Autosomal Dominant Tubulointerstitial Kidney Disease – UMOD
Synonyms: ADTKD-UMOD, Uromodulin Kidney Disease
Anthony J Bleyer, MD, MS, Kendrah Kidd, MS, Martina Živná, PhD, and Stanislav Kmoch, PhD.
Author Information and AffiliationsInitial Posting: January 12, 2007; Last Revision: December 23, 2021.
Estimated reading time: 25 minutes
Summary
Clinical characteristics.
Autosomal dominant tubulointerstitial kidney disease – UMOD (ADTKD-UMOD) is characterized by normal urinalysis and slowly progressive chronic kidney disease (CKD), usually first noted in the teen years and progressing to end-stage renal disease (ESRD) between the third and seventh decades. Hyperuricemia is often present from an early age, and gout (resulting from reduced kidney excretion of uric acid) occurs in the teenage years in about 8% of affected individuals and develops in 55% of affected individuals over time.
Management.
Treatment of manifestations: With allopurinol treatment, serum uric acid concentration returns to normal and gout attacks can be entirely prevented. Lifelong therapy with allopurinol is required for future gout prevention. Referral to a nephrologist to monitor kidney function; evaluate for manifestations of CKD, and prepare for renal replacement therapy when ESRD occurs. Renal replacement therapies such as hemodialysis and peritoneal dialysis replace kidney function but are associated with potential complications; kidney transplantation is curative.
Surveillance: Measurement of serum creatinine concentration at least annually in affected individuals, and more frequently in those with severe disease; measurement of serum uric acid concentration at least annually.
Agents/circumstances to avoid: Drugs known to be nephrotoxic; volume depletion and dehydration. Nonsteroidal anti-inflammatory drugs are generally discouraged but could be used for short-term administration for treatment of gout or similar painful conditions in early CKD (prior to Stage 3 CKD). Chronic daily use should be avoided.
Evaluation of relatives at risk: Asymptomatic at-risk relatives younger than age 18 years from a family with ADTKD-UMOD with an early age of onset of gout may benefit from testing for the familial UMOD pathogenic variant so that those with the variant can initiate treatment that would prevent gout.
It is appropriate to clarify the genetic status of apparently asymptomatic at-risk adult relatives in order to identify those with the familial UMOD pathogenic variant (and thus, at risk for CKD) who would be benefit from routine surveillance and awareness of agents/circumstances to avoid.
Any relative who is a potential kidney donor should be tested for the familial UMOD pathogenic variant so that only those without the variant are evaluated further for kidney donation.
Pregnancy management: The use of angiotensin-converting enzyme inhibitors even in early pregnancy can result in fetal damage and death. Use of allopurinol during pregnancy should be avoided.
Diagnosis
Consensus clinical diagnostic criteria for autosomal dominant tubulointerstitial kidney disease – UMOD (ADTKD-UMOD) have been published [Eckardt et al 2015] (full text).
Suggestive Findings
ADTKD-UMOD
should be suspected in individuals with the following findings and family history.
Autosomal dominant inheritance is a key finding in this disease with few distinctive clinical features. One should consider this diagnosis when both the patient and a parent have kidney disease, even if the parent's kidney disease is suspected of having another cause.
Slowly progressive chronic kidney disease (CKD) is often present from childhood, as manifested with an estimated glomerular filtration rate (eGFR) < 90 ml/min/1.73m2 [Kidd et al 2020].
Bland urinary sediment without blood or protein is present in almost all individuals. Urinary protein is <250 mg/24 hours except when CKD is advanced (e.g., eGFR <40 mL/min/1.73m2).
Hyperuricemia results from decreased renal excretion of uric acid and is found in almost all affected individuals. The fractional excretion of urate is <5% in most individuals with this disorder, but such a value can also be seen in healthy individuals [Stibůrková & Bleyer 2012]. In children with the disorder, serum creatinine levels may be normal but elevated serum urate levels are usually present.
Usually, hyperuricemia in an individual with normal kidney function corresponds to a serum concentration of uric acid >1 SD of the normal value for age and sex. It is important to use age-related norms for serum urate [Wilcox 1996] (see Table 1).
Table 1.
Serum Uric Acid Concentration in Individuals with Normal Renal Function
View in own window
| Age | Serum Concentration (mg/dL) |
|---|
| Males | Females |
|---|
| <5 years | 3.6±0.9 | 3.6±0.9 |
| 5-10 years | 4.1±1.0 | 4.1±1.0 |
| 12 years | 4.4±1.1 | 4.5±0.9 |
| 15 years | 5.6±1.1 | 4.5±0.9 |
| >18 years | 6.2±0.8 | 4.0±0.7 |
Gout due to hyperuricemia occurs in 55% of affected individuals [Kidd et al 2020]; 8% of affected children develop gout before age 18 years [Bleyer et al 2021].
Establishing the Diagnosis
NOTE: Kidney biopsy should NOT be performed because it is an invasive procedure with some risk, and pathologic findings are too nonspecific to reliably identify the causative disorder (see Clinical Description). Molecular genetic testing, the gold standard for diagnosis, is safer and less expensive than kidney biopsy.
The diagnosis of ADTKD-UMOD
is established in a proband with suggestive findings and a heterozygous pathogenic variant in UMOD identified by molecular genetic testing (see Table 2).
Note: Identification of a heterozygous UMOD variant of uncertain significance does not by itself establish or rule out the diagnosis of this disorder. The authors of this chapter have offered to review with clinicians UMOD variants of uncertain significance; see Molecular Pathogenesis, UMOD-specific laboratory technical considerations, and Chapter Notes.
Molecular genetic testing approaches can include gene-targeted testing (multigene panel) (Option 1) or comprehensive
genomic testing (exome sequencing, genome sequencing) (Option 2).
Option 1
A kidney disease multigene panel that includes UMOD and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Note: Single-gene testing of MUC1 is required to diagnose ADTKD-MUC1, a condition in the differential diagnosis of ADTKD-UMOD, as the techniques used in multigene panels and in genome sequencing will not detect variants in MUC1 [Kirby et al 2013].
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive
genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2.
Molecular Genetic Testing Used in ADTKD-UMOD
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
UMOD
| Sequence analysis 3 | >95% 4 |
| Gene-targeted deletion/duplication analysis 5 | None reported to date 6 |
- 1.
- 2.
- 3.
- 4.
Data derived from the subscription-based professional view of Human Gene Mutation Database [Stenson et al 2017]
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
No exon or whole-gene deletions or duplications have been reported as a cause of ADTKD-UMOD. This type of pathogenic variant would be an unlikely cause of this condition; thus, the clinical utility of such testing is unknown.
Clinical Characteristics
Clinical Description
Autosomal dominant tubulointerstitial kidney disease – UMOD (ADTKD-UMOD) is characterized by a normal urinalysis and slowly progressive chronic kidney disease (CKD), usually first noted in the teen years and progressing to end-stage renal disease (ESRD) between the third and seventh decades [Kidd et al 2020]. Hyperuricemia is often present from an early age, and gout (resulting from reduced kidney excretion of uric acid) occurs in the teenage years in about 8% of affected individuals and develops in 55% of affected individuals over time [Hart et al 2002, Bleyer et al 2021].
Slowly Progressive Chronic Tubulointerstitial Kidney Disease
Most commonly, the presenting sign is elevated serum creatinine in an individual with a family history of kidney disease. A mild elevation in serum creatinine may occur in childhood and is usually incidentally noted on laboratory testing for other reasons or when screening children of an affected parent.
Kidney disease usually progresses to ESRD between the third and seventh decades of life [Kidd et al 2020], rarely occurring in children [Wolf et al 2007]. The age at which ESRD occurs varies both between and within families [Kidd et al 2020, Olinger et al 2020]. A family may have a member who reaches ESRD at age 30 years, while another family member does not reach ESRD until age 70 years.
Hyperuricemia and Gout
Hyperuricemia occurs in the majority of individuals with this disorder, though there are exceptions [Kidd et al 2020]. Unlike other kidney disorders, hyperuricemia (due to decreased urinary excretion of uric acid) occurs prior to any decline in kidney function or elevation in serum creatinine [Moro et al 1991].
Fifty-five percent of individuals with ADTKD-UMOD will develop gout, with a median age of onset of 28 years [Kidd et al 2020]. While not all affected individuals have gout, almost all families with ADTKD-UMOD have family members who have gout. In individuals with a strong family history of ADTKD-UMOD, gout is usually diagnosed by family members who are familiar with its manifestations.
Onset of gout is acute with severe tenderness and redness of the affected joint –characteristically the big toe, other areas of the feet, the ankles, and/or the knees. As kidney function worsens, gout worsens, and the frequency of attacks increases. Gout may occur in children and may be precipitated by a sporting event. As gout is uncommon in childhood, this condition may elude diagnosis. Without treatment, tophi (large subcutaneous depositions of uric acid) and crippling arthritis can develop.
Kidney Biopsy
Note: The following information is provided in the event that some affected individuals (or their relatives) may have undergone kidney biopsy prior to consideration of ADTKD-UMOD as a diagnostic possibility.
Histologic examination reveals chronic interstitial fibrosis, with focal tubular atrophy and interstitial fibrosis, occasionally accompanied by lymphocytic infiltration. Accumulation of mutated uromodulin may be detected by PAS staining as polymorphic unstructured materials [Onoe et al 2021].
Penetrance
Penetrance appears to be complete, but age related. Some individuals (especially females) may not develop ESRD until the sixth or seventh decade.
Nomenclature
According to the 2015 nomenclature [Eckardt et al 2015], the term "autosomal dominant tubulointerstitial kidney disease" (ADTKD) refers to disorders characterized by the following:
Autosomal dominant inheritance
Slowly progressive chronic tubulointerstitial kidney disease resulting in ESRD in the third through seventh decade of life
Urinalysis revealing a bland urinary sediment (i.e., little blood or protein)
Renal ultrasound examination that is normal early in the disease course [
Bleyer et al 2010]
Terms previously used to refer to ADTKD-UMOD that are no longer in use:
Familial juvenile hyperuricemic nephropathy 1
Medullary cystic kidney disease 2 (MCKD2)
UMOD-associated kidney disease
Prevalence
ADTKD-UMOD accounts for approximately 0.2% of individuals with ESRD [Cormican et al 2019, Groopman et al 2019]. The prevalence is expected to be similar in all populations. Approximately 2,000 individuals have been identified with this disorder; however, these numbers are expected to increase significantly due to better recognition and genetic testing for this condition [Bleyer et al 2020].
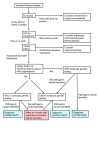
Differential Diagnosis
provides a diagnostic algorithm for inherited kidney disease.
Testing strategy for inherited kidney disease – 2015 update
Hereditary glomerulonephritis. Affected individuals usually have proteinuria and/or hematuria. If blood or protein is present in the urine, consider hereditary forms of glomerulonephritis or hematuria (e.g., Alport syndrome). Rarely, individuals with ADTKD-UMOD have had proteinuria; however, this is uncharacteristic.
Autosomal dominant polycystic kidney disease. If the urinary sediment is bland (i.e., with little blood or protein) in persons with kidney disease inherited in an autosomal dominant manner, one must exclude autosomal dominant polycystic kidney disease (ADPKD), in which a large number of cysts are seen on kidney ultrasound examination in affected individuals older than age 25 years.
Other forms of ADTKD. If the individual does not have ADPKD and the urinary sediment is bland, consider the following forms of autosomal dominant tubulointerstitial kidney disease:
ADTKD-MUC1 is associated with pathogenic variants in
MUC1. Affected individuals have slowly progressive CKD and minimal proteinuria. An important differentiating factor is that in ADTKD-
MUC1 gout usually only occurs in the setting of Stage 3 or later CKD. Affected individuals do not have anemia or other manifestations in childhood.
ADTKD-REN is associated with pathogenic variants in
REN [
Zivná et al 2009]. Like ADTKD-
UMOD, this condition is associated with early-onset gout and slowly progressive CKD. Persons with a
REN pathogenic variant also manifest hypoproliferative anemia in childhood.
Fabry disease, an X-linked disorder, results from deficient activity of the enzyme α-galactosidase (α-Gal) A and progressive lysosomal deposition of globotriaosylceramide (GL-3) in cells throughout the body. The classic form, occurring in males with <1% α-Gal A activity, usually has its onset in childhood or adolescence with periodic crises of severe pain in the extremities (acroparesthesias), the appearance of vascular cutaneous lesions (angiokeratomas), hypohidrosis, characteristic corneal and lenticular opacities, and proteinuria (which exceeds that seen in ADTKD-UMOD). Gradual deterioration of kidney function leads to ESRD, usually occurring in the third to fifth decade. Males with >1% α-Gal A activity have a cardiac or renal variant phenotype. Rarely, heterozygous carrier females may have symptoms as severe as those observed in males with the classic phenotype.
Table 3.
Monogenic Kidney Diseases in the Differential Diagnosis of ADTKD-UMOD
View in own window
| Gene(s) | DiffDx Disorder | MOI | Renal Phenotype | Distinguishing Features of DiffDx Disorder |
|---|
CEP290
INVS
IQCB1
NPHP1
NPHP3
NPHP4
TMEM67
(19 genes 1) | Isolated nephronophthisis | AR | TKD, often seen in childhood; may be assoc w/anemia & mild hypotension | Absence of affected family members in multiple generations. Anemia usually correlates w/level of kidney function (i.e., may not be present in childhood). Severity of kidney failure is usually much greater (usually requiring dialysis in the teens & early 20s). Hyperkalemia & acidemia are not as pronounced. |
COL4A3
COL4A4
COL4A5
| Alport syndrome (& other types of hereditary glomerulonephritis) | XL
AR
AD | Microscopic hematuria (microhematuria); proteinuria; progression to ESRD | Frequent cochlear & ocular manifestations; hematuria; males affected much more severely than females |
DNAJB11
GANAB
PKD1
PKD2
| Autosomal dominant polycystic kidney disease (ADPKD) | AD | Bland urinary sediment 2; large # of cysts in persons age >25 yrs | Numerous cysts seen on kidney ultrasound |
|
GLA
| Fabry disease, classic form | XL | Proteinuria (usually exceeding that seen in ADTKD-UMOD). Gradual deterioration of renal function to ESRD occurs in ~3rd-5th decade. 3 | Classic form (males w/<1% α-Gal A activity) usually has onset in childhood or adolescence w/periodic crises of severe pain in extremities (acroparesthesias), appearance of vascular cutaneous lesions (angiokeratomas), hypohidrosis, & characteristic corneal & lenticular opacities. |
|
MUC1
|
ADTKD-MUC1
| AD | Minimal proteinuria; slowly progressive CKD | Only clinical finding is CKD & its sequelae. 4 |
| DNAJB11 4 | Atypical ADPKD-ADTKD | AD | Slowly progressive CKD, multiple renal cysts | Numerous kidney cysts are common. |
|
HNF1B
| ADTKD-HNF1B | AD | Variable other manifestations incl MODY, hyperuricemia & gout, CKD, CAKUT, & unexplained liver function abnormalities | Incomplete penetrance for characteristic renal involvement & absence of the other variable manifestations |
|
mtDNA
| m.547A>T 5 | Mat | Chronic TKD | Absence of childhood anemia, hyperkalemia, & acidemia |
|
PAX2
|
PAX2-related disorder
| AD | Glomerular proteinuria, hematuria, CKD, & ocular coloboma | Absence of hematuria, proteinuria, & coloboma |
|
SEC61A1
| ADTKD-SEC61A1 | AD | Slowly progressive CKD, leukopenia, abscess formation, IUGR & postnatal growth restriction | Absence of leukopenia, abnormal growth |
α-Gal = α-galactosidase; AD = autosomal dominant; AR = autosomal recessive; CAKUT = congenital anomalies of the kidneys and urinary tract; CKD = chronic kidney disease; ESRD = end-stage renal disease; IUGR = intrauterine growth restriction; Mat = maternal; MODY = maturity-onset diabetes of the young; MOI = mode of inheritance; TKD = tubulointerstitial kidney disease; XL = X-linked
- 1.
Listed genes represent the most common genetic causes of isolated nephronophthisis. Other genes known to be associated with nephronophthisis are ANKS6, CEP164, CEP83, DCDC2, GLIS2, IFT172, NEK8, RPGRIP1L, SDCCAG8, TTC21B, WDR19, and ZNF423.
- 2.
Bland refers to urinary sediment with little blood or protein.
- 3.
Males with >1% α-Gal A activity have a cardiac or renal variant phenotype. Rarely, heterozygous carrier females may have symptoms as severe as those observed in males with the classic phenotype.
- 4.
- 5.
Management
Consensus management guidelines for autosomal dominant tubulointerstitial kidney disease, UMOD (ADTKD-UMOD) have been published [Eckardt et al 2015] (full text).
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with ADTKD-UMOD, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with ADTKD-UMOD
View in own window
| System/Concern | Evaluation |
|---|
|
Hypertension risk
| Measurement of blood pressure |
|
Anemia
| Hemoglobin level |
|
Kidney function
| Serum creatinine (part of basic metabolic panel) |
|
Gout risk
| Serum urate concentration |
|
Kidney structure
| Renal ultrasound exam |
|
Nephrology referral
| |
|
Genetic counseling
| By genetics professionals 1 to inform affected persons re nature, MOI, &implications of ADTKD-UMOD in order to facilitate medical & personal decision making |
- 1.
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
Care by a nephrologist is recommended.
Hyperuricemia/Gout
Prevention of gout attacks with allopurinol should be considered in individuals with gout. With allopurinol treatment, serum uric acid concentration returns to normal and gout attacks can be entirely prevented. Lifelong therapy with allopurinol is required for future gout prevention.
In individuals with allergies or intolerance to allopurinol, febuxostat may be considered; however, no data on the use of this medication in ADTKD-UMOD are available at present.
Acute gout typically responds well to prednisone, short-term nonsteroidal anti-inflammatory drugs (NSAIDs), or colchicine. One should avoid NSAIDs in the setting of CKD Stage 3 or greater.
Kidney Disease
Referral to a nephrologist is indicated to monitor kidney function, evaluate for manifestations of CKD, and prepare for renal replacement therapy when end-stage renal disease (ESRD) occurs.
Renal replacement therapies such as hemodialysis and peritoneal dialysis replace kidney function but are associated with potential complications.
Individuals with ADTKD-UMOD are excellent transplant candidates. Progression of CKD is usually slow, allowing time to find a living donor. Besides gout, there are no other associated systemic manifestations of ADTKD-UMOD, resulting in most individuals having low comorbidities prior to transplant. Kidney transplantation cures ADTKD-UMOD, as the transplanted kidney does not develop the disease.
There is insufficient evidence that allopurinol slows the progression of kidney disease. Some authors have suggested that it may slow progression, but these studies were small, not randomized, and with short-term follow-up. In addition, genetic diagnoses were not available at the time of these studies [Pirson et al 2000, Fairbanks et al 2002].
Surveillance
Appropriate surveillance includes the following:
Measurement of serum creatinine concentration at least annually in affected individuals, and more frequently in those with severe disease
Measurement of serum uric acid concentration at least annually
Agents/Circumstances to Avoid
Avoid use of the following:
Nonsteroidal anti-inflammatory drugs. NSAIDs are generally discouraged except for short-term treatment of gout or similar painful conditions in early CKD (prior to Stage 3 CKD). Chronic daily use should be avoided.
Drugs known to be nephrotoxic
The low sodium diet, which is typically prescribed in the treatment of CKD
Volume depletion, dehydration, and physical exertion under extreme conditions (e.g., in hot weather), as these may worsen hyperuricemia, leading to more frequent attacks of gout
Evaluation of Relatives at Risk
For early diagnosis and treatment
Asymptomatic at-risk relatives younger than age 18 years. Many experts believe that testing of asymptomatic children for genetic disorders for which there are no specific treatments is inadvisable and violates the child's autonomy. However, genetic testing of a child from a family with ADTKD-UMOD may be considered for the purpose of early diagnosis to initiate treatment that would prevent gout (a secondary manifestation of ADTKD-UMOD). For example, a child may belong to a family with an early age of onset of ESRD and the occurrence of gout in the teenage years. If the child is an athlete or dancer, testing could be considered in order to provide preventive therapy for gout and thus avoid the inopportune occurrence of gout prior to an important sporting event or performance. In contrast, testing of asymptomatic individuals younger than age 18 years should likely be avoided in families with a late age of onset of ESRD and no history of gout in childhood.
If performed, testing should only be done after a full discussion with the child and after obtaining the child's assent and the parents'/guardians' consent.
* Chronic kidney disease, one of the primary manifestations of this disorder, is often asymptomatic.
For kidney donation. Any relative who is a potential kidney donor should undergo molecular genetic testing to clarify his/her genetic status so that only those who do not have the UMOD pathogenic variant are evaluated further.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Pregnancy in women with ADTKD-UMOD is associated with fetal outcomes comparable to the general population. There is an increased risk of hospitalization for high blood pressure [Author, personal observation]. Acceleration of loss of kidney function can be seen in some kidney diseases during pregnancy; data specifically for ADTKD-UMOD are not available.
Women of childbearing age who are taking medications such as an angiotensin-converting enzyme (ACE) inhibitor or allopurinol should discuss their medication regimen with their physician prior to conception.
ACE inhibitors. The use of ACE inhibitors even in early pregnancy can result in fetal damage and death. Women who are taking ACE inhibitors prior to pregnancy or at the time of conception should be transitioned to another antihypertensive medication.
Allopurinol. Published data on the fetal risk associated with use of allopurinol during pregnancy is limited. While a number of pregnancies in which allopurinol was used resulted in the birth of healthy infants, the rare occurrence of a pattern of malformations similar to what is observed in women who use mycophenolate mofetil during pregnancy was reported in two infants born to women who took allopurinol throughout pregnancy [Kozenko et al 2011, Hoeltzenbein et al 2013].
This finding is concerning because the mechanism of action of allopurinol (inhibiting purine degradation) is similar to the mechanism of action of mycophenolate mofetil (inhibition of de novo purine biosynthesis).
Prednisone. Use of prednisone during pregnancy has been associated with fetal growth restriction. Use in the first trimester of pregnancy is associated with a slightly increased risk of orofacial clefting [Carmichael et al 2007].
Colchicine. Chronic use of colchicine that includes the immediate preconception and conception period has been associated with an increased risk of fetal chromosome abnormalities [Berkenstadt et al 2005]. However, the risk of adverse fetal outcome for short courses of colchicine during pregnancy outside of the periconeptional period is low.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
By definition, autosomal dominant tubulointerstitial kidney disease – UMOD (ADTKD-UMOD) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Sibs of a proband. The risk to the sibs of a proband depends on the genetic status of the proband's parents:
If a parent of the
proband has the
UMOD pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
The rate of chronic kidney disease progression and age of onset of end-stage renal disease (ESRD) can vary significantly between
heterozygous sibs and does not correlate well with the age of ESRD in the affected parent.
If the parents have not been tested for the
UMOD pathogenic variant but are clinically asymptomatic, sibs are still presumed to be at increased risk for ADTKD-
UMOD because of the possibility of later onset in a
heterozygous parent or the theoretic possibility of parental
germline mosaicism.
Offspring of a proband. Each child of an individual with ADTKD-UMOD has a 50% chance of inheriting the UMOD pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the UMOD pathogenic variant, his or her family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the UMOD pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Medline Plus
UMOD-Related Kidney Disease Registry
Dr. Anthony Bleyer has established a registry of individuals with UMOD pathogenic variants. Patient educational materials (including webinars) are available upon request; genetic testing can be offered as part of a research protocol. Please contact Dr. Bleyer (ableyer@wakehealth.edu) if interested in participation.
Email: ableyer@wakehealth.edu
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Autosomal Dominant Tubulointerstitial Kidney Disease -- UMOD: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
Autosomal dominant tubulointerstitial kidney disease – UMOD (ADTKD-UMOD) is an endoplasmic reticulum storage disease resulting in chronic kidney disease from deposition of abnormal uromodulin over time [Rampoldi et al 2003, Bleyer et al 2004, Williams et al 2009]. Uromodulin is produced only in the thick ascending limb of Henle's loop of the renal tubule; therefore, disease is limited to the kidney. While uromodulin is the most common protein found in normal human urine, its function is uncertain. Uromodulin is likely responsible for maintaining the integrity of the thick ascending limb of Henle's loop, in which the water permeability is remarkably low and salts are efficiently absorbed.
UMOD pathogenic variants are mostly missense variants [Kidd et al 2020]. The majority involve the addition or deletion of a cysteine residue or highly conserved polar residue that is likely to alter either disulfide bond formation, thereby disrupting the correct protein folding [Whiteman & Handford 2003] or hydrophobicity distribution responsible for protein spatial flexibility [Xu et al 1997].
Mechanism of disease causation. In heterozygotes, urinary excretion of uromodulin is much less than half the expected amount, likely resulting from a dominant-negative effect (e.g., interference with synthesis of the normal uromodulin by abnormal uromodulin). Accordingly, the abnormal expression of the mutated uromodulin in the thick ascending limb of Henle's loop of the renal tubule decreases NaCl reabsorption and subsequently induces a state of volume contraction known to promote the proximal reabsorption of urate [Renigunta et al 2011].
UMOD-specific laboratory technical considerations.
UMOD comprises 11 exons. Exon 1 is noncoding. Note: Exon numbering may vary in the literature; this GeneReview uses that of Williams et al [2009].
Pathogenic variants in UMOD have been identified in exons 3, 4, 5, and 7 [Williams et al 2009]. Most variants are in exons 3 and 4.
For interpretation of variants of uncertain significance:
Table 5.
Notable UMOD Pathogenic Variants
View in own window
| Reference Sequences | DNA Nucleotide Change | Predicted
Protein Change | Comment 1 |
|---|
# of
families | Median age
of ESRD |
|---|
NM_003361.4
NP_003352.2
| c.115G>A | p.Ala39Thr | (Later onset of ESRD) |
| 4 | 66 yrs 2 |
| c.263G>A | p.Gly88Asp | Common pathogenic variant
(Later onset of ESRD) |
| 8 | 65.5 yrs 2 |
| c.274T>G | p.Cys92Gly | (Early onset of ESRD) |
| 2 | 31.5 yrs 2 |
c.278_289delTCTGCCC
CGAAGinsCCGCCTCCT | p.Val93_Gly97del
insAlaAlaSerCys | Common pathogenic variant; assoc w/less frequent gout (Later onset of ESRD) 3 |
| 11 | 48 yrs 2 |
| c.317G>T | p.Cys106Phe | Common pathogenic variant (Later onset of ESRD) |
| 10 | 55 yrs 2 |
| c.529_555del27 | p.His177-Arg185del | Common pathogenic variant |
| 25 | 46 yrs 2 |
| c.533G>C | p.Arg178Pro | Common pathogenic variant (Later onset of ESRD) |
| 9 | 53 yrs 2 |
| c.744C>G | p.Cys248Trp | (Later onset of ESRD) |
| 3 | 71 yrs 2 |
ESRD = end-stage renal disease
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Comments shown in ( )s are possible associations that require further evidence.
- 2.
- 3.
Chapter Notes
Author Notes
Dr Bleyer pursues clinical and genetic research of autosomal dominant tubulointerstitial kidney disease (ADTKD).
Regarding ADTKD-UMOD:
In addition:
He is most interested in hearing about families with ADTKD in whom no causative variant has been identified on
molecular genetic testing of the genes known to cause ADTKD.
Single-
gene testing for
MUC1 pathogenic variants can be provided free of charge.
Please contact Dr Bleyer (ableyer@wakehealth.edu) with any questions about ADTKD.
Related website: www.wakehealth.edu
Author History
Anthony J Bleyer, MD, MS (2007-present)
Karn Gupta, MD; Wake Forest University School of Medicine (2007-2011)
P Suzanne Hart, PhD; National Human Genome Research Institute (2011-2021)
Kendrah Kidd, MS (2021-present)
Stanislav Kmoch, PhD (2016-present)
Martina Živná, PhD (2021-present)
Revision History
23 December 2021 (sw) Revision: nomenclature corrections (
Table 5)
8 April 2021 (bp) Comprehensive update posted live
30 June 2016 (ha) Comprehensive update posted live
12 September 2013 (me) Comprehensive update posted live
15 March 2011 (me) Comprehensive update posted live
12 January 2007 (me) Review posted live
3 August 2006 (ajb) Original submission
References
Published Guidelines / Consensus Statements
Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available
online. 2013. Accessed 12-21-21.
National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset conditions. Available
online. 2018. Accessed 12-21-21.
Literature Cited
Berkenstadt M, Weisz B, Cuckle H, Di-Castro M, Guetta E, Barkai G. Chromosomal abnormalities and birth defects among couples with colchicine treated familial Mediterranean fever.
Am J Obstet Gynecol. 2005;193:1513–6. [
PubMed: 16202748]
Bleyer AJ, Hart TC, Shihabi Z, Robins V, Hoyer JR. Mutations in the uromodulin gene decrease urinary excretion of Tamm-Horsfall protein.
Kidney Int. 2004;66:974–7. [
PubMed: 15327389]
Bleyer AJ, Kidd K, Robins V, Martin L, Taylor A, Santi A, Tsoumas G, Hunt A, Swain E, Abbas M, Akinbola E, Vidya S, Moossavi S, Bleyer AJ Jr, Živná M, Hartmannová H, Hodaňová K, Vyleťal P, Votruba M, Harden M, Blumenstiel B, Greka A, Kmoch S. Outcomes of patient self-referral for the diagnosis of several rare inherited kidney diseases.
Genet Med. 2020;22:142–9. [
PMC free article: PMC6946861] [
PubMed: 31337885]
Carmichael SL, Shaw GM, Ma C, Werler MM, Rasmussen SA, Lammer EJ. Maternal corticosteroid use and orofacial clefts.
Am J Obstet Gynecol. 2007;197:585.e1–585.e7. [
PubMed: 18060943]
Connor TM, Hoer S, Mallett A, Gale DP, Gomez-Duran A, Posse V, Antrobus R, Moreno P, Sciacovelli M, Frezza C, Duff J, Sheerin NS, Sayer JA, Ashcroft M, Wiesener MS, Hudson G, Gustafsson CM, Chinnery PF, Maxwell PH. Mutations in mitochondrial DNA causing tubulointerstitial kidney disease.
PloS Genet. 2017;13:e1006620. [
PMC free article: PMC5360345] [
PubMed: 28267784]
Cormican S, Connaughton DM, Kennedy C, Murray S, Živná M, Kmoch S, Fennelly NK, O'Kelly P, Benson KA, Conlon ET, Cavalleri G, Foley C, Doyle B, Dorman A, Little MA, Lavin P, Kidd K, Bleyer AJ, Conlon PJ. Autosomal dominant tubulointerstitial kidney disease (ADTKD) in Ireland.
Ren Fail. 2019;41:832–41. [
PMC free article: PMC6746258] [
PubMed: 31509055]
Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi L, Bleyer AJ. Autosomal dominant tubulointerstitial kidney disease.
Nat Rev Dis Primers. 2019;5:60. [
PubMed: 31488840]
Eckardt KU, Alper SL, Antignac C, Bleyer AJ, Chauveau D, Dahan K, Deltas C, Hosking A, Kmoch S, Rampoldi L, Wiesener M, Wolf MT, Devuyst O. Autosomal dominant tubulointerstitial kidney disease: Diagnosis, classification, and management- a KDIGO consensus report.
Kidney Int. 2015;88:676–683. [
PubMed: 25738250]
Fairbanks LD, Cameron JS, Venkat-Raman G, Rigden SP, Rees L. Van'T Hoff W, Mansell M, Pattison J, Goldsmith DJ, Simmonds HA. Early treatment with allopurinol in familial juvenile hyerpuricaemic nephropathy (FJHN) ameliorates the long-term progression of renal disease.
QJM. 2002;95:597–607. [
PubMed: 12205338]
Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, Li Y, Zhang J, Nestor J, Krithivasan P, Lam WY, Mitrotti A, Piva S, Kil BH, Chatterjee D, Reingold R, Bradbury D, DiVecchia M, Snyder H, Mu X, Mehl K, Balderes O, Fasel DA, Weng C, Radhakrishnan J, Canetta P, Appel GB, Bomback AS, Ahn W, Uy NS, Alam S, Cohen DJ, Crew RJ, Dube GK, Rao MK, Kamalakaran S, Copeland B, Ren Z, Bridgers J, Malone CD, Mebane CM, Dagaonkar N, Fellström BC, Haefliger C, Mohan S, Sanna-Cherchi S, Kiryluk K, Fleckner J, March R, Platt A, Goldstein DB, Gharavi AG. Diagnostic utility of exome sequencing for kidney disease.
N Engl J Med. 2019;380:142–51. [
PMC free article: PMC6510541] [
PubMed: 30586318]
Hart TC, Gorry MC, Hart PS, Woodard AS, Shihabi Z, Sandhu J, Shirts B, Xu L, Zhu H, Barmada MM, Bleyer AJ. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy.
J Med Genet. 2002;39:882–92. [
PMC free article: PMC1757206] [
PubMed: 12471200]
Hoeltzenbein M, Stieler K, Panse M, Wacker E, Schaefer C. Allopurinol use during pregnancy: Outcome of 31 prospectively ascertained cases and a phenotype possibly indicative for teratogenicity.
PLoS One. 2013;8:e66637. [
PMC free article: PMC3686712] [
PubMed: 23840514]
Kidd K, Vylet'al P, Schaeffer C, Olinger E, Živná M, Hodaňová K, Robins V, Johnson E, Taylor A, Martin L, Izzi C, Jorge SC, Calado J, Torres RJ, Lhotta K, Steubl D, Gale DP, Gast C, Gombos E, Ainsworth HC, Chen YM, Almeida JR, de Souza CF, Silveira C, Raposeiro R, Weller N, Conlon PJ, Murray SL, Benson KA, Cavalleri GL, Votruba M, Vrbacká A, Amoroso A, Gianchino D, Caridi G, Ghiggeri GM, Divers J, Scolari F, Devuyst O, Rampoldi L, Kmoch S, Bleyer AJ. Genetic and clinical predictors of age of ESKD in individuals with autosomal dominant tubulointerstitial kidney disease due to UMOD mutations.
Kidney Int Rep. 2020;5:1472–85. [
PMC free article: PMC7486199] [
PubMed: 32954071]
Kirby A, Gnirke A, Jaffe DB, Barešová V, Pochet N, Blumenstiel B, Ye C, Aird D, Stevens C, Robinson JT, Cabili MN, Gat-Viks I, Kelliher E, Daza R, DeFelice M, Hůlková H, Sovová J, Vylet'al P, Antignac C, Guttman M, Handsaker RE, Perrin D, Steelman S, Sigurdsson S, Scheinman SJ, Sougnez C, Cibulskis K, Parkin M, Green T, Rossin E, Zody MC, Xavier RJ, Pollak MR, Alper SL, Lindblad-Toh K, Gabriel S, Hart PS, Regev A, Nusbaum C, Kmoch S, Bleyer AJ, Lander ES, Daly MJ. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing.
Nat Genet. 2013;45:299–303. [
PMC free article: PMC3901305] [
PubMed: 23396133]
Kozenko M, Grynspan D, Oluyomi-Obi T, Sitar D, Elliott AM, Chodirker BN. Potential teratogenic effects of allopurinol: a case report.
Am J Med Genet A. 2011;155A:2247–52. [
PubMed: 21815259]
Mikkelsen WM, Dodge HJ, Valkenburg H. The distribution of serum uric acid values in a population unselected as to gout or hyperuricemia: Tecumseh, Michigan 1959-1960.
Am J Med. 1965;39:242–51. [
PubMed: 14320691]
Moro F, Ogg CS, Simmonds HA, Cameron JS, Chantler C, McBride MB, Duley JA, Davies PM. Familial juvenile gouty nephropathy with renal urate hypoexcretion preceding renal disease.
Clin Nephrol. 1991;35:263–9. [
PubMed: 1873940]
Olinger E, Hofmann P, Kidd K, Dufour I, Belge H, Schaeffer C, Kipp A, Bonny O, Deltas C, Demoulin N, Fehr T, Fuster DG, Gale DP, Goffin E, Hodaňová K, Huynh-Do U, Kistler A, Morelle J, Papagregoriou G, Pirson Y, Sandford R, Sayer JA, Torra R, Venzin C, Venzin R, Vogt B, Živná M, Greka A, Dahan K, Rampoldi L, Kmoch S, Bleyer AJ Sr, Devuyst O. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease due to mutations in UMOD and MUC1.
Kidney Int. 2020;98:717–731. [
PubMed: 32450155]
Onoe T, Hara S, Yamada K, Zoshima T, Mizushima I, Ito K, Mori T, Daimon S, Muramoto H, Shimizu M, Iguchi A, Kuma A, Ubara Y, Mitobe M, Tsuruta H, Kishimoto N, Imura J, Konoshita T, Kawano M. Significance of kidney biopsy in autosomal dominant tubulointerstitial kidney disease-UMOD: is kidney biopsy truly nonspecific?
BMC Nephrol. 2021;22:1. [
PMC free article: PMC7784305] [
PubMed: 33397327]
Pirson Y, Loute G, Cosyns JP, Dahan K, Verellen C. Autosomal-dominant chronic interstitial nephritis with early hyperuricemia.
Adv Nephrol Necker Hosp. 2000;30:357–69. [
PubMed: 11068651]
Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation.
Nat Genet. 2016;48:126–33. [
PMC free article: PMC4731925] [
PubMed: 26656846]
Rampoldi L, Caridi G, Santon D, Boaretto F, Bernascone I, Lamorte G, Tardanico R, Dagnino M, Colussi G, Scolari F, Ghiggeri GM, Amoroso A, Casari G. Allelism of MCKD, FJHN and GCKD caused by impairment of uromodulin export dynamics.
Hum Mol Genet. 2003;12:3369–84. [
PubMed: 14570709]
Renigunta A, Renigunta V, Saritas T, Decher N, Mutig K, Waldegger S. Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function.
J Biol Chem. 2011;286:2224–35. [
PMC free article: PMC3023518] [
PubMed: 21081491]
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med. 2015;17:405–24. [
PMC free article: PMC4544753] [
PubMed: 25741868]
Smith GD, Robinson C, Stewart AP, Edwards EL, Karet HI, Norden AG, Sandford RN, Karet Frankl FE. Characterization of a recurrent in-frame UMOD indel mutation causing late-onset autosomal dominant end-stage renal failure.
Clin J Am Soc Nephrol. 2011;6:2766–74. [
PMC free article: PMC3255364] [
PubMed: 22034507]
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies.
Hum Genet. 2017;136:665–77. [
PMC free article: PMC5429360] [
PubMed: 28349240]
Stibůrková B, Bleyer AJ. Changes in serum urate and urate excretion with age.
Adv Chronic Kidney Dis. 2012;19:372–6. [
PubMed: 23089271]
Whiteman P, Handford PA. Defective secretion of recombinant fragments of fibrillin-1: implications of protein misfolding for the pathogenesis of Marfan syndrome and related disorders.
Hum Mol Genet. 2003;12:727–37. [
PubMed: 12651868]
Wilcox WD. Abnormal serum uric acid levels in children.
J Pediatr. 1996;128:731–41. [
PubMed: 8648529]
Williams SE, Reed AAC, Galvanovskis J, Antignac C, Goodship T, Karet FE, Kotanko P, Lhotta K, Moriniere V, Williams P, Wong W, Rorsman P, Thakker RV. Uromodulin mutations causing familial juvenile hyperuricaemic nephropathy lead to protein maturation defects and retention in the endoplasmic reticulum.
Hum Mol Genet. 2009;18:2963–74. [
PMC free article: PMC2714724] [
PubMed: 19465746]
Wolf MT, Beck BB, Zaucke F, Kunze A, Misselwitz J, Ruley J, Ronda T, Fischer A, Eifinger F, Licht C, Otto E, Hoppe B, Hildebrandt F. The Uromodulin C744G mutation causes MCKD2 and FJHN in children and adults and may be due to a possible founder effect.
Kidney Int. 2007;71:574–81. [
PubMed: 17245395]
Xu D, Lin SL, Nussinov R. Protein binding versus protein folding: the role of hydrophilic bridges in protein associations.
J Mol Biol. 1997;265:68–84. [
PubMed: 8995525]
Zivná M, Hůlková H, Matignon M, Hodanová K, Vylet'al P, Kalbácová M, Baresová V, Sikora J, Blazková H, Zivný J, Ivánek R, Stránecký V, Sovová J, Claes K, Lerut E, Fryns JP, Hart PS, Hart TC, Adams JN, Pawtowski A, Clemessy M, Gasc JM, Gübler MC, Antignac C, Elleder M, Kapp K, Grimbert P, Bleyer AJ, Kmoch S. Dominant renin gene mutations associated with early-onset hyperuricemia, anemia, and chronic kidney failure.
Am J Hum Genet. 2009;85:204–13. [
PMC free article: PMC2725269] [
PubMed: 19664745]