Clinical Description
GRIN1-related neurodevelopmental disorder (GRIN1-NDD) is characterized by mild-to-profound developmental delay / intellectual disability in all affected individuals. Epilepsy (seen in 65%), muscular hypotonia (66%), and movement disorders (48%) are common manifestations.
To date, 72 individuals with GRIN1-NDD have been reported, including 64 individuals with de novo heterozygous pathogenic missense variants and eight individuals from four families with biallelic pathogenic missense or truncating variants [Firth et al 2009, Hamdan et al 2011, Allen et al 2013, Redin et al 2014, Farwell et al 2015, Ohba et al 2015, Zhu et al 2015, Bosch et al 2016, Halvardson et al 2016, Helbig et al 2016, Kobayashi et al 2016, Lemke et al 2016, Retterer et al 2016, Vanderver et al 2016, Chen et al 2017, Ortega-Moreno et al 2017, Rossi et al 2017, Tan et al 2017, Zehavi et al 2017, Dillon et al 2018, Fry et al 2018, Paderova et al 2018, Papa et al 2018, Pironti et al 2018, Staněk et al 2018]. The following description of the phenotypic spectrum associated with GRIN1-NDD is based on these reports. In 62 of the 72 reported individuals, clinical information was sufficient to draw conclusions on the overall phenotype (54 individuals heterozygous for a de novo missense variant and 8 individuals with homozygous variants). Of note, phenotypic data on 11 individuals with a heterozygous de novo variant comes from the DECIPHER database.
Developmental delay (DD) and intellectual disability (ID). All affected individuals have a variable degree of DD or ID (profound in 17%, severe in 71%, moderate in 7%, mild in 5%). No active speech has been noted in 48% of individuals.
Epilepsy. Seizures occurred in 65% of individuals. Some affected individuals presented with different seizure types over time. Where specified, seizures have been classified as epileptic spasms (13%), generalized seizures (68%), and focal seizures (20%). Seizure types reported among generalized and focal seizures comprise tonic, tonic-clonic, atonic, and/or myoclonic seizures, bilateral eyelid myoclonus, focal dyscognitive seizures, absence seizures, focal motor seizures, gelastic seizures, and status epilepticus.
Onset of seizures ranged from birth to 11 years with a median onset of 22.5 months. In 27 individuals on whom follow up or outcome on treatment with anti-seizure medication was available, 17 had refractory seizures and ten were well controlled with standard anti-seizure medication.
Other neurologic findings
Muscular hypotonia (in 66%)
Spasticity (40%)
Movement disorders (48%); where specified, affected individuals showed signs of dystonic (13%), dyskinetic (11%), and/or choreiform movements (15%).
Cortical visual impairment (34%)
Oculogyric crisis (11%)
Behavioral findings. Signs of autism spectrum disorder were observed in 22%. Other behavior issues included stereotypic movements (32%), self-injurious behavior (7%), and sleep disorder (15%).
Feeding difficulties / gastrointestinal abnormalities. Feeding difficulties were reported in 31% of individuals. Severe muscular hypotonia, gastroesophageal reflux, or oral-pharyngeal dysphagia with chewing and swallowing difficulty caused persistent feeding problems, requiring G-tube insertion in a subset of individuals.
Growth. Growth restriction or short stature was seen in 11% while microcephaly was documented in 27%.
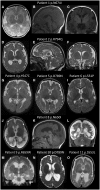
Neuroimaging. A malformation of cortical development (MCD) consisting of extensive diffuse bilateral polymicrogyria has been seen in 11 individuals [Fry et al 2018]. Polymicrogyria-affected brain regions comprised frontal, perisylvian, parietal, and temporal areas with some occipital sparing. Additional variable findings included increased extra-axial spaces, enlarged lateral ventricles, reduced white matter volume, thinning of the corpus callosum, and abnormal hippocampi. The MCD was similar in appearance to tubulinopathy-related or GRIN2B-related dysgyria [Platzer et al 2017]. See GRIN2B-Related Neurodevelopmental Disorder.
Other signs repeatedly noted in individuals without an MCD were generalized volume loss or cerebral atrophy (23%).
Signs of a leukoencephalopathy have been noted in two individuals with nonspecific hyperintensities of the white matter [Vanderver et al 2016, Pironti et al 2018].
Other
Prognosis. Psychomotor regression or loss of acquired skills has specifically been noted in one individual starting at age 3.5 years with loss of speech, impaired social interaction, drooling, and loss of sphincter control [Papa et al 2018].
It is unknown if life span in GRIN1-NDD is abnormal. Since many adults with disabilities have not undergone advanced genetic testing, it is likely that adults with this condition are underrecognized and underreported.