Prevention of Primary Manifestations
The principles of treatment are to correct the biochemical abnormalities – especially to control the elevated plasma homocysteine concentrations as much as possible, to prevent or at least reduce the complications of homocystinuria [Yap & Naughten 1998], and to prevent further complications such as thrombosis [Morris et al 2017].
The best results have been reported in those individuals identified by newborn screening and treated shortly after birth in whom the plasma free homocystine concentration is maintained below 11 µmol/L (preferably, ≤5 µmol/L) [Yap et al 2001b]. This corresponds to a plasma total homocysteine concentration below 120 µmol/L or, preferably, below 100 µmol/L [Morris et al 2017]. For B6-responsive individuals, the goal for plasma total homocysteine is below 50 µmol/L [Morris et al 2017].
These goals may need revision when very long-term data becomes available.
Measures used to control total plasma homocysteine concentration include vitamin B6 (pyridoxine) therapy (if shown to be B6 responsive), methionine-restricted diet, and folate and vitamin B12 supplementation. Betaine therapy is usually added to the therapeutic regimen; in adolescents and adults betaine may be the major form of treatment but it is preferable to remain on life-long metabolic diet. In those who have already had a vascular event, betaine therapy alone may prevent recurrent events [Lawson-Yuen & Levy 2010].
Details about each aspect of treatment follow.
Vitamin B6 (pyridoxine) therapy. In those who are shown to be B6 responsive, treatment with pyridoxine in a dose of approximately 200 mg/day or the lowest dose that produces the maximum biochemical benefit (i.e., lowest plasma homocysteine and methionine concentrations), as determined by measurement of total homocysteine and amino acid levels, should be given.
Pyridoxine may also be included in treatment despite evidence of B6 non-responsiveness, typically in doses of 100-200 mg daily (although some adults receive 500-1000 mg daily).
Dietary treatment. B6-non-responsive neonates or those only very poorly responsive to pyridoxine require a methionine-restricted diet with frequent metabolic monitoring. This diet should be continued indefinitely. Dietary treatment should be considered for clinically diagnosed individuals but often is not tolerated if begun in mid-childhood or later.
The majority of B6-responsive individuals also require a methionine-restricted diet for metabolic control.
The diet for homocystinuria is very complex and the skills of an experienced metabolic dietician must be utilized. Dietary treatment reduces methionine intake by restricting natural protein intake. However, to prevent protein malnutrition, a methionine-free amino acid formula supplying the other amino acids (as well as cysteine, which may be an essential amino acid in CBS deficiency) is provided. Breast feeding may be continued in combination with the methionine-free amino acid infant formula [MacDonald et al 2006]. The amount of methionine required is calculated by a metabolic dietician and supplied in natural food and special low-protein foods and monitored on the basis of plasma concentrations of total homocysteine as well as methionine.
Folate and vitamin B12 supplementation. Folate and vitamin B12 optimize the conversion of homocysteine to methionine by methionine synthase, thus helping to decrease the plasma homocysteine concentration. When the red blood cell folate concentration and serum B12 concentration are reduced, folic acid is given orally at 5 mg per day; and vitamin B12 is given as hydroxycobalamin at 1 mg IM per month.
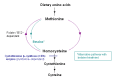
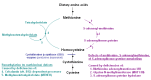
Betaine treatment. Treatment with betaine provides an alternate remethylation pathway to convert excess homocysteine to methionine (see ) and may help to prevent complications, particularly thrombosis [Yap et al 2001a, Lawson-Yuen & Levy 2010]. By converting homocysteine to methionine, betaine lowers plasma total homocysteine concentrations but raises the plasma concentration of methionine.
For children the initial betaine dose is 50 mg/kg twice daily, adjusted according to response (increased weekly by 50 mg/kg increments). For adults the initial dose is 3 g twice daily. The dose and frequency are adjusted according to biochemical response. There is unlikely to be any benefit in exceeding a dose of 150-200 mg/kg/day [Morris et al 2017].
Betaine may be added to the treatment regimen in individuals poorly compliant with dietary treatment or may become the major treatment modality in those intolerant of the diet. Individuals who are pyridoxine non-responsive who were unable to attain metabolic control with diet substantially reduced their plasma homocysteine concentrations when betaine was supplemented [Singh et al 2004].
Side effects of betaine are few. (1) Some affected individuals develop a detectable body odor, resulting in reduced compliance. (2) The increase in methionine produced by betaine is usually harmless; however, cerebral edema has occurred when hypermethioninemia is extreme (>1000 µmol/L) [Yaghmai et al 2002, Devlin et al 2004, Tada et al 2004, Braverman et al 2005]. Eliminating betaine resulted in rapid reduction of the hypermethioninemia and resolution of the cerebral edema.
Note: In a murine model for homocystinuria the effect of betaine treatment diminished significantly over time [Maclean et al 2012].
Surveillance
Affected individuals should be monitored at regular intervals to detect any clinical complications that may develop, to assess dietary compliance, and to measure plasma total homocysteine and methionine concentrations. Infants should be monitored monthly for the first six months of life and bimonthly until age one year, then every three months until age three years. Semiannual monitoring through the remainder of childhood and annual monitoring in adolescence and adulthood are indicated. Complications should be promptly addressed with appropriate therapy.
Plasma total homocysteine and methionine concentrations should be monitored in all persons receiving betaine (see Prevention of Primary Manifestations, Betaine treatment).
Vitamin B12 and folate levels should be monitored.
Regular ophthalmology assessments can identify eye complications such as progressive myopia and ectopia lentis and allow for early treatment and prevention of further complications such as retinal detachment.
DXA scans should be performed every three to five years following adolescence to monitor for osteoporosis [Morris et al 2017].
Pregnancy Management
Because women with homocystinuria may be at greater than average risk for thromboembolism, especially post partum, prophylactic anticoagulation during the third trimester of pregnancy and post partum is recommended. The usual regimen is injection of low molecular-weight heparin during the last two weeks of pregnancy and the first six weeks post partum [Pierre et al 2006]. Aspirin in low doses has also been given throughout pregnancy.
Maternal homocystinuria, unlike maternal phenylketonuria (see Phenylalanine Hydroxylase Deficiency), does not appear to have major teratogenic potential requiring additional counseling or, with respect to the fetus, more stringent management [Levy et al 2002, Vilaseca et al 2004]. Nevertheless, treatment with pyridoxine or methionine-restricted diet or both should be continued during pregnancy [Morris et al 2017]. Betaine may also be continued and appears not to be teratogenic [Yap et al 2001b, Levy et al 2002, Vilaseca et al 2004, Pierre et al 2006].