Clinical Description
NKX2-1-related disorders include benign hereditary chorea (BHC) and choreoathetosis, congenital hypothyroidism, and neonatal respiratory distress syndrome (collectively known as brain-lung-thyroid syndrome). Individuals can have one or more of several features associated with this spectrum (see ). To date, more than 120 individuals have been identified with a pathogenic variant in NKX2-1 [Gras et al 2012, Peall & Kurian 2015, Kharbanda et al 2017]. In a review of 46 affected individuals, Carré et al [2009] found that 50% had the full brain-lung-thyroid syndrome, 30% had only brain and thyroid involvement, and 13% had isolated chorea. The following description of the phenotypic features associated with this condition is based on these reports.
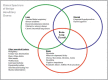
Phenotypic spectrum of NKX2-1-related disorders. NKX2-1-related disorders may manifest as abnormalities in a single organ system or as any combination of brain, thyroid, and lung involvement. "Brain-lung-thyroid syndrome" refers to involvement of all (more...)
Neurologic Manifestations
Chorea, an involuntary, random, irregular, jerk-like, and continuous movement, is a classic early finding in BHC and other NKX2-1-related disorders. The onset of chorea generally occurs during one of the following time periods:
Chorea progresses into the second decade, after which it remains static or may spontaneously remit [Kleiner-Fisman et al 2003, Peall & Kurian 2015]. Although originally referred to as "benign" hereditary chorea, the neurologic manifestations of NKX2-1-related disorders can be quite disabling, due to chorea as well as gait and balance abnormalities, hypotonia, and a variety of other motor and non-motor manifestations [Parnes et al 2018].
Chorea typically involves all body regions (e.g., face, tongue, neck, trunk, limbs) and may be associated with motor and gait abnormalities, possibly secondary to the choreiform movements. The movements are jerk-like and spread randomly from one body part to another; they often worsen with stress and may disappear with sleep. Children with BHC may fall frequently [Kleiner-Fisman et al 2003]. Although affected children may be delayed in walking, persistent gait abnormalities are rare [McMichael et al 2013]. Rosati et al [2015] described two unrelated children presenting with spontaneous falls without loss of consciousness preceding the development of chorea.
The prevalence of chorea in NKX2-1-related disorders is unknown. In one study, all 28 affected individuals from 13 families with a heterozygous NKX2-1 pathogenic variant had chorea and hypotonia [Gras et al 2012]. However, in a retrospective study of 21 individuals with NKX2-1-related disorders presenting with pulmonary dysfunction, at least two unrelated individuals and three members of one family did not have any neurologic symptoms [Hamvas et al 2013].
Other less common neurologic manifestations
Hypotonia, incoordination, and motor delays, which are common during the neonatal period / infancy but rarely reported in childhood/adulthood [
Peall et al 2014,
Veneziano et al 2014]
In one report, two sibs initially diagnosed with ataxic dyskinetic cerebral palsy were later found to have a pathogenic variant in NKX2-1 [McMichael et al 2013].
Neuropsychiatric manifestations including attention-deficit/hyperactivity disorder have been reported [Gras et al 2012]. Although psychiatric disorders are rare in individuals with an NKX2-1 pathogenic variant, schizophrenia [Glik et al 2008], postpartum psychosis [Salvatore et al 2010], and obsessive-compulsive disorder [Peall et al 2014] have been reported.
Neuroimaging is usually normal. Structural brain abnormalities have been rarely reported, including abnormal sella turcica [Krude et al 2002, Balicza et al 2018], agenesis of the corpus callosum [Carré et al 2009], cavum septum pellucidum, and microcephaly [Iwatani et al 2000]. Hypoplastic pallidum and lack of differentiation of medial and lateral components of the pallidum were reported in a single individual, and bilateral pallidal signal hyperintensities on T2-weighted MRI were described in another individual [Kleiner-Fisman & Lang 2007]. An expanding pituitary cyst was reported in two related individuals with a novel NKX2-1 pathogenic variant [Veneziano et al 2014]. Chiari I malformation was also reported in one female at age 24 months [Gonçalves et al 2019].
Single-photon emission computed tomography (SPECT) has demonstrated reduced cerebral blood flow to the basal ganglia bilaterally, and more specifically to the caudate nuclei, in three affected individuals [Uematsu et al 2012]. Subtle abnormalities in presynaptic dopamine transporter function utilizing positron emission tomography (PET) imaging have also been reported in two affected individuals [Konishi et al 2013].
Neuropathology. Autopsies of two individuals with NKX2-1-related disorders did not identify gross or microscopic abnormalities of the brain, but showed reduced number of striatal and neocortical interneurons consistent with a defect in neuronal migration and supporting the theory that these disorders are due to abnormalities in brain development rather than neurodegeneration [Kleiner-Fisman et al 2003].
Pulmonary Manifestations
Pulmonary dysfunction is the second most common manifestation of NKX2-1-related disorders. In a meta-analysis of 29 published reports of NKX2-1-related disorders, up to 49% (61/124) of affected individuals had pulmonary manifestations of varying severity [Gras et al 2012]. Clinical presentation and course vary among affected individuals.
The highest risk for respiratory distress is in the neonatal period. Affected infants often require mechanical ventilation [Carré et al 2009]. Although usually not fatal, NKX2-1-related disorders have resulted in death from respiratory failure in three infants in the immediate postnatal period to date [Maquet et al 2009, Kleinlein et al 2011, Gillett et al 2013]. However, in most infants, respiratory function typically improves over time [Young et al 2013].
As a result of pulmonary involvement, individuals with NKX2-1-related disorders are at increased risk for recurrent pulmonary infections and chronic interstitial lung disease [Carré et al 2009, Inzelberg et al 2011, Peca et al 2011]. High-resolution CT scan of six children with pathogenic variants in NKX2-1 identified ground-glass opacities and pulmonary consolidation [LeMoine et al 2019].
Pulmonary histologic abnormalities. In one restrospective study of individuals with known pulmonary dysfunction and a pathogenic variant in NKX2-1, histologic abnormalities included interstitial widening and pneumocyte hyperplasia, desquamative interstitial pneumonia, accumulation of foamy alveolar macrophages, and pulmonary alveolar proteinosis [Hamvas et al 2013].
Pulmonary carcinoma. The risk for pulmonary carcinoma is increased in young adults (early 20s) with NKX2-1-related disorders [Fernandez et al 2001, Willemsen et al 2005, Glik et al 2008]. Screening for pulmonary carcinoma in adolecence is recommended (see Surveillance).
Thyroid Manifestations
Thyroid dysfunction, which results from thyroid dysgenesis, can present as congenital hypothyroidism (i.e., reduced or absent production of thyroid hormone) or compensated hypothyroidism (i.e., low-to-normal thyroid hormone production with elevated thyroid-stimulating hormone) [Moya et al 2006, Montanelli & Tonacchera 2010, Gras et al 2012]. Of note, thyroid dysgenesis can manifest structurally as thyroid hypoplasia or hemiagenesis (11/31) or complete absence of the thyroid gland (3/31) [Carré et al 2009].
Thyroid dysfunction varies between individuals with NKX2-1-related disorders and within families of multiple affected individuals. In a meta-analysis of 46 individuals reported with NKX2-1-related disorders, 40 had documented overt or subclinical hypothyroidism, and only six had normal thyroid function [Carré et al 2009].
Currently, newborn screening for hypothyroidism is available in most countries and includes measuring levels of thyroid-stimulating hormone with or without measuring thyroxine levels. Of note, congenital hypothyroidism can be the only manifestation of an NKX2-1-related disorder.
Other Features
Other features reported in single individuals or families include the following:
Prognosis and Progression
Life expectancy in individuals with NKX2-1-related disorders is expected to be normal [Fernandez et al 2001].
A retrospective study describing 28 individuals with 13 novel NKX2-1 pathogenic variants over a mean duration of 24.5 years reported a homogeneous progression of neurologic manifestations. Hypotonia is present in the first year of life with or without delays in motor milestones or early chorea. Chorea is generally mild and improves until puberty through early adulthood, when it typically stabilizes. In some individuals, it resolves entirely in adulthood [Gras et al 2012].
There is limited information on the long-term prognosis of pulmonary and thyroid manifestations. Progression is rare and features may improve in adulthood [Gras et al 2012]. Individuals with lung involvement are at risk of respiratory failure in early infancy, as well as recurrent infections and asthma throughout life. Compensated hypothyroidism is typically well controlled in individuals with NKX2-1-related disorders.
Nomenclature
Before the molecular basis was known, the disorder now known to be caused by a heterozygous pathogenic variant in NKX2-1 [Inzelberg et al 2011] was referred to as benign hereditary chorea (BHC) based on the original description of non-progressive familial chorea in a five-generation family [Haerer et al 1967]. The broad phenotypic spectrum associated with pathogenic variants in NKX2-1 (including BHC and a variable combination of lung, thyroid, and neurologic abnormalities) led Willemsen et al [2005] to coin the term "brain-lung-thyroid syndrome." Considering the variable manifestations of individuals with pathogenic variants in NKX2-1, the authors suggest that these disorders be referred to as NKX2-1-related disorders.
Of note, NKX2-1 was previously known as TITF-1; thus, early literature describing the molecular basis of this disorder uses this gene designation [Breedveld et al 2002, Kleiner-Fisman et al 2003, Costa et al 2005, Devos et al 2006, Kleiner-Fisman & Lang 2007, Glik et al 2008].