Donohue Syndrome
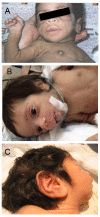
Donohue syndrome is characterized by severe insulin resistance, growth failure, hypotonia, developmental delay, characteristic facies, and organomegaly.
Endocrine manifestations. Insulin resistance with hyperinsulinemia (fasting hypoglycemia and postprandial hyperglycemia) is present from birth. There are currently no effective treatments for the insulin resistance. Postprandial hyperglycemia does not respond to insulin treatment [Semple et al 2010] or glucose-lowering therapies such as metformin [Musso et al 2004].
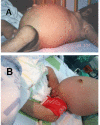
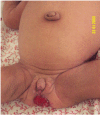
Enlargement of the external genitalia occurs in both males (enlargement of the penis) and females (labial hypertrophy and clitoral enlargement). Ovarian enlargement is characteristic; ovarian ultrasonography usually reveals multiple peripheral cysts, as seen in idiopathic polycystic ovary syndrome. Cysts may become very large and vulnerable to hemorrhage or torsion, and surgical removal may be required [Semple et al 2011]. Prominent nipples are seen in all individuals.
Central hypothyroidism has been reported [Baqir et al 2012, Falik Zaccai et al 2014].
Growth deficiency. Birth weight, length, and head circumference are below the third centile. However, weight and/or length are more affected than head circumference [Dagdeviren Cakir et al 2020]. Neonates continue to have poor weight gain and severe growth deficiency; the average weight at age one year is 4-5 kg [Longo et al 2002]. Reduced subcutaneous fat is present in all individuals.
Developmental delay / intellectual disability. Most infants have severe global developmental delay, including speech and motor as well as cognitive impairment [Falik Zaccai et al 2014]. Most individuals do not reach any significant developmental milestones prior to early demise [Falik Zaccai et al 2014, Joshi et al 2021]. Axial hypotonia and muscle atrophy are also observed [Baqir et al 2012]. Intellectual disability is thought to be a consequence of recurrent, severe hypoglycemic episodes [Ben Abdelaziz et al 2016].
Characteristic facies include proptosis, infraorbital folds, large, low-set, posteriorly rotated ears, thick vermilion of the upper and lower lips, and gingival hypertrophy [Grasso et al 2013] (see ). Macroglossia has been reported but may be secondary to prolonged therapy with insulin-like growth factor 1 (IGF-1) [Joshi et al 2021].
Recurrent infections. Recurrent bacterial infections including pneumonia and urinary tract infection are a common cause of death; health care-acquired infections may contribute to recurrent infections [Rojek et al 2023].
Integument. Hypertrichosis and hyperkeratosis are present at birth in all affected infants. Acanthosis nigricans can be apparent at birth or in early infancy [Musso et al 2004].
Cardiac manifestations. Hypertrophic cardiomyopathy is present in 30% of infants and is a major cause of death [Geffner et al 1987, Hovnik et al 2013, Falik Zaccai et al 2014].
Renal manifestations. Enlarged kidneys are common. Abnormalities of the renin-aldosterone system are reported, resulting in hypokalemia, hyperaldosteronism, and hyperreninemia [Grasso et al 2013]. Hypercalciuria and nephrocalcinosis are often seen during the first months of life and in some instances can be detected prenatally [Simpkin et al 2014]. Kidney function as measured by glomerular filtration rate and plasma creatinine concentration is normal [Grasso et al 2013, Simpkin et al 2014].
Liver/gastrointestinal manifestations. Cholestasis is common in the neonatal period [Hovnik et al 2013, Kawashima et al 2013]. Hepatomegaly occurs without liver dysfunction. Rectal prolapse or hypertrophy can be present, which sometimes requires colostomy [Weber et al 2014].
Dental findings include macrodontia and dental crowding.
Prognosis. Death usually occurs during the first year of life. The major causes of death are complications of hypoglycemia, recurrent bacterial infections [Elders et al 1982, de Bock et al 2012], and cardiomyopathy [Grasso et al 2013].
Rabson-Mendenhall Syndrome (RMS)
RMS, at the milder end of the spectrum, is characterized by severe insulin resistance with fluctuations in blood glucose levels, diabetic ketoacidosis, and microvascular complications. Findings can range from severe growth deficiency and intellectual disability to normal growth and development. Facial features can be milder than those of Donohue syndrome.
Endocrine manifestations. Insulin resistance with hyperinsulinemia is less severe than that of Donohue syndrome. Fluctuations in blood glucose levels occur with fasting hypoglycemia and postprandial hyperglycemia from birth. After age one year, insulin levels decline steadily, initially resulting in increased glucose levels and fewer hypoglycemic events, and subsequently resulting in increased risk for ketoacidosis. Morbidity in older individuals with RMS results from microvascular complications of prolonged hyperglycemia and hyperinsulinemia (e.g., proliferative retinopathy, peripheral neuropathy, and renal vascular complications) [Musso et al 2004, Carrasco de la Fuente et al 2010, Jiang et al 2011].
Enlargement of the external genitalia in both males and females typically appears later in childhood in individuals with RMS compared to those with Donohue syndrome. Enlarged ovaries are reported in some individuals and may be complicated by multiple cysts and development of tumors [Parker & Semple 2013]. Prominent nipples are also seen.
Thyroid abnormalities reported in RMS include high prevalence of thyroid nodules and thyromegaly [Kushchayeva et al 2019].
Growth is variable in individuals with RMS, from severe growth deficiency that includes reduction of weight, length, and head circumference [Dagdeviren Cakir et al 2020] to normal growth [Musso et al 2004]. In those with growth deficiency, linear growth does not improve with human growth hormone treatment even when the child's growth hormone levels are low [Musso et al 2004, Kim et al 2012, Brown et al 2013].
Developmental delay / intellectual disability. Developmental delay can range from severe to normal development [de Kerdanet et al 2015, Ben Abdelaziz et al 2016]. More than 50% of individuals with RMS are reported to have mild intellectual disability [Ben Abdelaziz et al 2016, Angelidi et al 2021].
Dysmorphic facial features in individuals with RMS can be more subtle than facial features characteristic of Donohue syndrome (e.g., proptosis, infraorbital folds, large, low-set, posteriorly rotated ears, thick vermilion of the upper and lower lips, and gingival hypertrophy). The face can show increased vertical growth, increased gonial angle, macroglossia, macrodontia, dental crowding, dental prematurity, and dysplastic dentition [Joshi et al 2021]. Early dental eruption, dental crowding, and low anterior hair line may be the only unique facial features [Jiang et al 2011].
Recurrent infections. Recurrent episodes of bacterial pneumonia are the most common infections in individuals with RMS [Iqbal et al 2022].
Integument. Cutaneous changes (hirsutism and acanthosis nigricans) typically appear later in childhood in individuals with RMS.
Cardiac manifestations. Hypertrophic cardiomyopathy is common and typically observed during the first decade of life. Neonatal hypertrophic cardiomyopathy was also reported in some individuals [Aftab et al 2022].
Renal manifestations. Enlarged kidneys are common. Hypercalcemia, nephrocalcinosis, and medullary sponge kidney are also reported as features of RMS [Chrzanowska et al 2023]. Nephrocalcinosis was reported in the vast majority of individuals [Simpkin et al 2014].
Liver/gastrointestinal manifestations. Hepatosplenomegaly occurs without liver or spleen dysfunction. Rectal prolapse was reported in some affected individuals [Joshi et al 2021].
Malignancy. Rare malignancies have been reported. Endometrial carcinoma was reported in a woman age 24 years treated with recombinant human IGF-1 (rhIGF-1) for severe insulin resistance [Jo et al 2013]. Ovarian granulosa cell tumor was reported in a girl age 35 months with severe insulin resistance treated with rhIGF-1 for 16 months [Weber et al 2014] and in a young girl who was untreated [Brisigotti et al 1993]. Because two of these individuals were treated with rhIGF-1, it is possible that the tumors resulted from an adverse effect of this treatment.
Prognosis. Complications of prolonged hyperglycemia and hyperinsulinemia are the major causes of death in individuals with RMS, usually during the second decade of life [Semple et al 2010]. However, survival can be into the third decade [Longo et al 2002, Musso et al 2004].