Molecular Pathogenesis
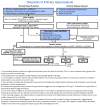
When alanine:glyoxylate aminotransferase (AGT) enzymatic activity is deficient, the substrate glyoxylate accumulates and is converted to oxalate by glycolate oxidase in peroxisomes or in the cytosol by lactate dehydrogenase [Holmes & Assimos 1998, Danpure 2001]. Oxalate forms insoluble calcium oxalate salts that the body cannot readily eliminate. In the most common pathogenic allele c.508G>A (p.Gly170Arg), the AGT enzyme is mistargeted to the mitochondria rather than to the peroxisomes, where the substrate is localized. The mistargeted AGT enzyme retains substantial enzymatic activity but has no contact with its substrate, and thus the functional consequences are the same as for other pathogenic variants that result in no enzymatic activity. Mistargeting and high residual activity are seen in heterozygotes and homozygotes for the p.Gly170Arg variant [Danpure 1998, Danpure 2001]. (See "Major" and "minor" AGXT alleles.)
Gene structure.
AGXT (NM_000030.2) spans approximately 10 kb and comprises 11 exons. For a detailed summary of gene and protein information, see Table A, Gene.
"Major" and "minor" AGXT alleles. Two common normal alleles of AGXT are known: the most frequent is termed the "major allele" (80% frequency in individuals of European origin) and the less frequent the "minor allele" (20% frequency in individuals of European origin, 2% in Japanese, 3% in South African blacks) [Danpure et al 1994b, Coulter-Mackie et al 2003].
The major allele is the haplotype defined by the transcript variant NM_000030.2, while the minor allele haplotype has two single amino acid substitutions, p.Pro11Leu and p.Ile340Met, among other genomic changes in strong disequilibrium [reviewed by Pey et al 2013].
In the minor allele, the only normal allelic variant of functional significance is p.Pro11Leu, which alters the amino acid sequence and creates a cryptic N-terminal mitochondrial targeting sequence [Purdue et al 1991, Fargue et al 2013a]. The mitochondrial targeting sequence of the minor allele is functionally ineffective due to protein conformation; only about 5% of AGT encoded by the minor allele is found in the mitochondria (see Pey et al [2013] and references therein). However, certain pathogenic variants on the minor allele disrupt AGT folding, thereby unmasking the mitochondrial targeting signal, resulting in efficient mislocalization of AGT. Therefore, when in cis configuration the minor allele acts synergistically with some pathogenic variants.
Other AGXT allelic haplotypes have been reported [Danpure et al 1994a, Tarn et al 1997, Coulter-Mackie et al 2003]. These normal variants may be useful intragenic markers for determination of phase of pathogenic variants.
Pathogenic variants. More than 190 AGXT pathogenic variants have been documented [Williams et al 2009, Hopp et al 2015]. Missense variants make up approximately 67% of PH1-causing variants [Hopp et al 2015].
There are four common pathogenic variants and a few with ethnic associations. The other pathogenic variants have been detected just once or in small number of families.
The four common pathogenic variants p.Gly170Arg, p.Phe152Ile, and p.Ile244Thr (which occur on the minor allele haplotype) and c.33dupC (p.Lys12GlnfsTer156) (on the major allele haplotype) together account for more than 65% of PH1-causing alleles.
An
AGXT minor
allele haplotype may exacerbate at least one copy of the
AGXT minor allele with one of the following common pathogenic variants in
cis configuration:
On the
AGXT major
allele haplotype:
Most missense variants have not had specific biochemical phenotypes associated with them other than degradation and loss of enzymatic activity [Coulter-Mackie & Lian 2006]. The pathogenic mechanism of a few of the rarer missense variants is known:
p.Gly82Glu (on the major
allele) apparently prevents binding of the essential cofactor pyridoxine (vitamin B
6) [
Lumb & Danpure 2000,
Cellini et al 2007]. Rather than an intrinsic inability to bind PLP, this is now thought to be due to an altered binding state of PLP and the AGT-PMP intermediate [
Cellini et al 2011].
In addition to the missense variants, splicing and nonsense variants and several small insertions and deletions are known (see databases in Table A; and Coulter-Mackie & Rumsby [2004], Williams et al [2009]).
Large documented deletions that are typically detected by gene-targeted deletion/duplication analysis include:
Table 5.
AGXT Variants Discussed in This GeneReview
View in own window
| Variant Classification | DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
|
Defines "minor AGXT allele"
| c.32C>T | p.Pro11Leu 1 |
NM_000030.2
NP_000021.1
|
| c.1020A>G | p.Ile340Met 1 |
|
Pathogenic
| c.33dupC | p.Lys12GlnfsTer156 |
| c.121G>A | p.Gly41Arg |
| c.245G>A | p.Gly82Glu |
| c.454T>A | p.Phe152Ile |
| c.466G>A | p.Gly156Arg |
| c.508G>A | p.Gly170Arg |
| c.560C>T | p.Ser187Phe |
| c.613T>C | p.Ser205Pro |
| c.697C>T | p.Arg233Cys |
| c.731T>C | p.Ile244Thr |
| c.738G>A | p.Trp246Ter |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Two of the variants that define the haplotype of the minor AGXT allele
Normal gene product. The mRNA (NM_000030.2) encodes a 392-amino-acid protein with a molecular mass of 43 kd. In humans, AGT is synthesized mainly in the liver and is normally located exclusively in the peroxisomes [Danpure 2001]. The enzyme is translated in the cytosol and transported into the peroxisomes. A C-terminal peroxisomal targeting signal is recognized by the peroxisomal receptor, Pex5p, allowing translocation into the peroxisome [Fodor et al 2012]. AGT is a key enzyme in the detoxification of glyoxylate, converting glyoxylate to glycine [Holmes & Assimos 1998, Danpure 2001]. In humans, glyoxylate is produced in the peroxisomes. PLP is an essential cofactor for AGT activity. The PLP site in AGT lies in a highly conserved amino acid sequence and is critical in the catalytic activity of the enzyme. The crystal structure of the normal AGT protein has been determined [Zhang et al 2003], allowing a delineation of the active site and the dimerization interface.
Note that AGXT encodes AGT (EC 2.6.1.44), whose activity is largely confined to peroxisomes in the liver. This protein also shows serine:pyruvate aminotransferase activity (SPT; EC 2.6.1.51) (OMIM 604285). AGT and SPT are two separate enzymatic activities on the same protein coded by AGXT. AGT is the major activity; when it is deficient, PH1 results.
Abnormal gene product. Approximately 50% of all individuals with PH1 show no AGT enzymatic activity and produce no immunologically detectable AGT protein.
Pathogenic variants resulting in nonsense codons, frameshifts, partial gene deletion, or splice junction variants are usually predicted to result in little or no functional protein.
Approximately 30% of affected individuals display a high level of residual AGT activity. Most of these individuals exhibit the mistargeting defect in which an otherwise functional AGT enzyme is synthesized in adequate amounts but is mislocalized to mitochondria instead of peroxisomes, where it is normally found and where the substrate glyoxylate remains. These individuals have classic PH1 despite the residual AGT enzymatic activity.
Pathogenic variants, apart from mistargeting ones, that cause true partial enzymatic activity appear to be rare but may be associated with late-onset or mild disease.
With many genetic diseases, it is now clear that a common consequence of pathogenic missense variants is protein misfolding and subsequent elimination by intracellular quality-control processes [Waters 2001]. This biologic instability of protein carrying a missense change has been documented in p.Ser205Pro [Nishiyama et al 1993] and with a variety of other pathogenic missense variants in AGT [Coulter-Mackie & Lian 2006, Coulter-Mackie & Lian 2008, Hopper et al 2008]. Biochemical studies of a broad range of individual pathogenic variants has revealed a diversity of effects both structural and functional, such as altered PLP or substrate binding, thermostability changes, altered interactions with peroxisomal targeting components, and misfolding with subsequent aggregation or degradation [Cellini et al 2007, Cellini et al 2012, Fodor et al 2012, Oppici et al 2012, Mesa-Torres et al 2013, Pey et al 2013]. The findings may provide clues to potential therapeutic strategies as well as clues to the response to PLP. For instance, p.Gly82Glu has been demonstrated to have a reduced affinity for the pyridoxal phosphate cofactor [Cellini et al 2007].
It has been reported recently that the protein encoded by four pathogenic variants that occur on the minor allele (p.Gly170Arg, p.Ile244Arg, p.Phe152Ile, and p.Gly41Arg) undergo mistargeting [Fargue et al 2013a]. It is speculated that this is a common feature of variants occurring on the minor allele. Variant AGT proteins with p.Gly170Arg, p.Ile244Thr, p.Ile244Arg, or p.Phe162Ile are able to dimerize and are catalytically active, although functionally ineffective if located in the mitochondria. The variant p.Gly41Arg tends to aggregate and is inactive.
The effect of a given pathogenic missense variant may be exacerbated if it occurs on the AGT minor allele. In vitro studies have shown increased stability and enzymatic activity for some pathogenic variants when expressed on a major allele haplotype compared to a minor allele [Williams & Rumsby 2007, Coulter-Mackie & Lian 2008, Williams et al 2009]. It has been speculated that some missense variants found on the minor allele in association with PH1 may not cause disease if they occurred on the major allele. However, some missense variants (e.g., p.Gly41Arg) found on both major and minor alleles cause disease in both instances.
The determination of a crystal structure for AGT [Zhang et al 2003] has permitted the rationalization of the functional consequences of selected missense pathogenic protein variants: p.Gly170Arg (mitochondrial mistargeting), p.Gly82Glu (prevention of cofactor binding), p.Gly41Arg (protein aggregation) [Danpure 2004, Danpure & Rumsby 2004, Danpure 2006], p.Gly47Arg (affects dimerization), and p.Ser81Leu (no effect on dimerization) [Robbiano et al 2010]. See Pathogenic variants for additional descriptions of abnormal proteins.