy [
Orthmann-Murphy et al 2009]
Relative preservation of deep gray nuclei and the thalami
Establishing the Diagnosis
The diagnosis of PMLD1 is established in a proband with suggestive clinical and neuroimaging findings and identification of biallelic pathogenic variants in GJC2 on molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel or single-gene testing) and genomic testing (comprehensive genomic sequencing) depending on the phenotype.
Gene-targeted testing requires the clinician to determine which gene(s) are likely involved, whereas genomic testing may not. Because the phenotype of PMLD1 is broad, children with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a mild phenotype indistinguishable from many other inherited leukodystrophies are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the clinical findings and brain MRI findings suggest the diagnosis of a PMLD1, molecular genetic testing approaches can include single-gene testing and use of a multigene panel:
A multigene panel that includes
GJC2 and other genes of interest (see
Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see
Table 1).
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Option 2
When the phenotype is indistinguishable from many other inherited leukodystrophies, molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) or comprehensive
genomic testing (when available). Comprehensive genomic testing includes exome sequencing and genome sequencing.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Pelizaeus-Merzbacher-Like Disease 1
View in own window
| Gene 1 | Method | Proportion of Probands with Pathogenic Variants 2 Detectable by Method |
|---|
|
GJC2
| Sequence analysis including promoter regions (first GJC2 noncoding exon) 3 | 53/79 4, 5 |
| Gene-targeted deletion/duplication analysis 6 | 26/79 4, 5 |
- 1.
- 2.
- 3.
- 4.
Uhlenberg et al [2004], Bugiani et al [2006], Wolf et al [2007], Henneke et al [2008], Orthmann-Murphy et al [2009], Wang et al [2010], Zittel et al [2012], Al-Yahyaee et al [2013], Biancheri et al [2013], Shimojima et al [2013], Abrams et al [2014]
- 5.
- 6.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
Clinical Characteristics
Clinical Description
Pelizaeus-Merzbacher-like disease 1 (PMLD1) is a slowly progressive leukodystrophy that typically presents in the neonatal period or early infancy with nystagmus, often complicated by hypotonia and developmental delay. Over time the hypotonia may evolve into spasticity, and extrapyramidal movement abnormalities may emerge. Older children often manifest significant motor impairments that can also effect communication. Cognition is relatively preserved. The following detailed description of clinical manifestations is based on findings in individuals with a molecularly proven diagnosis [Uhlenberg et al 2004, Bugiani et al 2006, Wolf et al 2007, Henneke et al 2008, Orthmann-Murphy et al 2009, Wang et al 2010, Zittel et al 2012, Al-Yahyaee et al 2013, Biancheri et al 2013, Shimojima et al 2013, Abrams et al 2014].
Nystagmus (either rotatory or horizontal) appears in early infancy and is not present in all individuals.
During later infancy, signs of central hypotonia and delayed acquisition of motor milestones become more apparent, with most children having speech delay and dysarthria as well.
Over time, progressive pyramidal tract involvement (manifest as spasticity, brisk deep tendon reflexes, and bilateral Babinski signs) affects the ability to walk. The lower limbs are often more involved than in the upper limbs. Most affected children become wheelchair dependent in their first decade.
Cerebellar signs including gait ataxia, dysmetria, intention tremor, head titubation, and dysdiadochokinesia frequently manifest during childhood.
Some develop extrapyramidal movement disorders (choreoathetosis and dystonia), which may contribute to the functional disability.
Motor impairments can lead to swallowing difficulty and orthopedic complications, including hip dislocation and scoliosis.
Cognitive function is relatively preserved: Most individuals have normal cognitive skills or mild intellectual disability that may be difficult to evaluate in the context of profound motor impairment. Dysarthria may severely impair communication in adolescents and young adults.
Other less common findings:
Onset is typically in infancy. Although connatal onset is thought to be very rare, one neonate with congenital nystagmus and severe neurologic impairment has been reported [Biancheri et al 2013]. Brain MRI revealed extensive white matter involvement and abnormal cervical spine white matter.
Neurophysiologic findings [Henneke et al 2010]
The following can be normal or delayed:
Electromyogram is usually normal.
Electroencephalography shows nonspecific findings or occasionally (multi)focal epileptiform activity.
Prevalence
The disease prevalence is not known.
Differential Diagnosis
Pelizaeus-Merzbacher disease
(PMD) is an X-linked disorder caused by a hemizygous pathogenic variant in PLP1. The clinical presentation and radiologic appearance are similar to PMLD1. Most individuals present with neonatal nystagmus, hypotonia, global developmental delay, spasticity, gait ataxia, and choreoathetosis. Diffuse hypomyelination on brain MRI is observed in most individuals. Note that MRI evidence of brain stem involvement is more characteristic of PMLD1 than PMD. Both PMD and PMLD1 can present as an isolated spastic paraparesis; see Genetically Related Disorders.
Hypomyelination with atrophy of the basal ganglia and cerebellum (see TUBB4A-Related Leukodystrophy) is caused by a heterozygous pathogenic variant in TUBB4A; affected individuals are typically simplex cases (i.e., a single occurrence in a family). Presentation is generally during infancy or early childhood with findings that overlap with PMLD1: psychomotor developmental delay, pyramidal signs, cerebellar signs, gait ataxia, extrapyramidal symptoms, and dysarthria. MRI demonstrates diffuse hypomyelination of the white matter typically associated with basal ganglia and cerebellar atrophy [Simons et al 2013].
4H syndrome (hypomyelination, hypodontia, and hypogonadotropic hypogonadism syndrome) (see POLR3-Related Leukodystrophy) is an autosomal recessive disorder caused by biallelic pathogenic variants in POLR1C, POLR3A, and POLR3B. 4H syndrome presents with a combination of motor findings (spasticity, gait ataxia and cerebellar tremor, extrapyramidal movement disorders, and generally mild spasticity) that are similar to those of PMLD1. Additional findings are abnormal dentition, severe myopia, and hypogonadotropic hypogonadism, which are not observed in PMLD1.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Pelizaeus-Merzbacher-like disease 1 (PMLD1), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis of Pelizaeus-Merzbacher-Like Disease 1
View in own window
| Organ System | Evaluation | Comment |
|---|
|
Eyes
| Ophthalmology | Assessment for optic atrophy & nystagmus |
|
ENT
| Audiology | Assessment for sensorineural hearing loss |
|
Gastrointestinal
| Consultation w/gastroenterologist | Consideration of swallowing study to assess for swallowing dysfunction & mgmt of constipation & gastroesophageal reflux |
|
Musculoskeletal
| Referral to an orthopedic surgeon for tone mgmt or orthopedic complications as indicated | Exam to assess for evidence of hip dislocation, joint contractures, & scoliosis |
|
Neurologic
| Consultation w/pediatric neurologist | Eval of movement disorders, tone, & seizures |
Miscellaneous/
Other
| Consultation w/nutritionist | Assessment of nutritional status & needs |
| Consultation w/developmental specialist | Assessment of developmental level & needs for supportive therapies |
| Consultation w/clinical geneticist &/or genetic counselor | Eval of underlying diagnosis & familial recurrence risk |
| Consultation w/rehab specialist | Assessment of functional disability & equipment needs/adjustments |
Treatment of Manifestations
There is no curative treatment for PMLD1; measures that can be taken to improve the individual's quality of life are summarized in Table 4 [Van Haren et al 2015].
Table 4.
Treatment of Manifestations in Individuals with Pelizaeus-Merzbacher-Like Disease 1
View in own window
| Manifestation | Treatment | Considerations/Other |
|---|
Developmental
delay & cognitive
dysfunction
| Accommodations in special classroom setting or w/aide | Recommendations from pediatric neurologist may be necessary to achieve maximum intellectual & functional abilities. |
|
Spasticity
| Oral GABA agonists (e.g., baclofen, diazepam) | For more focal spasticity, consider intramuscular injection of botulinum toxin. |
| Physical therapy | |
| Use of equipment such as walker & wheelchair | |
|
Dystonia
| When associated w/spasticity, mgmt of dystonia w/baclofen or intramuscular botulinum toxin;
trihexyphenidyl or tetrabenazine potentially helpful | In many cases, dystonia is refractory to medical mgmt. |
Scoliosis &
joint dislocation
| Mgmt or surgical intervention by orthopedist | |
Swallowing
dysfunction
| Consider swallowing eval & feeding therapy | Affected individuals are at risk of aspiration. |
| Nutrition plan & possible supportive feeding device to avoid malnutrition | |
|
Dysarthria
| Consider speech therapy to improve communication abilities | Augmentative communication approaches are often necessary. |
|
Seizures
| Standard antiepileptic drug therapy | |
|
Hearing loss
| Standard approaches to hearing loss incl augmentative communication approaches; no evidence exists for cochlear implants in this context | See Hereditary Hearing Loss and Deafness Overview. |
|
Optic atrophy
| Supportive approaches for the vision-impaired individual | |
Prevention of Secondary Complications
Table 5.
Prevention of Secondary Complications in Individuals with Pelizaeus-Merzbacher-Like Disease 1
View in own window
| Complication | Preventive Measure | Considerations/Other |
|---|
|
Constipation
| Dietary management, laxatives, stool softeners | |
|
Bone health
| Regular monitoring of serum vitamin D & calcium levels | If osteopenia is documented, consult w/bone health clinic to consider measures to avoid fracture. |
Community-
acquired
pneumonia
| Good hand hygiene; influenza & pneumococcal vaccines | Some affected individuals are reported to have deterioration of neurologic function w/febrile illness & infection. 1 |
Psychosocial
consequences
in caregiver
| Involvement of social worker | |
Surveillance
Table 6.
Recommended Surveillance for Individuals with Pelizaeus-Merzbacher-Like Disease 1
View in own window
| Organ System | Evaluation | Frequency/Comment |
|---|
|
Constitutional
| Monitoring of general health & growth; immunizations | Annually |
|
Eyes
| Ophthalmology | Biannually unless symptoms develop |
|
Neurologic
| Pediatric neurology assessment for disease progression, symptom control, & review of medications | Annually |
Miscellaneous/
Other
| Physiatrist & physical/occupational therapy assessments for functional capacity & equipment needs | Annually w/more frequent treatment visits once eval completed |
| Assessment of communication abilities | Annually |
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Pelizaeus-Merzbacher-like disease 1 (PMLD1) is inherited in an autosomal recessive manner.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the GJC2 pathogenic variants in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the GJC2 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
No specific resources for Pelizaeus-Merzbacher-Like Disease 1 have been identified by GeneReviews staff.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Pelizaeus-Merzbacher-Like Disease 1: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Gene structure.
GJC2 (previously known as GJA12) comprises two exons. The first exon is noncoding and contains the binding site for transcriptional factors; the second contains part of the 5′ UTR, the coding sequence, and the 3′ UTR. See Table A, Gene for a detailed summary of gene and protein information.
Pathogenic variants. See Table 7.
Table 7.
GJC2 Pathogenic Variants Discussed in This GeneReview
View in own window
DNA Nucleotide Change
(Alias 1) | Predicted Protein Change | Reference Sequences |
|---|
c.-167A>G
(-7899A>G relative to initiation codon) 2 | NA |
NM_020435.3
|
c.-170A>G
(-7902A>G relative to initiation codon) 3 | NA |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
- 2.
- 3.
Normal gene product.
GJC2 encodes the gap junction gamma-2 protein, a 439-amino acid protein referred to as connexin 47 (Cx47), which is a member of the connexin family of highly conserved integral membrane proteins [Schlierf et al 2006, Wang et al 2010, Gotoh et al 2014]. Cx47 is highly expressed in the brain and spinal cord, specifically in oligodendrocytes [Odermatt et al 2003, Menichella et al 2006].
Connexins form complex intercellular channels called gap junctions between adjacent cell membranes [Willecke et al 2002]. Gap junction channels enable coupling between adjacent oligodendrocytes, as well as with astrocytes, forming a glial syncytium [Rash et al 2001, Maglione et al 2010, Wasseff & Scherer 2011]. Because astrocytes express connexin proteins (Cx43 and Cx30) that differ from those of oligodendrocytes (Cx47 and Cx32), the gap junction channels between astrocytes and oligodendrocytes are necessarily heterotypic (i.e., Cx47/Cx43 and Cx32/Cx30), whereas the gap junction channels between adjacent oligodendrocytes are likely homotypic (i.e., Cx47/Cx47 and Cx32/Cx32) [Orthmann-Murphy et al 2007].
Gap junctions enable the transfer of ions and small molecules between adjacent cells. The function of oligodendrocyte/astrocyte coupling in particular is unknown but appears to be critical for proper myelin formation and maintenance.
Mice that do not express Cx47 have a normal phenotype, but have evidence of disrupted myelin (vacuole formation) on pathology. Mice deficient for both oligodendrocyte connexins (Cx47 and Cx32) exhibit a severe phenotype, characterized by seizures, tremor, and development of widespread vacuolated myelin on pathology [Menichella et al 2006].
Abnormal gene product. Pelizaeus-Merzbacher-like disease 1 (PMLD1)-associated GJC2 pathogenic variants result in loss of function of Cx47 [Uhlenberg et al 2004, Diekmann et al 2010, Kim et al 2013] which either fails to properly localize to the cell surface or mislocalizes to the endoplasmic reticulum [Orthmann-Murphy et al 2007]. The proteins of selected missense pathogenic variants had apparently normal location and distribution in gap-junction-deficient model cells, but showed no electric coupling in either the homopolymeric gap junction channel (Cx47/Cx47) or the heteropolymeric gap junction channel (Cx47/Cx43) [Kim et al 2013].
In a mouse model system, Tress et al [2011] showed that rather than dysfunctional Cx47, the critical outcome of PMLD1-associated GJC2 pathogenic variants was a decreased number of cells coupled within glial networks.
References
Literature Cited
Abrams CK, Scherer SS, Flores-Obando R, Freidin MM, Wong S, Lamantea E, Farina L, Scaioli V, Pareyson D, Salsano E. A new mutation in GJC2 associated with subclinical leukodystrophy.
J Neurol. 2014;261:1929–38. [
PMC free article: PMC4301586] [
PubMed: 25059390]
Al-Yahyaee SA, Al-Kindi M, Jonghe PD, Al-Asmi A, Al-Futaisi A, Vriendt ED, Deconinck T, Chand P. Pelizaeus-Merzbacher-like disease in a family with variable phenotype and a novel splicing GJC2 mutation.
J Child Neurol. 2013;28:1467–73. [
PubMed: 23143715]
Biancheri R, Rosano C, Denegri L, Lamantea E, Pinto F, Lanza F, Severino M, Filocamo M. Expanded spectrum of Pelizaeus-Merzbacher-like disease: literature revision and description of a novel GJC2 mutation in an unusually severe form.
Eur J Hum Genet. 2013;21:34–9. [
PMC free article: PMC3533259] [
PubMed: 22669416]
Bugiani M, Al Shahwan S, Lamantea E, Bizzi A, Bakhsh E, Moroni I, Balestrini MR, Uziel G, Zeviani M. GJA12 mutations in children with recessive hypomyelinating leukoencephalopathy.
Neurology. 2006;67:273–9. [
PubMed: 16707726]
Combes P, Kammoun N, Monnier A, Gonthier-Guéret C, Giraud G, Bertini E, Chahnez T, Fakhfakh F, Boespflug-Tanguy O, Vaurs-Barrière C. Relevance of GJC2 promoter mutation in Pelizaeus-Merzbacher-like disease.
Ann Neurol. 2012;71:146–8. [
PubMed: 21246605]
Diekmann S, Henneke M, Burckhardt BC, Gärtner J. Pelizaeus-Merzbacher-like disease is caused not only by a loss of connexin47 function but also by a hemichannel dysfunction.
Eur J Hum Genet. 2010;18:985–92. [
PMC free article: PMC2987409] [
PubMed: 20442743]
Ferrell RE, Baty CJ, Kimak MA, Karlsson JM, Lawrence EC, Franke-Snyder M, Meriney SD, Feingold E, Finegold DN. GJC2 missense mutations cause human lymphedema.
Am J Hum Genet. 2010;86:943–8. [
PMC free article: PMC3032064] [
PubMed: 20537300]
Gotoh L, Inoue K, Helman G, Mora S, Maski K, Soul JS, Bloom M, Evans SH, Goto YI, Caldovic L, Hobson GM, Vanderver A. GJC2 promoter mutations causing Pelizaeus-Merzbacher-like disease.
Mol Genet Metab. 2014;111:393–8. [
PMC free article: PMC4183365] [
PubMed: 24374284]
Henneke M, Combes P, Diekmann S, Bertini E, Brockmann K, Burlina AP, Kaiser J, Ohlenbusch A, Plecko B, Rodriguez D, Boespflug-Tanguy O, Gärtner J. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease.
Neurology. 2008;70:748–54. [
PubMed: 18094336]
Henneke M, Gegner S, Hahn A, Plecko-Startinig B, Weschke B, Gärtner J, Brockmann K. Clinical neurophysiology in GJA12-related hypomyelination vs Pelizaeus-Merzbacher disease.
Neurology. 2010;74:1785–9. [
PubMed: 20513814]
Kammoun Jellouli N, Salem IH, Ellouz E, Louhichi N, Tlili A, Kammoun F, Triki C, Fakhfakh F, et al. Molecular confirmation of founder mutation c.-167A>G in Tunisian patients with PMLD disease.
Gene. 2013;513:233–8. [
PubMed: 23142375]
Kim MS, Gloor GB, Bai D. The distribution and functional properties of Pelizaeus-Merzbacher-like disease-linked Cx47 mutations on Cx47/Cx47 homotypic and Cx47/Cx43 heterotypic gap junctions.
Biochem J. 2013;452:249–58. [
PubMed: 23544880]
Maglione M, Tress O, Haas B, Karram K, Trotter J, Willecke K, Kettenmann H. Oligodendrocytes in mouse corpus callosum are coupled via gap junction channels formed by connexin47 and connexin32.
Glia. 2010;58:1104–17. [
PubMed: 20468052]
Menichella DM, Majdan M, Awatramani R, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Genetic and physiological evidence that oligodendrocyte gap junctions contribute to spatial buffering of potassium released during neuronal activity.
J Neurosci. 2006;26:10984–91. [
PMC free article: PMC6674647] [
PubMed: 17065440]
Meyer E, Kurian MA, Morgan NV, McNeill A, Pasha S, Tee L, Younis R, Norman A, van der Knaap MS, Wassmer E, Trembath RC, Brueton L, Maher ER. Promoter mutation is a common variant in GJC2-associated Pelizaeus-Merzbacher-like disease.
Mol Genet Metab. 2011;104:637–43. [
PubMed: 21959080]
Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Büssow H, Schilling K, Steinhäuser C, Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS.
J Neurosci. 2003;23:4549–59. [
PMC free article: PMC6740816] [
PubMed: 12805295]
Orthmann-Murphy JL, Freidin M, Fischer E, Scherer SS, Abrams CK. Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins.
J Neurosci. 2007;27:13949–57. [
PMC free article: PMC6673504] [
PubMed: 18094232]
Orthmann-Murphy JL, Salsano E, Abrams CK, Bizzi A, Uziel G, Freidin MM, Lamantea E, Zeviani M, Scherer SS, Pareyson D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations.
Brain. 2009;132:426–38. [
PMC free article: PMC2640216] [
PubMed: 19056803]
Osaka H, Hamanoue H, Yamamoto R, Nezu A, Sasaki M, Saitsu H, Kurosawa K, Shimbo H, Matsumoto N, Inoue K. Disrupted SOX10 regulation of GJC2 transcription causes Pelizaeus-Merzbacher-like disease.
Ann Neurol. 2010;68:250–4. [
PubMed: 20695017]
Parikh S, Bernard G, Leventer RJ, van der Knaap MS, van Hove J, Pizzino A, McNeill NH, Helman G, Simons C, Schmidt JL, Rizzo WB, Patterson MC, Taft RJ, Vanderver A, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies.
Mol Genet Metab. 2015;114:501–15. [
PMC free article: PMC4390485] [
PubMed: 25655951]
Rash JE, Yasumura T, Dudek FE, Nagy JI. Cell-specific expression of connexins and evidence of restricted gap junctional coupling between glial cells and between neurons.
J Neurosci. 2001;21:1983–2000. [
PMC free article: PMC1804287] [
PubMed: 11245683]
Schlierf B, Werner T, Glaser G, Wegner M. Expression of connexin47 in oligodendrocytes is regulated by the Sox10 transcription factor.
J Mol Biol. 2006;361:11–21. [
PubMed: 16822525]
Shimojima K, Tanaka R, Shimada S, Sangu N, Nakayama J, Iwasaki N, Yamamoto T. A novel homozygous mutation of GJC2 derived from maternal uniparental disomy in a female patient with Pelizaeus-Merzbacher-like disease.
J Neurol Sci. 2013;330:123–6. [
PubMed: 23684670]
Simons C, Wolf NI, McNeil N, Caldovic L, Devaney JM, Takanohashi A, Crawford J, Ru K, Grimmond SM, Miller D, Tonduti D, Schmidt JL, Chudnow RS, van Coster R, Lagae L, Kisler J, Sperner J, van der Knaap MS, Schiffmann R, Taft RJ, Vanderver A. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum.
Am J Hum Genet. 2013;92:767–73. [
PMC free article: PMC3644625] [
PubMed: 23582646]
Steenweg ME, Vanderver A, Blaser S, Bizzi A, de Koning TJ, Mancini GM, van Wieringen WN, Barkhof F, Wolf NI, van der Knaap MS. Magnetic resonance imaging pattern recognition in hypomyelinating disorders.
Brain. 2010;133:2971–82. [
PMC free article: PMC3589901] [
PubMed: 20881161]
Tress O, Maglione M, Zlomuzica A, May D, Dicke N, Degen J, Dere E, Kettenmann H, Hartmann D, Willecke K. Pathologic and phenotypic alterations in a mouse expressing a connexin47 missense mutation that causes Pelizaeus-Merzbacher-like disease in humans.
PLoS Genet. 2011;7:e1002146. [
PMC free article: PMC3131295] [
PubMed: 21750683]
Uhlenberg B, Schuelke M, Rüschendorf F, Ruf N, Kaindl AM, Henneke M, Thiele H, Stoltenburg-Didinger G, Aksu F, Topaloğlu H, Nürnberg P, Hübner C, Weschke B, Gärtner J. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease.
Am J Hum Genet. 2004;75:251–60. [
PMC free article: PMC1216059] [
PubMed: 15192806]
Van Haren K, Bonkowsky JL, Bernard G, Murphy JL, Pizzino A, Helman G, Suhr D, Waggoner J, Hobson D, Vanderver A, Patterson MC, et al. Consensus statement on preventive and symptomatic care of leukodystrophy patients.
Mol Genet Metab. 2015;114:516–26. [
PubMed: 25577286]
Wang J, Wang H, Wang Y, Chen T, Wu X, Jiang Y. Two novel gap junction protein alpha 12 gene mutations in two Chinese patients with Pelizaeus-Merzbacher-like disease.
Brain Dev. 2010;32:236–43. [
PubMed: 19423250]
Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Güldenagel M, Deutsch U, Söhl G. Structural and functional diversity of connexin genes in the mouse and human genome.
Biol Chem. 2002;383:725–37. [
PubMed: 12108537]
Wolf NI, Cundall M, Rutland P, Rosser E, Surtees R, Benton S, Chong WK, Malcolm S, Ebinger F, Bitner-Glindzicz M, Woodward KJ. Frameshift mutation in GJA12 leading to nystagmus, spastic ataxia and CNS dys-/demyelination.
Neurogenetics. 2007;8:39–44. [
PubMed: 16969684]
Zittel S, Nickel M, Wolf NI, Uyanik G, Gläser D, Ganos C, Gerloff C, Münchau A, Kohlschütter A. "Pelizaeus-Merzbacher-like disease" presenting as complicated hereditary spastic paraplegia.
J Neurol. 2012;259:2498–500. [
PubMed: 22833003]
Chapter Notes
Revision History
17 January 2019 (av) Revision: edited
21 December 2017 (bp) Review posted live
26 July 2016 (av) Original submission