Genetic Hearing Loss Overview
A Eliot Shearer, MD, PhD, Michael S Hildebrand, PhD, Amanda M Schaefer, MS, LGC, and Richard JH Smith, MD.
Author Information and AffiliationsInitial Posting: February 14, 1999; Last Revision: September 28, 2023.
Estimated reading time: 40 minutes
Summary
The purpose of this overview is to increase the awareness of clinicians regarding the diagnosis, management, and genetic counseling of common causes of genetic hearing loss.
The goals of this overview on genetic hearing loss are the following.
Goal 2.
Review the causes of genetic hearing loss.
Goal 5.
Review the management of genetic hearing loss.
Goal 6.
Inform the genetic counseling of an individual with genetic hearing loss and their family members.
1. Audiometric and Clinical Aspects of Hearing Loss
Hearing loss can be characterized by type, onset, severity, and frequency.
Type
Conductive hearing loss, due to abnormalities of the external ear and/or the ossicles of the middle ear
Sensorineural hearing loss due to malfunction of the inner ear structures (i.e., cochlea or auditory nerve, with potential for vestibular dysfunction)
Mixed hearing loss, a combination of conductive and sensorineural hearing loss
Central auditory dysfunction, due to damage or dysfunction at the level of the eighth cranial nerve (auditory nerve), the auditory brain stem (medulla, pons, and midbrain), or the cerebral cortex
Onset
Congenital (present at birth)
Prelingual (occurring before the acquisition of speech)
Postlingual (occurring after the acquisition of speech)
Adult (occurring after age 18 years)
Presbycusis (age-related hearing loss that typically occurs after middle age)
Severity. Hearing is measured in decibels (dB). The threshold or 0 dB mark for each frequency refers to the level at which normal young adults perceive a tone burst 50% of the time. Hearing is considered normal if an individual's thresholds are within 15 dB of normal thresholds. Severity of hearing loss is graded as shown in Table 1.
Table 1.
Severity of Hearing Loss in Decibels (dB)
View in own window
| Severity | Hearing Threshold in dB |
|---|
| Slight | 12-25 dB |
| Mild | 26-40 dB |
| Moderate | 41-60 dB |
| Moderately severe | 61-70 dB |
| Severe | 71-90 dB |
| Profound | >90 dB |
Frequency (See Table 2.)
Table 2.
Human Hearing by Frequency of Sound
View in own window
| Frequency Level | Hertz (Hz) |
|---|
| Low | <500 Hz |
| Medium | 501-2,000 Hz |
| High | >2,000 Hz |
Laterality
Bilateral symmetric
Bilateral asymmetric
Unilateral
Configuration or shape or audioprofile (degree and pattern of hearing loss across frequencies)
Low frequency, upsloping, or rising
Mid-frequency, "cookie bite"
High frequency, downsloping
Flat
Assessment of the auditory system. Formal hearing assessment is performed by audiologists who are trained to adapt their clinical evaluation to the ability of the individual being tested. The method of assessment of hearing depends on the individual's age or developmental age and neurologic status (i.e., ability to respond to queries from the audiologist).
Audiometric measures based on the individual's age or ability to respond to queries from the audiologist include:
Visual response audiometry: age <2.5 years
Conditioned play audiometry: age 2.5-5 years
Conventional audiometry: age >5 years
Objective measures, which do not require participation of the individual, are typically used in young children or individuals who are unable to participate in other audiometric testing. They include:
Auditory brain stem response (ABR, also called brain stem auditory evoked response, or BAER), which measures physiologic response of the auditory nerve, brain stem, and brain to varying auditory stimuli. Automated ABR may be used in newborn hearing screening. Diagnostic ABR is used for assessment of transmission of auditory stimuli from the ear to the brain.
Otoacoustic emissions (OAE), which measure the response of the cochlea to auditory stimulus. OAE may be used in newborn hearing screening.
Conduction tests compare sound conduction through air and through bone to differentiate middle ear dysfunction (conductive hearing loss) from inner ear dysfunction (sensorineural hearing loss).
Air conduction tests measure responses from auditory stimuli presented through the external auditory canal.
Bone conduction tests measure responses from auditory stimuli transmitted through the bone near the inner ear.
Tympanometry assesses the function of the tympanic membrane and middle ear and is part of routine audiometric evaluation performed by an audiologist. The goal is to differentiate conductive hearing loss from sensorineural hearing loss.
Vestibular function testing evaluates the integrity of the peripheral vestibular system. This testing is typically performed by audiologists with specialized training and evaluates the ability of the inner ear to maintain balance and coordinate the vestibular-ocular pathways that are integral in maintenance of head position in space. Imbalance, dizziness, and vertigo are common among individuals with hearing loss given that the cochlea (responsible for hearing) and the vestibular organs (the saccule, utricle, and semicircular canals) share much of the same physiology and developmental origin.
Definitions of hearing loss and deafness
Hearing loss. Auditory
phenotype characterized by any degree of loss of the ability to hear
deaf (small "d"). In this
GeneReview, "deaf" describes an auditory
phenotype characterized by a total or near total loss of the ability to hear.
Deaf (always a capital "D"). A community with a distinct culture and visual language shared by the experience of being deaf or hard of hearing. The Deaf can include persons who are deaf, hard of hearing, and hearing. In the United States, members of the Deaf community use American Sign Language (ASL). Similar communities and sign languages exist in other countries. Members of the Deaf community (i.e., the Deaf) do NOT consider themselves to be hearing "impaired," nor do they feel that they have a hearing "loss." Their deafness is not considered a pathology or disease to be treated or cured.
Hard of hearing. Auditory
phenotype characterized by a partial loss of the ability to hear. It is sometimes used by the Deaf to signify that a person has some usable hearing – anything from mild to severe hearing loss.
2. Causes of Genetic Hearing Loss
Hearing loss is not a diagnosis; rather, hearing loss is a symptom of an underlying pathologic change to the auditory system.
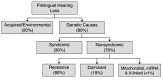
Eighty percent of prelingual hearing loss is attributed to genetic causes; likewise, a genetic cause can be identified in a signification proportion of individuals with adult-onset hearing loss (see ). Determining the genetic cause of hearing loss provides information for individuals and their families, including prognosis (e.g., whether hearing loss is static or progressive), whether hearing loss is nonsyndromic or syndromic (i.e., associated with other medical issues), habilitation options and other supportive care, availability of disease-specific therapies, and genetic counseling regarding chance of recurrence. Habilitation for hearing loss includes improved access to sound through hearing aids or cochlear implants and, when desired, exposure to and teaching of American Sign Language.
Causes of prelingual hearing loss in developed countries [Morton & Nance 2006, Sheffield & Smith 2019]
Nonsyndromic hearing loss is not associated with visible abnormalities of the external ear or related medical findings; however, it can be associated with abnormalities of the middle ear and/or inner ear.
Syndromic hearing loss is associated with any combination of malformations of the external ear or malformations or medical findings involving other organs or organ systems.
Nonsyndromic hearing loss "mimics" refers to syndromic hearing loss that "mimics" nonsyndromic hearing loss in the early presentation period, when other organ system involvement may not be evident on medical history and/or physical examination. Of note, approximately 20% of children with hearing loss as their only initial clinical feature (i.e., who apparently have nonsyndromic hearing loss) will subsequently be diagnosed with syndromic hearing loss [Sloan Heggen et al 2016, Downie et al 2020].
Nonsyndromic Hearing Loss
As of this writing, more than 125 genes are associated with nonsyndromic hearing loss (a regularly updated, comprehensive list of identified nonsyndromic hearing loss genes is available at the Hereditary Hearing Loss Homepage).
Approximately 70% of prelingual genetic hearing loss is nonsyndromic (see ) [Smith et al 2005, Sheffield & Smith 2019] (full text). In most individuals with nonsyndromic genetic hearing loss (80%), hearing loss is associated with biallelic pathogenic variants and inherited in an autosomal recessive manner. Nonsyndromic hearing loss may also be inherited in an autosomal dominant manner (19%) or, rarely, associated with mitochondrial or X-linked inheritance (<1%). Although comparable data are not available for postlingual nonsyndromic genetic hearing loss, autosomal dominant inheritance is most common.
Note: Nonsyndromic hearing impairment may be referred to by the gene involved (e.g., OTOF-related deafness) or by the genetic locus (e.g., DFNB9). Nonsyndromic deafness loci are designated DFN (for DeaFNess) and further classified by mode of inheritance (DFNA: autosomal dominant; DFNB: autosomal recessive; DFNX: X-linked) and a number indicating the numeric locus assigned in the course of gene mapping.
Autosomal Recessive Nonsyndromic Hearing Loss
As of this writing, more than 70 genes have been associated with autosomal recessive nonsyndromic hearing loss.
In general, autosomal recessive nonsyndromic hearing loss is prelingual and severe to profound. However, there is a spectrum of degrees of hearing loss, and exceptions to this generalization and/or distinctive features associated with selected genes are summarized in Table 3.
Note: Table 3 lists selected genes associated with distinctive clinical features; for a current, comprehensive list of all identified autosomal recessive nonsyndromic hearing loss genes, see Hereditary Hearing Loss Homepage.
Table 3.
Autosomal Recessive Nonsyndromic Hearing Loss: Distinctive Features Associated with Selected Genes
View in own window
| Gene | Distinctive Features |
|---|
|
BDP1
| HL can be progressive & postlingual. |
|
CDH23
| Assoc w/phenotypic spectrum incl AR nonsyndromic HL & AR syndromic HL (See Usher Syndrome Type I.) |
|
EPS8L2
| HL can be progressive & postlingual. |
|
GJB2
| GJB2-related AR nonsyndromic HL is the most common genetic cause of severe-to-profound AR nonsyndromic HL in Asian & White populations. In contrast, biallelic GJB2 pathogenic variants essentially do not contribute to genetic HL in sub-Saharan African populations. 1 |
|
LOXHD1
| HL can be progressive & postlingual. |
|
MYO7A
|
|
|
PCDH15
| Assoc w/phenotypic spectrum incl AR nonsyndromic HL & AR syndromic HL (See Usher Syndrome Type I.) |
|
SLC26A4
|
|
|
STRC
|
|
|
TECTA
| Assoc w/phenotypic spectrum incl AR & AD nonsyndromic HL |
|
TMC1
|
|
TMPRSS3
| HL can be progressive & postlingual. |
|
USH1C
| Assoc w/phenotypic spectrum incl AR nonsyndromic HL & AR syndromic HL (See Usher Syndrome Type I.) |
|
WHRN
| Assoc w/phenotypic spectrum incl AR nonsyndromic HL & AR syndromic HL (See Usher Syndrome Type II.) |
Autosomal Dominant Nonsyndromic Hearing Loss
As of this writing, 50 genes have been associated with autosomal dominant nonsyndromic hearing loss.
In general, autosomal dominant nonsyndromic hearing loss is postlingual, progressive, and high frequency. Exceptions to this generalization and/or distinctive features associated with selected genes are summarized in Table 4.
Note: Table 4 lists selected genes associated with distinctive clinical features; for a current, comprehensive list of all identified autosomal dominant nonsyndromic hearing loss genes, see Hereditary Hearing Loss Homepage.
Table 4.
Autosomal Dominant Nonsyndromic Hearing Loss: Distinctive Features Associated with Selected Genes
View in own window
| Gene | Distinctive Features |
|---|
|
COL11A2
| HL is low frequency or mid-frequency. |
|
DIAPH1
|
|
GJB2
| HL is prelingual. (See GJB2-Related AR Nonsyndromic HL.) |
|
MYO7A
| Assoc w/phenotypic spectrum incl AD & AR nonsyndromic HL & AR syndromic HL (See Usher Syndrome Type I.) |
|
TECTA
|
|
|
TMC1
| Assoc w/phenotypic spectrum incl AR & AD nonsyndromic HL |
|
WFS1
| HL is low frequency or mid-frequency. |
X-Linked Nonsyndromic Hearing Loss
As of this writing, five genes (AIFM1, COL4A6, POU3F4, PRPS1, and SMPX) are associated with X-linked nonsyndromic prelingual or postlingual hearing loss.
Distinctive features associated with selected genes are summarized in Table 5.
Table 5.
X-Linked Nonsyndromic Hearing Loss: Distinctive Features Associated with Selected Genes
View in own window
Mitochondrial DNA (mtDNA) Hearing Loss
Mitochondrial DNA-associated hearing loss is inherited in a maternal manner. Most mtDNA hearing loss is syndromic and involves a broad spectrum of multisystem disorders. Exceptions include the genes MT-RNR1 and MT-TS1, in which hearing loss is nonsyndromic and likely due to increased sensitivity to cellular damage caused by aminoglycoside antibiotics and other ototoxic drugs [McDermott et al 2022]. See Nonsyndromic Hearing Loss and Deafness, Mitochondrial.
Syndromic Hearing Loss
At the time of this writing, Online Mendelian Inheritance in Man (OMIM) lists 665 entries for syndromic forms of sensorineural hearing loss (SNHL). See Table 6a for select common causes of autosomal dominant syndromic hearing loss. See Table 6b for select common causes of autosomal recessive syndromic hearing loss. See Table 6c for select common causes of X-linked syndromic hearing loss.
Syndromic hearing loss accounts for about 20% of prelingual genetic hearing loss. The contribution of syndromic hearing loss to later-onset hearing loss is an active focus of research but is not currently known.
Table 6a.
Select Common Causes of Autosomal Dominant Syndromic Hearing Loss
View in own window
| Gene(s) | Syndrome | Hearing Loss | Comment |
|---|
| Type | Onset | Severity |
|---|
|
CHD7
|
CHD7 disorder (incl CHARGE syndrome)
| Conductive, SNHL | Variable | Variable | Cranial nerve VIII dysfunction/anomaly: SNHL &/or vestibular dysfunction Cochlear implantation may be difficult or impossible due to altered cochleovestibular anomalies. Other cranial neuropathies may also be present. Ear malformations such as ossicular malformations assoc w/complex mixed HL
|
COL2A1
COL11A1
COL11A2
| AD Stickler syndrome | Conductive, SNHL | Variable | Variable | 40% have some degree of sensorineural hearing impairment (typically high tone, often subtle). Conductive HL can be seen & may be secondary to recurrent ear infections that are often assoc w/cleft palate &/or may be secondary to a defect of the ossicles of the middle ear.
|
EDN3
EDNRB
KITLG
MITF
PAX3
SNAI2
SOX10
| Waardenburg syndrome (WS) (See Waardenburg Syndrome Type I.) | SNHL | Congenital | Variable | Hearing loss, pigmentary anomalies (white forelock), & dystopium canthorum (widely spaced medial canthus) are most commonly seen clinical features. Various temporal bone abnormalities have been identified in persons w/WS1 & HL. WS2, WS3, & WS4 can be inherited in an AD or AR manner.
|
EYA1
SIX1
SIX5
| Branchiootorenal spectrum disorder (BOR) | Conductive, SNHL, mixed | Variable | Variable | Kidney abnormalities Branchial anomalies (branchial cleft tags, pits, cysts) Malformations of outer, middle, & inner ear (e.g., preauricular pits)
|
|
NF2
|
Neurofibromatosis 2
| SNHL | Average: age 18-24 yrs | Generally unilateral & gradual; can be bilateral & sudden | Bilateral vestibular schwannomas w/assoc symptoms of tinnitus, HL, & balance dysfunction Vestibular schwannomas are a rare, potentially treatable type of HL.
|
Table 6b.
Select Common Causes of Autosomal Recessive Syndromic Hearing Loss
View in own window
| Gene(s) | Syndrome | Hearing Loss | Comment |
|---|
| Type | Onset | Severity |
|---|
ADGRV1
USH2A
WHRN
|
Usher syndrome (USH) type II
| SNHL | Congenital | Mild to severe |
|
|
BTD
|
Biotinidase deficiency
| SNHL | Variable | Variable; some degree of HL is present in ≥75% of children who become symptomatic. | If not recognized & corrected by daily addition of biotin to diet, affected persons develop neurologic features (e.g., seizures, hypotonia, DD, ataxia, vision findings, HL, & cutaneous abnormalities. |
CDH23
MYO7A
PCDH15
USH1C
USH1G
|
Usher syndrome (USH) type I
| SNHL | Congenital | Severe to profound | Usher syndrome overall is the most common type of AR syndromic HL; USH1 is the most common type of Usher syndrome. Nonsyndromic HL mimic: HL is congenital; RP begins in late adolescence or early adulthood. Unless fitted w/cochlear implant, affected persons do not typically develop speech. Severe vestibular dysfunction or vestibular areflexia. Imbalance is assoc w/HL & is defining feature of USH1. Children typically walk later than usual, at age ~18 mos-2 yrs. CDH23, PCDH15, & USH1C are also assoc w/AR nonsyndromic HL. MYO7A is also assoc w/AD & AR nonsyndromic HL.
|
|
CLRN1
| Usher syndrome type III (OMIM PS276900) | SNHL | Congenital | Variable SNHL severity, progressive | Usher syndrome overall is the most common type of AR syndromic HL. |
COL9A1
COL9A2
COL9A3
| AR Stickler syndrome | Conductive, SNHL | Variable | Variable | 40% have some degree of sensorineural hearing impairment (typically high tone, often subtle). Conductive hearing loss can be seen & may be secondary to recurrent ear infections that are often assoc w/cleft palate &/or may be secondary to a defect of the ossicles of the middle ear.
|
KCNE1
KCNQ1
|
Jervell & Lange-Nielsen syndrome
| SNHL | Congenital | Profound | HL & cardiac conduction anomalies Classic presentation is a child who experiences syncopal episodes during periods of stress, exercise, or fright. 50% of affected persons had cardiac events before age 3 yrs.
|
PEX1
PEX2
PEX3
PEX5
PEX6
PEX10
PEX11B
PEX12
PEX13
PEX14
PEX16
PEX19
PEX26
| Zellweger spectrum disorder (ZSD) | SNHL | Variable | Variable | ZSD is due to peroxisome disorder & incl hearing & vision loss, hypotonia, & other clinical features. Milder ZSD may first come to attention due to failed hearing screening.
|
PEX7
PHYH
|
Adult Refsum disease
| SNHL | Variable | Severe, progressive | RP, anosmia, neuropathy, ataxia, & HL Auditory nerve involvement (auditory neuropathy) may be evident on testing of auditory brain stem evoked responses. Persons w/auditory nerve involvement may experience hearing difficulty even in presence of normal audiogram.
|
| SLC26A4 1 | Pendred syndrome (PDS) | SNHL | Congenital or childhood onset | Usually (but not invariably) severe to profound | HL, vestibular dysfunction, & thyroid goiter HL may fluctuate but often progresses. Vestibular dysfunction should be suspected in infants w/delayed walking. Temporal bones are abnormal radiologically in all persons w/PDS & most commonly incl incomplete partition type II anomaly & enlarged vestibular aqueduct. SLC26A4 is also assoc w/AR nonsyndromic HL.
|
- 1.
Digenic inheritance, in which an affected individual has double heterozygosity for a pathogenic variant in SLC26A4 and a pathogenic variant in FOXI1 [Yang et al 2007], or double heterozygosity for a pathogenic variant in SLC26A4 and a pathogenic variant in KCNJ10 has also been observed in Pendred syndrome.
Table 6c.
Select Common Causes of X-Linked Syndromic Hearing Loss
View in own window
| Gene(s) | Syndrome | Hearing Loss | Comment |
|---|
| Type | Onset | Severity |
|---|
|
COL4A5
| Alport syndrome 1 | SNHL | Typically after age 10 yrs | Varying severity, progressive | |
|
TIMM8A
| Deafness-dystonia-optic neuronopathy syndrome (Mohr-Tranebjaerg syndrome) | SNHL | Early childhood | Progressive, pre- or postlingual | Nonsyndromic HL mimic: HL is always presenting manifestation. Slowly progressive dystonia or ataxia develops in teens, & slowly progressive decreased visual acuity from optic atrophy develops at age ~20 yrs. Dementia develops at age ~40 yrs. |
SNHL = sensorineural hearing loss
- 1.
3. Differential Diagnosis of Genetic Hearing Loss
In developed countries, approximately 65% of prelingual hearing loss is attributed to genetic causes and approximately 35% to acquired/environmental causes (see ).
The most common acquired/environmental cause of prelingual hearing loss is congenital cytomegalovirus (cCMV) infection (the overall birth prevalence of cCMV is approximately 0.64% [Kenneson & Cannon 2007]). Testing for cCMV, typically performed by PCR from saliva or urine, is most specific for cCMV when performed in infants who are younger than three weeks of age.
About 10% of infants with cCMV are symptomatic with findings that can include a characteristic rash, hearing loss (in ~50%), neurologic deficits (e.g., seizures, spasticity), and/or hepatic insufficiency. Some infants succumb to their complications.
About 90% of infants with cCMV are considered "asymptomatic." Of these up to 10% develop unilateral or bilateral hearing loss. This variability is characteristic of cCMV-related hearing loss.
Acquired/environmental hearing loss in children may also result from:
Prenatal infections caused by "TORCH" organisms (i.e., toxoplasmosis, rubella, cytomegalovirus, and herpes);
Postnatal infections, particularly bacterial meningitis caused by Neisseria meningitidis, Haemophilus influenzae, or Streptococcus pneumoniae. Other causative organisms include Escherichia coli, Listeria monocytogenes, Streptococcus agalactiae, and Enterobacter cloacae.
Acquired/environmental hearing loss in adults is most often attributed to factors such as noise and medications. It is likely that complex environmental-genetic interactions affect the age of onset and severity of age-related hearing loss (presbycusis) and noise-induced hearing loss [Ahmadmehrabi et al 2021].
4. Evaluation Strategy
Identification of Children with Hearing Loss
Newborns. Universal newborn hearing screening (NBHS) using physiologic screening (either otoacoustic emissions [OAE] or automated ABR [AABR]), required by law or rule in all 50 states in the United States, is performed on >98% of children in the US typically within days after birth (see CDC Annual Data). Universal NBHS in the US identifies about 6,000 children with hearing loss each year; note that NBHS, which is designed to detect moderate-to-profound hearing loss, may miss infants with mild hearing loss.
Other children referred for complete diagnostic assessment regardless of NBHS result often include:
Children considered to have a high chance of hearing loss for reasons such as a family history of hearing loss, a syndrome associated with hearing loss (e.g., trisomy 21 or cleft lip and palate), or exposure to specific environmental risk factors (e.g., care in a neonatal intensive care unit [NICU], exposure to aminoglycoside antibiotics);
School-age children who typically have milder hearing loss that was not detected on NBHS and are often identified following: (1) parental concern regarding hearing; (2) delayed speech or language development; and/or (3) hearing screening as part of a well-child examination or school program [
Wake et al 2006].
Identification of Adults with Hearing Loss
Adults with hearing loss – at the time of presentation – typically are symptomatic with either diminished hearing or tinnitus (ringing in the ears), which may indicate hearing loss.
Initial Diagnostic Evaluation
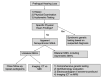
Initial diagnostic evaluation of any individual with hearing loss (see ) should include genetic testing (see Molecular Genetic Testing) as well as the following:
Diagnostic algorithm for presumed hereditary nonsyndromic sensorineural hearing loss
Family history of
hearing loss, particularly early-onset hearing loss, and presence of features suggestive of the syndromes included in
Tables 6a,
6b, and
6c (select common syndromes with hearing loss organized by
mode of inheritance)
Evaluation of vestibular function is indicated because about 50% of hearing loss is associated with peripheral vestibular dysfunction. The finding of vestibular dysfunction may guide physical therapy, particularly in children with delayed motor milestones.
Consideration of
imaging (CT and/or MRI) if hearing loss is unilateral or asymmetric and/or sudden onset.
Other ancillary testing/evaluation. Electrocardiogram (EKG) is often performed in children with severe-to-profound hearing loss to evaluate for prolonged QT interval, which may be life-threatening. An ophthalmology evaluation may be recommended, as individuals with hearing loss may depend more on vision for sensory input, as well as to evaluate for retinitis pigmentosa.
Timing and order of diagnostic evaluation is individual specific and depends on the age of the individual and the anticipated habilitation options. For instance, a child with severe-to-profound SNHL will typically require imaging (CT or MRI) prior to consideration of cochlear implantation. In clinical practice, the diagnostic evaluation is often performed simultaneously with molecular genetic testing given the turnaround time (1-3 months) for genetic testing.
Molecular Genetic Testing
Molecular genetic testing is the standard of care in evaluation of individuals with hearing loss. See Liming et al [2016] (full text) and Li et al [2022] (full text).
The diagnosis of a specific genetic cause of hearing loss is established in a proband with suggestive findings and pathogenic (or likely pathogenic) variant(s) in the causative gene identified by molecular genetic testing.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include any likely pathogenic variants. (2) The identification of variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis.
Recommended molecular genetic testing approaches include use of a multigene hearing loss panel and/or genomic testing.
Note: Single-gene testing (sequence analysis of a given gene, followed by gene-targeted deletion/duplication analysis) is rarely useful and typically NOT recommended. Isolated chromosomal microarray testing in an individual with apparent nonsyndromic hearing loss also has a low diagnostic yield.
A multigene hearing loss panel can often identify the cause of genetic hearing loss while limiting identification of variants of
uncertain significance and pathogenic variants in genes that are irrelevant to the underlying
phenotype. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome or genome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Genomic testing does not require the clinician to determine which
gene is likely involved.
Exome sequencing is most commonly used.
Genome sequencing is also increasingly being used in the clinical setting as the cost of testing decreases; diagnostic yield is higher, as noncoding regions are interrogated for pathogenic regulatory variants.
For an introduction to comprehensive
genomic testing click
here. More detailed information for clinicians ordering genomic testing can be found
here.
5. Management
Management of hearing loss in children is structured around the goals of the parents and/or care providers for speech and language. Families may want their child to attain oral speech and language and to participate in mainstream schooling. Members of the Deaf community may prefer to rely on visual communication (typically American Sign Language) to fulfill communication goals or may prefer to incorporate total communication. (Total communication is a holistic approach to communication that promotes the use of all modes of communication including sign language, spoken language, gestures, facial expressions, and environmental cues such as pictures and sounds.)
Discussion between health care providers and parents/caregivers to solicit the goals and preferences of the family is the first step in establishing a plan for management.
Management of hearing loss in adults also takes into account the affected individual's preference for communication. Adults who communicate with spoken language most often seek to restore hearing using hearing aids, cochlear implants, or a combination of both. It is becoming increasingly clear that hearing loss in older adults leads to isolation and increases risk for dementia [Thomson et al 2017]. These risks may be mitigated by addressing hearing loss.
Multidisciplinary Supportive Treatment of Hearing Loss
Children. Multidisciplinary supportive treatment recommended for hearing loss includes an otolaryngologist, an audiologist experienced in the assessment of hearing loss in children, a speech-language pathologist, a clinical geneticist, a genetic counselor, and a pediatrician. In some instances, other specialists such a neuropsychologist and/or an educator of the Deaf may be required. If a syndromic diagnosis is made, then clinicians with expertise in other areas may be required (e.g., an ophthalmologist for a child diagnosed with Usher syndrome).
Adults. Management of hearing loss in adults typically focuses on continued integration into a hearing society primarily by use of verbal communication and often other abilities, such as use of a telephone and enjoyment of music. Hearing loss in older adults is associated with increased risk of dementia [Thomson et al 2017]. Improved quality of life is possible with early treatment with hearing aids or cochlear implants [Andries et al 2021].
Habilitation
Early auditory intervention can include sound amplification with hearing aids, otologic surgery to implant bone conduction devices, and/or cochlear implantation [Smith et al 2005]. Early identification of hearing loss in children is critical to developing speech and language [Yoshinaga-Itano et al 2018]. Conversely, delayed identification of hearing loss and delayed exposure to language is associated with poor reading performance, poor communication skills, and poor speech production; subsequent intervention may be insufficient to completely remediate these deficiencies [Tobey et al 2013].
Habituation options include the following:
Hearing aids (sound amplification), customized by an audiologist to the degree and frequency of hearing loss, can be used in individuals with mild-to-severe hearing loss.
Bone conduction devices (bone-anchored hearing systems), surgically implanted devices, are used for some types of conductive hearing loss or unilateral hearing loss.
Cochlear implantation
Cochlear implantation can be considered in children with severe-to-profound hearing loss who are older than age nine months. Children who undergo cochlear implantation early (before age two years) may achieve – by school age – oral speech and language that is indistinguishable from their normal-hearing peers [
Loy et al 2010,
Langereis & Vermeulen 2015].
In adults, cochlear implant performance may be compromised when the auditory nerve itself is affected; however, further research is needed in this area [
Shearer et al 2017].
Although research has focused on cochlear implant performance based on the
gene involved, given the genetic heterogeneity of hearing loss, large sample sizes are difficult to obtain for performance on a per-gene basis. However, data are clear that individuals with
GJB2-related hearing loss (see
GJB2-Related Autosomal Recessive Nonsyndromic Hearing Loss) have excellent cochlear implant outcomes that are significantly better than those of individuals with hearing loss due to an environmental cause [
Yoshida et al 2013,
Abdurehim et al 2017].
Communication and Identity Development
On initial evaluation of individuals with hearing loss, the goals for communication must be established with a focus on equipping individuals with language and appropriate access to language.
Early access to sign language for children with hearing loss often provides a "head start" to auditory communication skills that are used later by such children who will proceed to communicate using spoken language [Blanco-Elorrieta et al 2018].
A speech-language therapist as well as a neuropsychologist/psychologist for children with hearing loss provides access to resources within the community to aid in speech and language and D/deaf and hard of hearing (DHH) identity development.
Education
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech-language, and feeding therapy as well as specialized D/deaf and hard of hearing (DHH) services to support DHH identity development in the child and family. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education program (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician and/or neuropsychologist/psychologist is recommended to ensure the involvement of appropriate community, state, and federal educational agencies and to maximize quality of life for the individual with hearing loss and their family. Some issues to consider:
IEP services:
An IEP provides specially designed instruction and related services to children who qualify.
IEP services will be reviewed annually to determine whether any changes are needed.
Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
Physical, occupational, and speech-language therapy services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered.
When developmentally appropriate, self-advocacy skills for DHH children can be fostered in a safe and supportive school environment. Access to a DHH mentor and/or DHH adult as well as social-emotional learning are tools that support a student's self-expression, which can lead to positive social interactions with peers and educators.
DHH specialty services should be provided, especially if a child is in a mainstream setting, to ensure appropriate hearing support and promote positive DHH identity development.
As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For some of those receiving IEP services, the public school district is required to provide services until age 21.
Should an individual not qualify for an IEP, a 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time, modified assignments, and enlarged text.
Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Targeted Medical Therapy
Specific medical therapy is available for select disorders (see Table 7).
Table 7.
Specific Medical Therapy for Select Genetic Disorders
View in own window
| Disorder Name | Gene(s) | Therapy | Comments |
|---|
|
Biotinidase deficiency
|
BTD
| Administration of biotin | Goal is to treat biotin deficiency & prevent progression of hearing loss. |
| Hypoparathyroidism, sensorineural deafness, renal dysplasia (HDR syndrome; Barakat syndrome) |
GATA3
| Oral calcium & calcitriol, parathyroid hormone injection | Goal is to treat underlying hypoparathyroidism & subsequent hypocalcemia. |
| NLRP3-related disorders 1 |
NLRP3
| Injection w/anakinra, rilonacept, or canakinumab | Goal is to reduce or reverse autoimmune-mediated hearing loss. |
| Riboflavin transporter deficiency (Brown-Vialetto-Van Laere syndrome) |
SLC52A2
SLC52A3
| High-dose supplementation of riboflavin | Goal is to treat riboflavin deficiency & prevent progression of hearing loss. |
- 1.
Cryopyrin-associated periodic syndrome (CAPS), familial cold autoinflammatory syndrome, chronic infantile neurologic cutaneous articular syndrome (CINCA), neonatal-onset multisystem inflammatory disease (NOMID), Muckle-Wells syndrome
Surveillance
Regular follow up is recommended for all individuals with genetic hearing loss in order to:
Table 8.
Recommended Follow Up for Select Genetic Causes of Hearing Loss by Progression Type
View in own window
| Progression Type | Gene | Recommended Testing Intervals |
|---|
| Generally stable or very slowly progressive |
OTOG
STRC
| Yearly or biannually |
| Progressive |
COCH
GSDME
KCNQ4
MYO7A
MYO15A
SLC26A4
USH2A
WFS1
| At least yearly; consider biannually for children |
| Fluctuating |
OTOF
SLC26A4
|
Agents/Circumstances to Avoid
Noise exposure is a well-recognized environmental cause of hearing loss. Since this risk can be minimized by avoidance, persons with documented hearing loss should be counseled appropriately and repeated overexposure to loud noises should be avoided. There is no established "safe" noise level, but the US Occupational Safety and Health Administration (OSHA) and National Institute for Occupational Safety and Health (NIOSH) have identified a permissible noise exposure limit of 85 dB over an eight-hour workday.
One of the primary sources of loud noise exposure in our environment is sound from headphones and earbuds. An 85-dB limit on earbuds and headphones may provide a reasonable way to reduce loud noise exposure. The headphone safety feature built into most smartphones can be set to limit noise level.
Headphone/earbud safety features can be found in the phone settings menu:
In iPhones, under Settings > Sounds & Haptics > Headphone Safety
In Android phones, under Settings > Sounds & Vibrations > Volume > Media Volume Limit
Also see these general resources on noise reduction:
For genetic disorders with specific agents/circumstances to avoid, see Table 9.
Table 9.
Agents/Circumstances to Avoid: Select Genetic Disorders
View in own window
| Gene | Disorder | Concern | Circumstance to Avoid |
|---|
KCNE1
KCNQ1
|
Jervel and Lange-Nielsen syndrome
| Prolonged QT interval | Anesthetic agents & medications that may further prolong the QT interval. This is particularly important for children undergoing cochlear implantation. Avoid activities that may precipitate syncopal episodes: competitive sports, amusement parks, jumping into cold water. |
| MT-RNR1 1 | Mitochondrial SNHL (See Nonsyndromic Hearing Loss and Deafness, Mitochondrial.) | Sensitivity to aminoglycosides predisposes to hearing loss | Exposure (even intermittent) to aminoglycosides |
| MT-TS1 2 |
|
POU3F4
| X-linked deafness (OMIM 304400) | ↑ risk that middle ear surgery can result in complete loss of hearing | Conductive hearing loss in this disorder is caused by stapedial fixation. Fenestration or removal of the stapes footplate at the time of middle ear surgery that results in an abnormal communication between the CSF & perilymph can lead to fluid leakage ("perilymphatic gusher") and complete loss of hearing. |
CSF = cerebrospinal fluid; SNHL = sensorineural hearing loss
- 1.
Virtually all persons w/m.1095T>C, m.1494C>T, or m.1555A>G pathogenic variants will have hearing loss after even intermittent exposure to aminoglycosides. Sensitivity to aminoglycosides related to several other variants in this gene are unknown. See McDermott et al [2022].
- 2.
The m.7444G>A pathogenic variant located on the boundary of MT-CO1 and MT-TS1 has been reported to be causative for nonsyndromic hearing loss and aminoglycoside-induced hearing loss.
6. Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Autosomal Recessive Nonsyndromic Hearing Loss – Risk to Family Members
See Table 3. Autosomal Recessive Nonsyndromic Hearing Loss: Distinctive Features Associated with Selected Genes.
Parents of a proband
If a
pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the
proband occurred as a
de novo event in the proband or as a
postzygotic de novo event in a mosaic parent [
Jónsson et al 2017]. If the proband appears to have
homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
Sibs of a proband
If both parents are known to be
heterozygous for an
autosomal recessive hearing loss-related
pathogenic variant, each sib of the
proband has at conception a 25% chance of having hearing loss, a 50% chance of having no hearing loss and being a
carrier, and a 25% chance of having no hearing loss and not being a carrier.
Offspring of a proband. The offspring of an individual with autosomal recessive hearing loss are obligate heterozygotes (carriers of a hearing loss-related pathogenic variant).
Other family members. For each sib of the proband's parents, the probability of being a carrier of a hearing loss-related pathogenic variant is 50%.
Carrier detection. Carrier testing for relatives who may have a hearing loss-related pathogenic variant requires prior identification of the pathogenic variants in the family.
Autosomal Dominant Nonsyndromic Hearing Loss – Risk to Family Members
See Table 4. Autosomal Dominant Nonsyndromic Hearing Loss: Distinctive Features Associated with Selected Genes.
Parents of a proband
Rarely, an individual with hearing loss has hearing loss as the result of a
de novo pathogenic variant. The proportion of individuals with hearing loss caused by a
de novo pathogenic variant varies depending on the
gene involved. In a recent study, approximately 2% of
autosomal dominant nonsyndromic hearing loss could be attributed to
de novo pathogenic variants [
Klimara et al 2022].
If a molecular diagnosis has been established in the
proband and the proband appears to be the only family member with hearing loss, targeted
molecular genetic testing for the
pathogenic variant identified in the proband is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence counseling.
If the
pathogenic variant identified in the
proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
Sibs of a proband. The likelihood of hearing loss in sibs depends on the genetic status of the proband's parents:
If one of the
proband's parents has the
pathogenic variant identified in the proband, sibs have a 50% chance of inheriting the pathogenic variant and having hearing loss.
Depending on the specific diagnosis, clinical severity and
phenotype may differ between sibs with the same
pathogenic variant; thus, age of onset and/or progression cannot be reliably predicted.
Offspring of a proband. Individuals with autosomal dominant hearing loss have a 50% chance of transmitting the pathogenic variant to each child.
Other family members. If a parent has the pathogenic variant identified in the proband, the parent's family members may also have the pathogenic variant.
X-Linked Nonsyndromic Hearing Loss – Risk to Family Members
See Table 5. X-Linked Nonsyndromic Hearing Loss: Distinctive Features Associated with Selected Genes.
Note: For the purposes of this GeneReview, the terms "male" and "female" are narrowly defined as the individual's biological sex at birth as it determines clinical care [Caughey et al 2021].
Parents of a male proband
In a family with more than one individual with
X-linked hearing loss, the mother of a male with hearing loss is an
obligate heterozygote. Note: If a woman has more than one child with hearing loss and no other relatives with hearing loss and if the
pathogenic variant cannot be detected in her leukocyte DNA, she most likely has
germline mosaicism.
Parents of a female proband
Detailed evaluation of the parents and review of the extended family history may help distinguish probands with a
de novo pathogenic variant from those with an inherited pathogenic variant. Molecular genetic testing of the mother (and possibly the father, or subsequently the father) can determine if the pathogenic variant was inherited.
Sibs of a male proband. The likelihood of hearing loss in sibs depends on the genetic status of the mother:
If the mother of the
proband has the hearing loss-related
pathogenic variant, the chance of the mother transmitting it in each pregnancy is 50%.
If the
proband is the only family member known to have hearing loss and if the
pathogenic variant cannot be detected in the leukocyte DNA of either parent, the likelihood of hearing loss in sibs is presumed to be low but greater than that of the general population because of the possibility of maternal
germline mosaicism.
Depending on the specific diagnosis, clinical severity and
phenotype may differ between individuals with the same
pathogenic variant; thus, age of onset and/or progression may not be predictable.
Sibs of a female proband. The likelihood of hearing loss in sibs depends on the genetic status of the parents:
If the mother of the
proband has the hearing loss-related
pathogenic variant, the chance of transmitting it in each pregnancy is 50%.
Females who inherit the
pathogenic variant will be heterozygotes and may or may not have hearing loss.
Offspring of a male
proband. Males with X-linked hearing loss transmit the hearing loss-related pathogenic variant to all of their daughters and none of their sons.
Offspring of a female
proband. Females with X-linked hearing loss have a 50% chance of transmitting the pathogenic variant to each child.
Other family members. If a parent has the pathogenic variant identified in the proband, the parent's family members may also have the pathogenic variant.
Heterozygote detection. Heterozygote testing for female relatives requires prior identification of the pathogenic variant in the family.
Empiric Risk to Family Members
If a specific diagnosis cannot be established (and/or the mode of inheritance cannot be established in a person with a positive family history of deafness or hearing loss), the following empiric figures can be used for families of northern European ancestry.
The subsequent offspring of a hearing couple with one deaf child and an otherwise negative family history of deafness have an 18% empiric probability of deafness in future children [Green et al 1999].
If the deaf child does not have
GJB2-related
autosomal recessive nonsyndromic hearing loss, the chance of recurrence is 14% for deafness unrelated to connexin 26 [
Green et al 1999].
If the hearing couple is
consanguineous or comes from a highly consanguineous community, the subsequent offspring have close to a 25% probability of deafness because of the high likelihood of
autosomal recessive inheritance.
The offspring of a deaf person and a hearing person have a 10% empiric probability of deafness [Green et al 1999].
The child of a nonconsanguineous deaf couple in whom autosomal dominant deafness has been excluded has an approximately 15% empiric probability of deafness [Green et al 1999].
The child of a hearing sib of a deaf proband (presumed to have autosomal recessive nonsyndromic deafness) and a deaf person has a one in 200 (0.5%) empiric probability for deafness, or five times the general population probability.
GJB2 molecular genetic testing can clarify if the probability is higher. If the hearing sib is a carrier of a GJB2 deafness-causing variant and the hearing sib's reproductive partner has GJB2-related autosomal recessive nonsyndromic hearing loss, the chance of having a deaf child is 50%.
Prenatal Testing and Preimplantation Genetic Testing
Once the pathogenic variant(s) have been identified in the family, prenatal and preimplantation genetic testing for genetic hearing loss are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
American Society for Deaf Children
Phone: 800-942-2732 (ASDC)
Email: info@deafchildren.org
Medical Home Portal
MedlinePlus
National Association of the Deaf
Phone: 301-587-1788 (Purple/ZVRS); 301-328-1443 (Sorenson); 301-338-6380 (Convo)
Fax: 301-587-1791
Email: nad.info@nad.org
Alexander Graham Bell Association for the Deaf and Hard of Hearing
Phone: 866-337-5220 (toll-free); 202-337-5221 (TTY)
Fax: 202-337-8314
Email: info@agbell.org
American Speech-Language-Hearing Association (ASHA)
Phone: 800-638-8255; 301-296-5650 (TTY)
Fax: 301-296-8580
BabyHearing.org
This site, developed with support from the National Institute on Deafness and Other Communication Disorders, provides information about newborn hearing screening and hearing loss.
Hands & Voices
Newborn Screening in Your State
Health Resources & Services Administration
Chapter Notes
Author Notes
A Eliot Shearer, MD, PhD, is a pediatric otolaryngologist at Boston Children's Hospital and Harvard Medical School. He is actively involved in clinical research regarding individuals with pediatric hearing loss and methods for genetic diagnosis for syndromic and nonsyndromic hearing loss. He would be happy to communicate with persons who have any questions regarding diagnosis of hearing loss or other considerations. Lab website: www.hearing-genomics.org
Richard JH Smith, MD, is a pediatric otolaryngologist and human geneticist at the University of Iowa. He directs the Molecular Otolaryngology and Renal Research Laboratories (MORL), which offers comprehensive genetic testing for hearing loss. For questions about hearing loss and gene therapy for hearing loss, email ude.awoiu@lrom. Lab website: morl.lab.uiowa.edu
Amanda M Schaefer, MS, LGC, is a licensed and board-certified genetic counselor who specializes in hearing loss and deafness genetics at the University of Iowa Hospitals and Clinics. She works with the Molecular Otolaryngology and Renal Research Laboratories (MORL), which offers genetic testing for hearing loss. Amanda is actively involved in translational research studies for hearing loss including genotype-phenotype studies, genetic counseling for persons with hearing loss, and implementation of genetic evaluation for hearing loss.
Acknowledgments
Thank you to Nicole Salamy, MS, CCC-SLP, Rachel Landsman, PsyD, and Kaitlyn Fitzpatrick, AuD, from the Deaf and Hard of Hearing Program at Boston Children's Hospital for their contributions to the Communication and Identity Development section.
This work was supported in part by NIDCDs R01's DC002842, DC012049, and DC017955 to RJHS.
Author History
Glenn Edward Green, MD; University of Arizona (1999-2005)
Michael S Hildebrand, PhD (2010-present)
Amanda M Schaefer, MS, LGC (2023-present)
A Eliot Shearer (2012-present)
Richard JH Smith, MD (1999-present)
Guy Van Camp, PhD; University of Antwerp (1999-2017)
Revision History
28 September 2023 (aes) Revision: removed references to DFNA3; added recommendations for
reducing loud noise exposure from headphones and earbuds
29 June 2023 (rjs) Revision: removed
CIB2 from
Table 6b6 April 2023 (bp) Comprehensive update posted live
27 July 2017 (bp) Comprehensive update posted live
14 October 2010 (me) Comprehensive update posted live
28 October 2008 (me) Comprehensive update posted live
30 December 2005 (me) Comprehensive update posted live
3 November 2003 (me) Comprehensive update posted live
24 April 2001 (me) Comprehensive update posted live
14 February 1999 (pb) Overview posted live
30 October 1998 (rjs) Original overview submission [Supported in part by grants 1RO1DC02842 and 1RO1DC03544 (RJHS) and Belgian National Fonds voor Wetenschappelijk Onderzoek (GVC).]
References
Published Guidelines / Consensus Statements
Li MM, Tayoun AA, DiStefano M, Pandya A, Rehm HL, Robin NH, Schaefer AM, Yoshinaga-Itano C, et al. Clinical evaluation and etiologic diagnosis of hearing loss: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2022;24:1392-406. [
PubMed]
Literature Cited
Abdurehim
Y, Lehmann
A, Zeitouni
AG. Predictive value of GJB2 mutation status for hearing outcomes of pediatric cochlear implantation.
Otolaryngol Head Neck Surg.
2017;157:16-24.
[
PubMed: 28322114]
Ahmadmehrabi
S, Brant
J, Epstein
DJ, Ruckenstein
MJ, Rader
DJ. Genetics of postlingual sensorineural hearing loss.
Laryngoscope.
2021;131:401-9.
[
PubMed: 32243624]
Andries
E, Gilles
A, Topsakal
V, Vanderveken
OM, Van de Heyning
P, Van Rompaey
V, Mertens
G. Systematic review of quality of life assessments after cochlear implantation in older adults.
Audiol Neurootol.
2021;26:61-75.
[
PubMed: 32653882]
Caughey
AB, Krist
AH, Wolff
TA, Barry
MJ, Henderson
JT, Owens
DK, Davidson
KW, Simon
MA, Mangione
CM. USPSTF approach to addressing sex and gender when making recommendations for clinical preventive services.
JAMA. 2021;326:1953-61.
[
PubMed: 34694343]
Chan
DK, Chang
KW. GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype.
Laryngoscope.
2014;124:E34-53.
[
PubMed: 23900770]
Corriols-Noval
P, López Simón
EC, Cadiñanos
J, Diñeiro
M, Capín
R, González Aguado
R, Costales Marcos
M, Morales Angulo
C, Cabanillas Farpón
R. Clinical impact of genetic diagnosis of sensorineural hearing loss in adults.
Otol Neurotol.
2022;43:1125-36.
[
PubMed: 36190904]
Downie
L, Halliday
J, Burt
R, Lunke
S, Lynch
E, Martyn
M, Poulakis
Z, Gaff
C, Sung
V, Wake
M, Hunter
MF, Saunders
K, Rose
E, Lewis
S, Jarmolowicz
A, Phelan
D, Rehm
HL, Amor
DJ, et al.
Exome sequencing in infants with congenital hearing impairment: a population-based cohort study.
Eur J Hum Genet.
2020;28:587-96.
[
PMC free article: PMC7171096] [
PubMed: 31827275]
Green
GE, Scott
DA, McDonald
JM, Woodworth
GG, Sheffield
VC, Smith
RJ. Carrier rates in the Midwestern United States for GJB2 mutations causing inherited deafness.
JAMA. 1999;281:2211-6
[
PubMed: 10376574]
Jónsson
H, Sulem
P, Kehr
B, Kristmundsdottir
S, Zink
F, Hjartarson
E, Hardarson
MT, Hjorleifsson
KE, Eggertsson
HP, Gudjonsson
SA, Ward
LD, Arnadottir
GA, Helgason
EA, Helgason
H, Gylfason
A, Jonasdottir
A, Jonasdottir
A, Rafnar
T, Frigge
M, Stacey
SN, Th Magnusson
O, Thorsteinsdottir
U, Masson
G, Kong
A, Halldorsson
BV, Helgason
A, Gudbjartsson
DF, Stefansson
K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland.
Nature.
2017;549:519-22.
[
PubMed: 28959963]
Kenneson
A, Cannon
MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection.
Rev Med Virol.
2007;17:253-76.
[
PubMed: 17579921]
Klimara
MJ, Nishimura
C, Wang
D, Kolbe
DL, Schaefer
AM, Walls
WD, Frees
KL, Smith
RJH, Azaiez
H. De novo variants are a common cause of genetic hearing loss.
Genet Med.
2022;24:2555-67.
[
PMC free article: PMC9729384] [
PubMed: 36194208]
Langereis
M, Vermeulen
A. School performance and wellbeing of children with CI in different communicative–educational environments.
Int J Pediatr Otorhinolaryngol.
2015;79: 834-9
[
PubMed: 25840945]
Li
MM, Tayoun
AA, DiStefano
M, Pandya
A, Rehm
HL, Robin
NH, Schaefer
AM, Yoshinaga-Itano
C, et al.
Clinical evaluation and etiologic diagnosis of hearing loss: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG).
Genet Med.
2022;24:1392-406.
[
PubMed: 35802133]
Liming
BJ, Carter
J, Cheng
A, Choo
D, Curotta
J, Carvalho
D, Germiller
JA, Hone
S, Kenna
MA, Loundon
N, Preciado
D, Schilder
A, Reilly
BJ, Roman
S, Strychowsky
J, Triglia
JM, Young
N, Smith
RJ. International Pediatric Otolaryngology Group (IPOG) consensus recommendations: hearing loss in the pediatric patient.
Int J Pediatr Otorhinolaryngol.
2016; 90:251-8.
[
PubMed: 27729144]
Loy
B, Warner-Czyz
AD, Tong
L, Tobey
EA, Roland
PS. The children speak: an examination of the quality of life of pediatric cochlear implant users.
Otolaryngol Head Neck Surg.
2010;142:247-53
[
PMC free article: PMC2852181] [
PubMed: 20115983]
McDermott
JH, Wolf
J, Hoshitsuki
K, Huddart
R, Caudle
KE, Whirl-Carrillo
M, Steyger
PS, Smith
RJH, Cody
N, Rodriguez-Antona
C, Klein
TE, Newman
WG. Clinical Pharmacogenetics Implementation Consortium Guideline for the use of aminoglycosides based on MT-RNR1 genotype.
Clin Pharmacol Ther.
2022;111:366-72.
[
PMC free article: PMC8613315] [
PubMed: 34032273]
Morton
CC, Nance
WE. Newborn hearing screening--a silent revolution.
N Engl J Med.
2006;354:2151-64.
[
PubMed: 16707752]
Rahbari
R, Wuster
A, Lindsay
SJ, Hardwick
RJ, Alexandrov
LB, Turki
SA, Dominiczak
A, Morris
A, Porteous
D, Smith
B, Stratton
MR, Hurles
ME, et al.
Timing, rates and spectra of human germline mutation.
Nat Genet.
2016;48:126-33.
[
PMC free article: PMC4731925] [
PubMed: 26656846]
Richards
S, Aziz
N, Bale
S, Bick
D, Das
S, Gastier-Foster
J, Grody
WW, Hegde
M, Lyon
E, Spector
E, Voelkerding
K, Rehm
HL, et al.
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med.
2015;17:405-24.
[
PMC free article: PMC4544753] [
PubMed: 25741868]
Rudman
JR, Kabahuma
RI, Bressler
SE, Feng
Y, Blanton
SH, Yan
D, Liu
XZ. The genetic basis of deafness in populations of African descent.
J Genet Genomics.
2017; 44:285-94.
[
PubMed: 28642064]
Seligman
KL, Shearer
AE, Frees
K, Nishimura
C, Kolbe
D, Dunn
C, Hansen
MR, Gantz
BJ, Smith
RJH. Genetic causes of hearing loss in a large cohort of cochlear implant recipients.
Otolaryngol Head Neck Surg.
2022;166:734-37.
[
PMC free article: PMC9128025] [
PubMed: 34154485]
Shearer
AE, Eppsteiner
RW, Frees
K, Tejani
V, Sloan-Heggen
CM, Brown
C, Abbas
P, Dunn
C, Hansen
MR, Gantz
BJ, Smith
RJH. Genetic variants in the peripheral auditory system significantly affect adult cochlear implant performance.
Hear Res.
2017;348:138-42
[
PMC free article: PMC5527292] [
PubMed: 28213135]
Sloan-Heggen
CM, Bierer
AO, Shearer
AE, Kolbe
DL, Nishimura
CJ, Frees
KL, Ephraim
SS, Shibata
SB, Booth
KT, Campbell
CA, Ranum
PT, Weaver
AE, Black-Ziegelbein
EA, Wang
D, Azaiez
H, Smith
RJ. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss.
Hum Genet.
2016;135:441-50.
[
PMC free article: PMC4796320] [
PubMed: 26969326]
Smith
RJH, Bale
JF, White
KR. Sensorineural hearing loss in children
Lancet.
2005;365:879-90.
[
PubMed: 15752533]
Tobey
EA, Thal
D, Niparko
JK, Eisenberg
LS, Quittner
AL, Wang
NY, et al.
Influence of implantation age on school-age language performance in pediatric cochlear implant users.
Int J Audiol.
2013;52:219-29.
[
PMC free article: PMC3742378] [
PubMed: 23448124]
Wake
M, Tobin
S, Cone-Wesson
B, Dahl
HH, Gillam
L, McCormick
L, Poulakis
Z, Rickards
FW, Saunders
K, Ukoumunne
OC, Williams
J. Slight/mild sensorineural hearing loss in children.
Pediatrics.
2006;118:1842-51.
[
PubMed: 17079553]
Yang
T, Vidarsson
H, Rodrigo-Blomqvist
S, Rosengren
SS, Enerback
S, Smith
RJ. Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4).
Am J Hum Genet.
2007;80:1055-63.
[
PMC free article: PMC1867094] [
PubMed: 17503324]
Yoshida
H, Takahashi
H, Kanda
Y, Usami
S. Long term speech perception after cochlear implant in pediatric patients with GJB2 mutations.
Auris Nasus Larynx.
2013; 40:435-9
[
PubMed: 23477838]
Yoshinaga-Itano
C, Sedey
AL, Wiggin
M, Mason
CA. Language outcomes improved through early hearing detection and earlier cochlear implantation.
Otol Neurotol.
2018;39:1256-63.
[
PubMed: 30444842]