Clinical Description
Ellis-van Creveld (EVC) syndrome is characterized by postaxial polydactyly of the hands, disproportionate short stature, features of ectodermal dysplasia, congenital heart disease, and radiologic abnormalities (such as short ribs and short tubular bones) [Da Silva et al 2023]. Other less common and more variable features include postaxial polydactyly of the feet, nonspecific dysmorphic facial features, and developmental delay. To date, approximately 250 individuals with EVC syndrome have been identified with pathogenic variants in one of the genes listed in Table 1 [Aubert-Mucca et al 2023, Da Silva et al 2023]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Ellis-van Creveld Syndrome: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
|
Postaxial polydactyly (hands)
| 98% | Bilateral in >95% |
|
Limb shortening
| 83% | Prenatal limb shortening in 36% |
|
Nail dystrophy/hypoplasia
| 78% | |
|
Short stature
| 73% | |
|
Congenital heart disease
| 66% | Atrial septal defect in >80% |
|
Thoracic narrowing
| 66% | Typically symmetric |
|
Dental anomalies
| 59% | |
|
Brachydactyly
| 35% | |
|
Postaxial polydactyly (feet)
| 34% | Bilateral in >90% |
|
Upper lip defect
| 28% | |
|
Developmental delay
| 9% | |
Growth deficiency. Disproportionate short stature is common in individuals with EVC syndrome. About one third of individuals manifest growth deficiency in the perinatal setting, with shortened long bones evident on prenatal ultrasound, as well as short stature and/or limb shortening at birth [Da Silva et al 2023]. Most children present with growth failure and consequent short stature. Body segments are disproportionate, with a long, narrow thorax, usually accompanied by shortening of all four limbs. Limb segments are typically affected, with increasing severity from proximal to distal (rhizomelia > mesomelia > acromelia) [Baujat & Le Merrer 2007]. Adult height is most often below the midparental height and ranges between 109 and 152 cm.
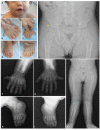
Musculoskeletal manifestations. The most common feature in individuals with EVC syndrome is bilateral postaxial polydactyly of the hands (see and ) [Da Silva et al 2023]. Almost all individuals have hexadactyly of each hand, although a smaller subset have heptadactyly (seven digits) on each hand. Postaxial polydactyly of the feet is less common and is almost always bilateral. Polydactyly can sometimes be identified on prenatal ultrasound.
The thorax is usually long and narrow due to underdevelopment of the rib cage / short ribs, which may lead to lung hypoplasia during development or restrictive lung disease after birth. However, respiratory insufficiency is rarely severe [Huber & Cormier-Daire 2012]. Less commonly, pectus carinatum can be observed.
Other common skeletal abnormalities include shortening of long bones, brachydactyly (see and ), carpal and metacarpal fusions (see ), and genu valgum (due to a delay in ossification of the lateral tibial metaphysis) (see ) [Handa et al 2020, Da Silva et al 2023]. The ulnae and radii usually have a distinct appearance due to the presence of abnormally bulbous ends. Bone age is usually delayed [Baujat & Le Merrer 2007]. Clinodactyly and syndactyly have also been reported.
The skull and spine are usually normal [Handa et al 2020]. Muscle strength is also normal.
Dental and oral manifestations. Anomalies of the teeth are frequent and varied. The most common findings are delayed eruption and hypodontia. Some individuals have natal teeth or early eruption of the primary dentition during the first two months of life and dental dysmorphism (e.g., cone-shaped teeth). Less frequent findings include enamel hypoplasia, dental transposition, and taurodontism [Peña-Cardelles et al 2019]. Frenulum anomalies, with multiple frenula and adhesions, are also common [Da Silva et al 2023].
Nail dystrophy. Nails are usually hypoplastic, and all nails show at least mild dystrophy, including discoloration, brittleness, pitting, ingrown nails, and deep-set nails (see ).
Congenital heart defects are relatively frequent and can be detected prenatally. When present, they almost always include an atrial septal defect that can be isolated or coexist with other malformations. Ventricular septal defects, single atrium, and left superior vena cava are also rather common. Just under half of individuals with congenital heart defects develop some degree of heart failure [Hills et al 2011]. Other types of malformations (e.g., hypoplastic left ventricle, pulmonary valve stenosis/atresia, aortic coarctation) are rare [Hills et al 2011].
Craniofacial features are variable and nonspecific, and there is no specific facial gestalt for EVC syndrome. Upper lip defects (see ) frequently manifest with partial cleft lip (without cleft palate); disruption of the superior gingivolabial groove due to adhesions is frequently seen with the lip defect. Additional reported facial features described in less than one in four individuals include hypertelorism, short broad nose (see ), long philtrum, and postnatal microcephaly [Da Silva et al 2023].
Development. Intellectual disability is not typical of EVC syndrome. Individuals usually exhibit normal intelligence. A small percentage of individuals exhibit developmental delay and/or intellectual disability that is mostly mild or moderate [Öztürk et al 2021, Zaka et al 2021, Qian et al 2022]. Motor skills are most often affected, which may be secondary to the musculoskeletal abnormalities.
Other. Other rare features (described in <5% of individuals with EVC syndrome) include:
Genitourinary abnormalities (epispadias, hypospadias, cryptorchidism, hydrometrocolpos, kidney malformations, kidney cysts)
Central nervous system malformations (Dandy-Walker malformation, corpus callosum hypoplasia, cerebellar hypoplasia)
Sensorineural deafness
Phenotype Correlations by Gene
EVC2. A detailed assessment showed increased frequency of some manifestations in individuals with EVC2-related EVC syndrome compared to those with pathogenic variants in EVC [Da Silva et al 2023], but the phenotype in those with EVC-related EVC syndrome is most often clinically indistinguishable from that of EVC2-related EVC syndrome. Individuals with biallelic EVC2 pathogenic variants have increased shortening and thickening of the tubular bones and lower weight than individuals with EVC pathogenic variants. This suggests (but does not confirm) increased severity for EVC2-related EVC syndrome.
DYNC2H1, DYNC2LI1, GLI1, PRKACA, PRKACB, SMO,
and
WDR35. EVC syndrome due to pathogenic variants in DYNC2H1, DYNC2LI1, GLI1, PRKACA, PRKACB, SMO, or WDR35 are rare, so phenotype correlations by gene cannot be definitively established. However, postaxial polydactyly of the feet seems to be more common (and present in most individuals) with DYNC2H1-, DYNC2LI1-, GLI1-, SMO-, and WDR35-related EVC syndrome [Caparrós-Martín et al 2015, Palencia-Campos et al 2017, Niceta et al 2018, Aubert-Mucca et al 2023, Piceci-Sparascio et al 2023].
Genotype-Phenotype Correlations
EVC syndrome caused by large intragenic deletions or duplications affecting EVC and/or EVC2 are associated with more atypical clinical presentations, with a decrease in the proportion of musculoskeletal findings and an increase in the frequency of dysmorphic features [Da Silva et al 2023].
EVC
No definitive genotype-phenotype correlations have been identified for DYNC2H1-, DYNC2LI1-, GLI1-, PRKACA-, PRKACB-, SMO-, or WDR35-related EVC syndrome.