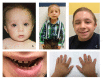
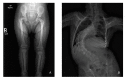
Clinical Description
Microcephalic osteodysplastic primordial dwarfism type II (MOPDII), the most common of the microcephalic primordial dwarfism syndromes, is characterized by extreme short stature and microcephaly along with distinctive facial features. Associated features that differentiate it from other forms of primordial dwarfism and which may necessitate treatment include: abnormal dentition, a slender bone skeletal dysplasia with hip deformity and/or scoliosis, insulin resistance / diabetes mellitus, chronic kidney disease, cardiac malformations, and global vascular disease. The latter includes neurovascular disease such as moyamoya vasculopathy and intracranial aneurysms (which can lead to strokes), coronary artery disease (which can lead to premature myocardial infarctions), and renal vascular disease. Hypertension, which is also common, can have multiple underlying causes given the complex comorbidities [Bober & Jackson 2017, Duker et al 2021].
Anticipated life expectancy is shortened due to the associated comorbidities, which when untreated can lead to early death, predominately in early adulthood (age range 7-41 years) [Duker et al 2021].
More than 150 individuals with a molecularly confirmed diagnosis have been identified [Bober & Jackson 2017].
The following description of the phenotypic features associated with this condition is based on the most recent review of 47 individuals with molecularly confirmed MOPDII [Duker et al 2021].
Table 2.
Features of Microcephalic Osteodysplastic Primordial Dwarfism Type II
View in own window
| Feature | % of Persons
w/Feature | Comment |
|---|
| Extreme pre- & postnatal growth restriction | ~100% | IUGR, severe short stature |
| Microcephaly | ~100% | |
| Skeletal dysplasia | ~100% | Can develop hip deformity &/or scoliosis in addition to osteochondrodysplasia. Dysplasia may be difficult to recognize in newborn period. |
| Small, loosely rooted teeth | ~100% | Frequency not definitively evaluated in large studies, but typically secondary teeth are more affected than primary teeth. |
| Hematologic | Anemia | >75% | Asymptomatic |
| Thrombocytosis | >75% | Asymptomatic |
| Cerebrovascular | Aneurysms | ~50% | Risk appears to be lifelong. |
| Moyamoya vasculopathy | ~50% | Risk mainly in younger ages, starting in utero |
| Aneurysms & moyamoya disease | 36% | |
| Cardiovascular | Hypertension | 43% | Median age 13 yrs |
| Hypercholesterolemia | >32% | Median age 18 yrs |
| Cardiac malformations | 28% | ASD, VSD, PFO |
| Coronary artery disease w/premature MIs | 17% | Median age of MI 24 yrs |
| Renal | Chronic kidney disease | 32% | Renal transplantation documented in 2 persons |
| Accessory renal arteries | 15% | All known affected persons have been male. |
| Renal vascular disease | 4% | Renal artery stenosis, aneurysm |
| Genital | Cryptorchidism / retractile testes | 44% of males | |
| Hypospadias | 8% of males | |
| Endocrine | Insulin resistance &/or diabetes mellitus | >38% | Median age 11 yrs |
| Cognitive ability | Borderline/low-normal intellectual function | Most | More impairment in those who have had strokes |
| ADHD | Most | Not yet definitively evaluated in large studies |
ADHD = attention-deficit/hyperactivity disorder; ASD = atrial septal defect; IUGR = intrauterine growth restriction; MI = myocardial infarction; PFO = patent foramen ovale; VSD = ventricular septal defect
Growth restriction and microcephaly. All individuals have extreme growth restriction. This has been identified as early as the first trimester of pregnancy and continues lifelong. At birth, average length was 7.0 standard deviations (SD) below the mean, weight 3.9 SD below the mean, and head circumference 8.5 SD below the mean. At skeletal maturity, average height was 10.3 SD below the mean, weight 14.3 SD below the mean, and head circumference 8.5 SD below the mean. Average adult height is approximately 100 cm.
Body habitus evolves with time. Infants and young children have decreased subcutaneous fat; truncal obesity tends to develop through puberty. In contrast to weight gain in age-related peers, the average expected daily weight gain in individuals with MOPDII is 2 g/day throughout the life span, from infancy to skeletal maturity. Appropriate weight gain expectations are paramount to avoid excessive and unnecessary nutritional interventions [Bober et al 2012, Bober & Jackson 2017, Duker et al 2017] (see Management).
Skeletal dysplasia. Mesomelia is present at birth and appears to become more prominent over time. Slender long bones are noted at birth with an epiphyseal ossification delay and progressive widening of the metaphyses. Radial head can dislocate or sublux in childhood, leading to limited range of motion at the elbow.
Ivory and cone-shaped phalangeal epiphyses have been described; fifth finger clinodactyly and brachymesophalangy are common.
Iliac wings are small with flat acetabular angles.
Scoliosis may occur and can rapidly progress in late childhood / puberty, leading to the need for spinal fusion [Hall et al 2004, Bober & Jackson 2017].
Associated hip pathology includes coxa vara, coxa valga, developmental dysplasia, dislocation/subluxation, proximal femoral epiphysiolysis, and avascular necrosis. Eight of 12 individuals had hip pathology, some unilaterally (equaling 50% of the total hips). Developmental coxa vara was the most common finding, often beginning with a slipped capital femoral epiphysis and progressing to severe coxa vara, typically identified between ages two and five years [Karatas et al 2014] (see Table 5 and ).
Dental abnormalities. Primary teeth are small for age but may appear proportionate to the mouth. Primary teeth can have deficient enamel and can be abnormally shaped. Hypodontia can occur.
Secondary teeth tend to be more affected. They are disproportionately small, dysplastic, and can have enamel hypoplasia. They are typically poorly rooted, and chewing can be affected. In many individuals, secondary teeth either prematurely shed or are extracted, and are replaced with dentures and/or implants [Hall et al 2004, Kantaputra et al 2011, Bober & Jackson 2017] (see ).
Hematologic abnormalities. Asymptomatic thrombocytosis, leukocytosis, and anemia are common [Unal et al 2014, Bober & Jackson 2017, Duker et al 2021].
Cerebrovascular. From birth onward, 25 of 47 individuals were diagnosed with intracranial aneurysms (with 30 having more than one aneurysm), 22 were diagnosed with moyamoya vasculopathy, 17 had both moyamoya vasculopathy and intracranial aneurysms, and 17 had neither. Approximately half of those diagnosed with moyamoya vasculopathy and/or aneurysms ultimately had a stroke (ischemic or hemorrhagic, respectively). Aneurysm risk appeared to be relatively steady through childhood, whereas moyamoya vasculopathy risk was higher prior to age five years [Duker et al 2021].
Cardiovascular. Elevated blood pressure was observed in almost 50% of teens and young adults. The many intertwined comorbidities that contribute to hypertension include moyamoya vasculopathy, renal disease, coronary artery disease, dyslipidemia, and insulin resistance.
In a cohort of 47 individuals, eight had myocardial infarctions as young adults (range 17-33 years; mean age 24±5.2 years; median age 24 years); multiple individuals had more than one acute coronary event.
Approximately 25% of individuals had cardiac malformations, including atrial septal defect, ventricular septal defect, and persistent patent foramen ovale. One individual had multiple cardiac rhabdomyomas [Duker et al 2021].
Other vasculopathies. MOPDII is associated with global vascular disease. Besides involvement of cerebral vasculature as well as renal and coronary vasculature, the external carotid artery has been stenotic as well as the site of an aneurysm. At least six individuals have had difficulties with femoral artery occlusion after catheterization procedures [Duker et al 2021].
Renal. Among 47 individuals, one third had chronic kidney disease (cause not specified). Mean age of the diagnosis of Stage III chronic kidney disease was 22 years. Additionally, two individuals had a kidney transplant, at ages 21 years and 23 years, one of the two had a renal artery aneurysm.
Another individual who had renal artery stenosis in addition to a renal artery aneurysm ultimately had a partial infarct of a kidney.
Six individuals had nephrolithiasis in early adulthood; all were on anti-hypertensive medications at the time of the diagnosis of nephrolithiasis.
Congenital anomalies included accessory/duplicated renal arteries in seven males and no females [Duker et al 2021].
Genitourinary. Two of 25 males had hypospadias and 11 had cryptorchidism or retractile testes [Duker et al 2021].
Insulin resistance and/or diabetes mellitus. Eighteen of 21 individuals older than age four years had insulin resistance or diabetes [Huang-Doran et al 2011].
Seventeen of 46 individuals (in a different but overlapping cohort) had insulin resistance and/or diabetes, typically detected in adolescence [Duker et al 2021].
These issues can also lead to dyslipidemia and fatty liver [Huang-Doran et al 2011].
Cognitive ability. Despite the significant microcephaly, intellectual development is generally in the typical to borderline range, and social skills are excellent. Many individuals have been described as hyperactive with easy distractibility in childhood [Hall et al 2004; Authors, personal observation]. However, those who have had strokes due to vascular disease have had more impairment [Bober & Jackson 2017]. Attending typical schools is expected, and some have attended college. However, it can be challenging for adults to live independently [Hall et al 2004].
Additional features
Skin:
Café au lait patches have been described, with areas of hypopigmentation later in life.
"Wizened hands" with multiple creases developing in childhood have been noted (see ).
Subglottic stenosis, described in some individuals, required tracheostomy in at least two affected individuals [
Hall et al 2004; Authors, unpublished data].
Craniosynostosis has been infrequently described, at times requiring surgical intervention. One child had bilateral coronal synostosis recognized in the neonatal period which required repair [
Hall et al 2004;
Abdel-Salam et al 2020; Authors, unpublished data].