Clinical Description
Pain is the predominant symptom of SCN9A neuropathic pain syndromes. Triggers for episodes of pain vary with the specific syndrome. Pain may be accompanied by signs of redness, warmth, or autonomic dysfunction. Erythromelalgia (EM) and paroxysmal extreme pain disorder (PEPD) typically begin in infancy or childhood, whereas small fiber neuropathy (SFN) typically begins in adulthood, and the prevalence increases with age. All three disorders have been reported with intrafamilial variability in age of onset and severity.
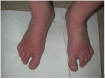
Erythromelalgia (EM) is characterized by recurrent attacks of intense pain, redness, warmth, and swelling involving the feet and, less frequently, the hands [Dib-Hajj et al 2013, McDonnell et al 2016]. Warmth is an essential part of the syndrome. During the attacks, the extremities appear red or purple and may be swollen. Commonly, the attacks occur in the evening or at night and so may not be observed by a physician. The individual may seek medical advice for painful extremities but neglect to mention the characteristic warmth or redness (especially if limited to the soles of the feet). The symptoms are usually bilateral and symmetric. Within a family, the manifestations of the disorder may vary considerably.
Onset of SCN9A-EM is usually in childhood or adolescence; however, it has been recognized in infants in families who are familiar with the disorder. While rare, later onset (age >20 years) has been reported in some individuals and families [Choi et al 2010].
Initially, the symptoms involve the soles of the feet and the hands; with age, the lower legs and the arms may become involved. In individuals with advanced disease, symptoms may occur many times a day and last hours, especially at night, or become constant and unremitting.
At the onset, the episodes are triggered by exposure to warmth. A pathognomonic feature is triggering of episodes by warm or hot ambient temperature and relief with cooling of the extremities.
Less consistent precipitating factors include exercise, tight shoes, wearing socks, alcohol, spicy foods, and other vasodilating agents.
Some individuals have allodynia (pain evoked by a normally innocuous stimulus) and hyperalgesia (increased sensitivity to a painful stimulus).
The episodes may be disabling, interfere with sleep, and severely limit normal activities such as walking, participation in sports, wearing socks and shoes, and attending school or going to work. Individuals tend to limit their activities in warm weather and to stay in air-conditioned environments. Some individuals move from hot, humid climates to cooler climates.
Affected individuals prefer to wear open-toed shoes and to sleep with their feet uncovered. Swimming can be helpful because it keeps the limbs cool during exercise.
Neurologic examination is typically normal, although reduced ankle reflexes and decreased distal sensation can be seen.
Histopathologic examination of skin biopsy in individuals with erythromelalgia shows nonspecific thickening of blood vessel basement membrane, perivascular edema and mononuclear infiltrate, and reduced density of the autonomic nerve plexuses.
Paroxysmal extreme pain disorder (PEPD) is characterized by episodes of rectal, ocular, or submandibular pain accompanied by erythema. The onset is in early infancy, and may occur prenatally [Fertleman et al 2007]. Infants may appear stiff and red at the time of delivery, but recover within minutes. Attacks are triggered by defecation, wiping the perineum, taking rectal temperature, or feeding. Pain is inferred from the infant's grimacing and crying.
Older children and adults describe the pain as excruciating, and burning or sharp. The attacks are always accompanied by erythematous flushing of the skin, which usually corresponds to the affected region. In severe attacks, the pain spreads and become generalized.
The number of rectal attacks can decrease with age, whereas the ocular and jaw attacks can increase with age [Fertleman et al 2007].
In PEPD, biopsies of rectum or colon have been reported as normal or only nonspecific findings [Fertleman et al 2007]
Inherited small fiber neuropathy (SFN) is characterized by symmetric, distal burning pain, numbness, and paresthesias. Autonomic manifestations such as dry eyes or mouth, orthostatic dizziness, change in sweating, and bowel or bladder dysfunction may be present. The prevalence of SFN increases with age.
Pathogenic missense variants in SCN9A were reported in eight of 28 adults (age ≥18 years) with inherited small fiber neuropathy (I-SFN) in whom other known causes for small fiber neuropathy (e.g., diabetes mellitus, impaired glucose tolerance, hyperlipidemia) were excluded, and who met strict diagnostic criteria for I-SFN with reduced intraepidermal nerve fiber density on skin biopsy or abnormal quantitative sensory testing [Faber et al 2012].
Of note, in SFN, the intraepidermal nerve fiber density (IENFD) in a skin biopsy is quantitatively reduced compared with age- and sex-adjusted normative values [Faber et al 2012], although I-SFN can occur in persons with normal IENFD [Eijkenboom et al 2019].