Clinical Description
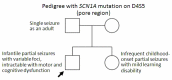
The natural history of SCN1A seizure disorders is strongly influenced by seizure phenotype, which can range from simple febrile seizures and generalized epilepsy with febrile seizures plus (GEFS+) at the mild end to Dravet syndrome and intractable childhood epilepsy with generalized tonic-clonic seizures (ICE-GTC) at the severe end [Kimura et al 2005, Mantegazza et al 2005, Fujiwara 2006, Gennaro et al 2006]. The phenotype varies even among family members with the same pathogenic variant (see ). As a result of this variable expressivity, long-term prognosis is difficult to determine.
Findings in a family illustrating variable expressivity among individuals with the same pathogenic variant. The proband, a boy (arrow) with febrile convulsions since age seven months, had frequent, difficult-to-control partial seizures beginning at age (more...)
Features associated with poor cognitive outcome include early myoclonic and absence seizures [Ragona et al 2011].
Phenotypes with intractable seizures (e.g., Dravet syndrome) usually cause epileptic encephalopathy, a form of progressive dementia. The root cause of the encephalopathy is unknown: the effects on cognition of seizures, the most obvious explanation, cannot be separated from the effects of medication or of an SCN1A pathogenic variant [Riva et al 2009].
In addition to having seizures in response to strong environmental stimuli, individuals with SCN1A seizure disorders often have an ADHD-like phenotype characterized by impulsivity, inattentiveness, and distractibility. Possibly related to the inability of the GABA system to provide negative feedback on extraneous sensory input, these symptoms tend to be less responsive to conventional stimulant medications.
The phenotypes in SCN1A seizure disorders include the following (see Table 2).
Table 2.
Seizure Phenotypes in SCN1A Seizure Disorders
View in own window
| Seizure Phenotypes | % of Individuals w/Phenotype & Identified SCN1A Pathogenic Variant |
|---|
| Intractable childhood epilepsy w/generalized tonic-clonic seizures (ICE-GTC) | 70% 1 |
| Dravet syndrome | 33%-90% 2 |
| Generalized epilepsy w/febrile seizures plus (GEFS+) | 5%-10% 3 |
| Febrile seizures plus (FS+) | Unknown |
| Simple febrile seizures | Unknown |
Intractable childhood epilepsy with generalized tonic-clonic seizures (ICE-GTC). This phenotype is defined as generalized seizures including absence seizures and generalized tonic-clonic seizures with onset in infancy or childhood. However, partial seizures can occur in up to 13% of affected individuals [Bonanni et al 2004]. Localized epilepsy, either alternating hemiconvulsive or complex partial seizures, may also be seen. Children with frequent generalized tonic-clonic seizures often develop cognitive impairment. The distinction between ICE-GTC and Dravet syndrome is not clear, and the former is not included in the ILAE classification system.
Dravet syndrome.
Wirrell et al [2017] published guidelines for the clinical diagnosis of Dravet syndrome. Presentation is between age one and 18 months after a period of normal development. Seizures are often prolonged and include recurrent generalized tonic-clonic or hemiconvulsive seizures. Myoclonic seizures are typically seen by age two years. Obtundation status, focal dyscognitive seizures, and atypical absences are often seen after age two years. The seizures are often triggered by hyperthermia (e.g., a hot bath, physical exertion, fever following vaccination), light stimuli, or sodium channel-blocking anti-seizure medication. Status epilepticus is common, and pharmacologic management is difficult. Seizures tend to lessen in severity after puberty; however, they rarely resolve completely.
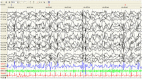
The initial EEGs are often normal or show nonspecific changes such as generalized slowing, but over time epileptiform activity appears. Patterns can include generalized spike and wave discharges, multiple spike and wave (also referred to as polyspike and wave) discharges, and multifocal spikes (see ). Brain MRI is typically normal or may show mild generalized atrophy and/or hippocampal sclerosis.
Individuals with Dravet syndrome often have an unusual seizure type that frequently will manifest as obtundation status epilepticus. The EEG during these difficult-to-classify seizures shows an alternation of generalized and focal discharges with variable (more...)
The myoclonic seizures that tend to appear later in the course often coincide with the appearance of cognitive dysfunction, ataxia, and psychomotor regression. Some degree of cognitive impairment is always seen, ranging from moderate to severe, often with marked inattention, impulsivity, and distractibility. Anxiety, obsessive personality traits, and autism spectrum disorder are common. Crouched gait, hypotonia, incoordination, and impaired dexterity are typically evident by age three to four years. Parkinsonian features of bradykinesia, tremor, and antecollis have been reported in adults with Dravet syndrome [Rilstone et al 2012, Aljaafari et al 2017].
Individuals with Dravet syndrome often develop a crouched gait. In spite of the gait being commonly described as "ataxic," affected individuals are more mobile than one would expect from how crouched they appear. The gait changes tend to be more prevalent in older children. In one study these changes were absent before age five years, but present in 5/10 children ages 6-12 years and in 8/9 children age 13 years or older [Rodda et al 2012]. In one cohort, 5/10 adults with Dravet syndrome had crouched gait [Rilstone et al 2012]. Decreased passive knee extension, increased external tibial torsion, and pes planovalgus all progressed [Rodda et al 2012]. Hip internal rotation did not show age-related changes. In one study antecollis was present in 9/14 and parkinsonian gait in 8/14 individuals with Dravet syndrome [Aljaafari et al 2017]. The degree of ataxia in affected individuals is greater than would be expected by the use of anticonvulsant medications alone. Pathogenic variants affecting the pore region appear to be more associated with gait changes [Kanai et al 2004, Rilstone et al 2012].
Severe myoclonic epilepsy, borderline (SMEB). This description is sometimes used for children who have some but not all of the features of Dravet syndrome [Fukuma et al 2004].
Generalized epilepsy. This phenotype is otherwise indistinguishable from idiopathic generalized epilepsy with onset in childhood or adolescence. Generalized epilepsies caused by SCN1A pathogenic variants are most often tonic, clonic, tonic-clonic, myoclonic, or absence.
Generalized epilepsy with febrile seizures plus (GEFS+). This term refers to the findings in a family rather than an individual [Arzimanoglou et al 2004]. In a family with GEFS+, epilepsy with variable expressivity and incomplete penetrance is inherited in an autosomal dominant manner. It implies a spectrum from mild (such as febrile seizure alone) to severe (including medically treatable generalized epilepsy, intractable generalized epilepsy, or Dravet syndrome). Intermediate phenotypes with myoclonic epilepsy, absence epilepsy, or focal epilepsy are also included. Individuals with GEFS+ often have febrile seizures (or FS+) in early childhood, followed by occasional tonic, clonic, myoclonic, or absence seizures that respond to medication and remit by late childhood or early adolescence. The proportion of children with GEFS+ whose first seizure occurs in the context of immunization appears to be greater than the proportion of children with febrile seizures unrelated to FS+ and GEFS+.
Febrile seizures plus (FS+). This subset of febrile seizures (simple or complex) is characterized by any of the following features:
Onset before age one year
Persistence beyond age six years
Unusual severity (including status epilepticus)
Occurrence of unprovoked (i.e., afebrile) seizures of any kind
Febrile seizures. These childhood seizures occur only in association with fever. The epidemiologic definition requires the following:
Onset on or after age six months
Resolution by age five years
Fever higher than 38°C (without other evidence of CNS infection)
No other identifiable cause
Febrile seizures are divided into simple febrile seizures and complex febrile seizures. Febrile seizures are considered complex if any of the following is present:
Duration longer than 15 minutes
Occurrence of more than one seizure within 24 hours
Presence of any partial (focal) features during the seizure
Febrile seizures with the following criteria are associated with a higher risk for developing Dravet syndrome [Hattori et al 2008]:
Febrile seizure onset before age seven months
Five or more febrile seizures
Prolonged seizure(s) lasting more than ten minutes
The febrile seizure characteristics include hemiconvulsions, partial seizures, myoclonic seizures, and hot water-induced seizures.
Infantile partial seizures with variable foci, also referred to as migrating partial seizures of infancy, cryptogenic focal epilepsy, or severe infantile multifocal epilepsy [Harkin et al 2007]. Multifocal partial seizures are often the first manifestation; however, in some children the first manifestation is febrile seizures. Severity varies and pharmacoresistance is common, but not absolute. Myoclonic seizures are rare but may be precipitated by administration of medications that inactivate the sodium channel, including phenytoin, carbamazepine, or lamotrigine. Cognitive deterioration may occur, especially when seizure control is incomplete. Electroencephalography shows multifocal independent spikes; generalized spike and wave discharges may be seen.
Less common phenotypes associated with SCN1A pathogenic variants include the following:
Myoclonic-astatic epilepsy (MAE, also called Doose syndrome). This phenotype is defined as the combination of myoclonic, atonic, and atypical absence seizures. Onset is usually after age two years (range: 7 months - 8 years). Although isolated myoclonic seizures as well as tonic seizures can occur, they are not characteristic of this syndrome (which distinguishes them from Lennox-Gastaut syndrome). Development prior to seizure onset is often normal. The course can range from spontaneous seizure resolution without cognitive impairment to intractable seizures with severe intellectual disability [
Arzimanoglou et al 2004].
Lennox-Gastaut syndrome (LGS). This phenotype is defined as slow spike-waves on EEG, developmental delay, and multiple types of generalized seizures (particularly atypical absence, tonic, and atonic seizures). LGS usually begins during childhood (ages 2-14 years). Any type of seizure can be seen in this syndrome; status epilepticus is common [
Arzimanoglou et al 2004]. Only a minority of persons with the LGS phenotype have an
SCN1A pathogenic variant, usually in the context of a family in which Dravet syndrome occurs [
Singh et al 2001]. This subset remains poorly characterized. It is unclear whether
SCN1A-associated LGS differs phenotypically from LGS of other etiologies.
Infantile spasms. This phenotype is defined as clustered seizures that show brief (<1 second) axial contractions associated with a slow-wave transient on EEG, often followed by generalized attenuation of the background. Both findings may be intermixed with fast activity. The resting EEG (between seizures) shows high-voltage slowing and a multifocal spike pattern known as hypsarrhythmia [
Arzimanoglou et al 2004]. Association of an
SCN1A pathogenic missense variant with infantile spasms has been reported once [
Wallace et al 2003]. The single individual represents fewer than 1% of reported cases, although publication bias makes it difficult to estimate the actual proportion.
Vaccine-related encephalopathy and seizures. This phenotype is defined as sudden onset of seizures and encephalopathy in infants 48 hours after immunization.
Berkovic et al [2006] identified an
SCN1A pathogenic variant in 11/14 children diagnosed with post-vaccine encephalopathy.
Tro-Baumann et al [2011] reported that 19 of 70 individuals with an SCN1A pathogenic variant and the Dravet phenotype had a history of seizures following vaccination.
Imaging. Brain MRI is most often normal early in the course of the disease; however, it often evolves to show cortical atrophy, cerebellar atrophy, white matter hyperintensity, ventricular enlargement, hippocampal sclerosis, or cortical dysplasia [Striano et al 2007]. Individuals with a more severe phenotype early in life often have more atrophic changes seen on MRI later in life.