Summary
Clinical characteristics.
The spectrum of COL4A1-related disorders includes: small-vessel brain disease of varying severity including porencephaly, variably associated with eye defects (retinal arterial tortuosity, Axenfeld-Rieger anomaly, cataract) and systemic findings (kidney involvement, muscle cramps, cerebral aneurysms, Raynaud phenomenon, cardiac arrhythmia, and hemolytic anemia). On imaging studies, small-vessel brain disease is manifest as diffuse periventricular leukoencephalopathy, lacunar infarcts, microhemorrhage, dilated perivascular spaces, and deep intracerebral hemorrhages. Clinically, small-vessel brain disease manifests as infantile hemiparesis, seizures, single or recurrent hemorrhagic stroke, ischemic stroke, and isolated migraine with aura. Porencephaly (fluid-filled cavities in the brain detected by CT or MRI) is typically manifest as infantile hemiparesis, seizures, and intellectual disability; however, on occasion it can be an incidental finding. HANAC (hereditary angiopathy with nephropathy, aneurysms, and muscle cramps) syndrome usually associates asymptomatic small-vessel brain disease, cerebral large vessel involvement (i.e., aneurysms), and systemic findings involving the kidney, muscle, and small vessels of the eye. Two additional phenotypes include isolated retinal artery tortuosity and nonsyndromic autosomal dominant congenital cataract.
Management.
Treatment of manifestations: Supportive care tailored to the individual’s specific medical needs and including practical help and emotional support for affected individuals and their families. Hypertension should be treated to reduce the overall risk of stroke.
Prevention of primary and secondary complications: Avoiding head trauma and anticoagulant exposure may decrease the risk for intracerebral hemorrhage.
Surveillance: Depends on the severity and type of symptoms.
Agents/circumstances to avoid: Smoking and hypertension because these factors increase the risk for stroke; sustained head pressure or physical activities that may cause head trauma; anticoagulant use.
Genetic counseling.
COL4A1-related disorders are inherited in an autosomal dominant manner. Most individuals diagnosed with a COL4A1-related disorder have an affected parent. The proportion of cases caused by a de novo pathogenic variant is estimated to be at least 27%. Each child of an individual with a COL4A1-related disorder has a 50% chance of inheriting the pathogenic variant. Prenatal testing for a pregnancy at increased risk is possible if the pathogenic variant in the family is known.
GeneReview Scope
View in own window
| COL4A1-Related Disorders: Included Phenotypes 1 |
|---|
Autosomal dominant familial porencephaly Autosomal dominant brain small-vessel disease with hemorrhage Hereditary angiopathy with nephropathy, aneurysms, and muscle cramps (HANAC) syndrome Tortuosity of retinal arteries
|
Diagnosis
COL4A1-related disorders cover a spectrum of overlapping phenotypes characterized by a small-vessel brain disease of varying severity including porencephaly, variably associated with eye defects (congenital cataract, retinal arterial tortuosity, eye anterior segment anomaly of Axenfeld-Rieger type) and systemic findings (muscle cramps and/or serum creatine kinase (CK) elevation, kidney involvement, cerebral aneurysms, Raynaud phenomenon, cardiac arrhythmia, and hemolytic anemia).
Suggestive Findings
A COL4A1-related disorder should be suspected in individuals with any of the following phenotypes, which have overlapping features:
Clinical features of COL4A1-related disorders vary widely and can include the following:
Neurologic
Infantile hemiplegia
Developmental delay
Migraines with or without aura
Seizures
Dementia
Intellectual disability
Intracerebral hemorrhage at any age including antenatal, neonatal, and recurrent episodes
Ischemic stroke
Typically on neuroimaging: features of brain small-vessel disease ()
Ophthalmic
Transient visual loss caused by retinal hemorrhage
Cataract, glaucoma, microphthalmia/anophthalmia
Other systemic findings
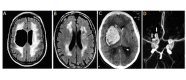
Spectrum of brain imaging abnormalities in COL4A1-related disorders A. Axial FLAIR showing right paraventricular porencephalic cyst and extensive white-matter abnormalities [van der Knaap et al 2006]
Family history is consistent with autosomal dominant inheritance. Findings and age of onset vary within and between families.
Establishing the Diagnosis
The diagnosis of a COL4A1-related disorder is established in a proband with suggestive findings and a heterozygous pathogenic variant in COL4A1 identified by molecular testing (see Table 1).
Molecular testing approaches can include single-gene testing, use of a multigene panel, and more comprehensive
genomic testing:
Single-gene testing. Sequence analysis of
COL4A1 is performed first followed by gene-targeted
deletion/duplication analysis if no
pathogenic variant is found. To date, no deletions or duplications involving
COL4A1 as causative of
COL4A1-related disorders have been reported. Because these disorders usually result from a
COL4A1 pathogenic
missense variant that disrupts the collagen triple helix (see
Molecular Genetics), a screening test for large duplications/deletions may have a very low yield.
A multigene panel that includes
COL4A1 and other genes of interest (see
Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused
exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
More comprehensive genomic testing (when available) including
exome sequencing and
genome sequencing may be considered if single-
gene testing (and/or use of a
multigene panel that includes
COL4A1) fails to confirm a diagnosis in an individual with features of a
COL4A1-related disorder. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation). For an introduction to comprehensive genomic testing click
here. More detailed information for clinicians ordering genomic testing can be found
here.
Table 1.
Molecular Genetic Testing Used in COL4A1-Related Disorders
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
COL4A1
| Sequence analysis 3 | 100% |
| Deletion/duplication analysis 4 | Unknown 5 |
- 1.
- 2.
- 3.
- 4.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range pf techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 5.
Clinical Characteristics
Clinical Description
COL4A1-related disorders cover a spectrum of overlapping phenotypes characterized by a small-vessel brain disease of varying severity including porencephaly, variably associated with eye defects (congenital cataract, retinal arterial tortuosity, eye anterior segment anomaly of Axenfeld-Rieger type) and systemic findings (muscle cramps and/or serum CK elevation, kidney involvement, cerebral aneurysms, Raynaud phenomenon, cardiac arrhythmia, hemolytic anemia).
Autosomal Dominant Familial Porencephaly
Autosomal dominant familial porencephaly related to COL4A1 pathogenic variants has been reported in more than 50 individuals [Gould et al 2005, Breedveld et al 2006, van der Knaap et al 2006, Yoneda et al 2013, Meuwissen et al 2015]. This condition is characterized by the presence of fluid-filled cavities in the brain, caused by antenatal or perinatal parenchymal hemorrhage and detected by either brain CT, MRI (), or fetal ultrasound imaging. In addition to porencephalic cavities, brain imaging shows various degrees of periventricular leukoencephalopathy, microbleeds, lacunar infarct, and calcifications [Vahedi et al 2007, Livingston et al 2011, Ayrignac et al 2015, Meuwissen et al 2015].
The spectrum of neurologic clinical symptoms varies in degree of severity and age of onset, with wide intrafamilial heterogeneity. Typically, affected individuals may present with infantile hemiparesis, seizures, intellectual disability, dystonia, stroke, and migraine. First manifestations (including intracerebral hemorrhages) may occur in previously asymptomatic adults, and MRI brain anomalies can be clinically silent.
Congenital cataract is frequently observed in affected individuals. Retinal arteriolar tortuosity is more rarely associated with porencephaly [van der Knaap et al 2006].
Autosomal Dominant Brain Small-Vessel Disease with Hemorrhage
Autosomal dominant brain small-vessel disease with hemorrhage differs from autosomal dominant familial porencephaly by the absence of porencephalic cavities, while brain imaging demonstrates characteristic brain small-vessel involvement, including diffuse periventricular leukoencephalopathy (), lacunar infarcts, microbleeds, dilated perivascular spaces, deep intracerebral hemorrhages, and intracerebral calcifications () [Vahedi et al 2007, Yoneda et al 2013, Meuwissen et al 2015].
Neurologic manifestations are also heterogeneous within families, and vary from infantile hemiparesis with seizure to isolated migraine with aura to absence of clinical symptoms. Single or recurrent intracerebral hemorrhage may occur in non-hypertensive adults who are younger than age 50 years. Such hemorrhages can occur spontaneously, after trauma, or as a result of anticoagulant use; some are fatal. Antenatal intracerebral and intraventricular hemorrhage may be observed using fetal ultrasound examination. Mild cognitive impairment has also been reported in one family [Sibon et al 2007]; however, it is unclear whether this is a separate finding or a manifestation of recurrent stroke.
Concomitant eye anomalies including retinal arteriolar tortuosity, congenital cataract, and/or anterior segment anomalies of the Axenfeld-Rieger type may be observed [Sibon et al 2007, Coupry et al 2010].
More rarely, systemic symptoms including serum CK elevation with or without muscle cramps [Yoneda et al 2013, Meuwissen et al 2015] and renal involvement including hematuria, unilateral renal atrophy [John et al 2015], renal cysts, and hemolytic anemia [Meuwissen et al 2015] are observed.
HANAC Syndrome
HANAC (hereditary angiopathy with nephropathy, aneurysms, and muscle cramps) syndrome has been well characterized in six families [Plaisier et al 2007, Plaisier et al 2010].
The small-vessel brain disease of HANAC is usually clinically asymptomatic [Alamowitch et al 2009]. By contrast, the systemic symptoms usually observed in HANAC – including muscle cramps, renal involvement, retinal arterial tortuosity, and Raynaud phenomenon – are rarely reported in COL4A1-related porencephaly or small-vessel brain disease [Gould et al 2006, Meuwissen et al 2015].
Brain involvement
Half of affected individuals have: cerebral small-vessel disease characterized by leukoencephalopathy affecting subcortical, periventricular, or pontine regions; dilated perivascular spaces; lacunar infarcts; and microbleeds. None have porencephaly. Only two of the 14 affected individuals have clinical cerebrovascular symptoms: a minor ischemic stroke and a mild post-traumatic intracerebral hemorrhage while on anticoagulants [
Alamowitch et al 2009].
Single or multiple intracranial aneurysms, all located on the carotid siphon, have been observed in six individuals, with no rupture episode ().
Renal manifestations
Abdominal MRI showing bilateral renal cysts in a patient with HANAC syndrome [Plaisier et al 2007]
Muscle cramps involving a variety of muscles usually occur in affected individuals, with first episodes occurring before age three years. Muscle strength was slightly affected in only two individuals. Electromyography does not show a specific abnormality, and muscle biopsy, available in one person only, is normal. All affected individuals have persistent elevation of serum CK concentration.
Bilateral retinal arteriolar tortuosity is observed in all individuals with HANAC syndrome (see Ocular manifestations below).
Other manifestations. Raynaud phenomenon, supraventricular arrhythmia, and liver cyst are variably reported.
Additional Findings in COL4A1-Related Disorders
Ocular manifestations are variably observed in COL4A1-related disorders. Three distinct ocular features have been reported:
Bilateral retinal arterial tortuosity is variably present in individuals with small-vessel brain disease with hemorrhage [
Gould et al 2006] or with porencephaly [
van der Knaap et al 2006], whereas all individuals with HANAC syndrome exhibited this finding [
Plaisier et al 2007]. Additionally, one
COL4A1 pathogenic variant has been characterized in individuals with
familial retinal arteriolar tortuosity without cerebral or systemic symptoms [
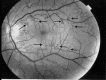
Zenteno et al 2014]. Fundus examination shows marked tortuosity of second- and third-order retinal arteries, with normal first-order arteries and retinal veins (). No leakage or staining is observed on fluorescein angiography. Affected individuals experience episodic transient visual loss as a result of retinal hemorrhage occurring spontaneously or after minor stress or trauma. Visual prognosis has been excellent, without retinal sequelae.
Anterior segment anomaly of Axenfeld-Rieger type comprises a spectrum of ocular findings affecting the anterior chamber including
congenital iris abnormalities, posterior embryotoxon, microcornea, increased intraocular pressure, and glaucoma. It has been described in families presenting with small-vessel brain disease and porencephaly [
Sibon et al 2007,
Meuwissen et al 2015].
Fluorescein angiography showing typical retinal arteriolar tortuosity (arrows) in a patient with HANAC syndrome [Plaisier et al 2007]
Renal manifestations
Bilateral renal cysts are very frequently observed in individuals with HANAC, and more rarely in affected individuals with small-vessel brain disease. In one family, renal cysts represent the main clinical manifestation, together with hematuria and glomerular filtration rate decrease [
Gale et al 2016].
Hematuria has been reported in individuals with porencephaly, small-vessel brain disease, and HANAC [
Meuwissen et al 2015].
Unilateral renal atrophy has been reported in two individuals [
John et al 2015].
Glomerular filtration rate decrease (usually affecting individuals age >40 years) has been observed in individuals with HANAC, as well as in affected individuals from one family presenting with renal cysts and hematuria without brain or extracerebral involvement [
John et al 2015,
Gale et al 2016].
Muscle manifestations. Serum CK elevation and painful muscle cramps, seen in all individuals with HANAC syndrome [Plaisier et al 2010], are only variably present in other affected individuals [Meuwissen et al 2015]. Muscle biopsy was performed in one affected individual with myopathic defects [Yoneda et al 2013].
Cardiac manifestations. Mitral valve prolapse and supraventricular arrythmia have been characterized in four patients [Plaisier et al 2010, Meuwissen et al 2015].
Ultrastructural basement membrane abnormalities have been demonstrated in skin vessels, at the dermo-epidermal junction, and in kidney.
In individuals with HANAC syndrome with hematuria, ultrastructural examination disclosed irregular and abnormal thickening of the basement membranes of the tubules, Bowman’s capsule, and interstitial capillaries [
Plaisier et al 2007]. In the skin, similar alterations including
duplication of the basement membrane are seen at the dermo-epidermal junction and in dermal arterioles; vascular smooth muscle cells are dissociated due to abnormal spreading of basement membrane [
Plaisier et al 2010].
Genotype-Phenotype Correlations
The number of COL4A1 pathogenic variants is too small to explore genotype-phenotype relationships.
However, all six COL4A1 pathogenic variants associated with hereditary angiopathy, nephropathy, aneurysms, and muscle cramps (i.e., HANAC syndrome) are localized in exons 24 and 25 [Plaisier et al 2007, Plaisier et al 2010]. They affect glycine residues localized in a short 30-amino acid region of the protein, whereas all but one pathogenic variant responsible for more severe brain disease, including porencephaly and small-vessel brain disease, are mostly distributed through exons 25 to 51 [Meuwissen et al 2015].
Penetrance
Penetrance of COL4A1-related disorders is probably close to 100%, with expression varying in age of onset and severity of the clinical symptoms, even in the same family; however, these data need verification in larger cohort studies.
Prevalence
Prevalence of COL4A1-related disorders cannot be established as fewer than 100 families have been described.
No data on the prevalence of COL4A1 pathogenic variants in persons with microscopic hematuria or renal cysts are available.
To date pathogenic variants have been reported in individuals of Dutch, Italian, French, German, American, Chinese, Spanish, and Japanese origin.
Differential Diagnosis
COL4A2-related porencephaly and intracerebral hemorrhages. Seven heterozygous COL4A2 pathogenic variants have been characterized in individuals with either porencephaly (porencephaly type 2; OMIM 614483) or intracerebral hemorrhage (OMIM 614519). Neurologic presentation of individuals with porencephaly type 2 was similar to that observed in COL4A1-related porencephaly [Yoneda et al 2012, Meuwissen et al 2015]. Four patients presented with adult-onset intracerebral hemorrhage [Jeanne et al 2012]. Variably present extracerebral symptoms included cerebellar and optic atrophy, cataracts, intracranial aneurysms, nephropathy, and myopathy [Verbeek et al 2012, Gunda et al 2014].
CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) is characterized by a history of migraine headaches with aura (30%-40% of individuals), mid-adult (30s-60s) onset of cerebrovascular disease, mood disturbance, apathy, cognitive disturbance progressing to dementia, and diffuse white-matter lesions and subcortical infarcts on neuroimaging. The pathologic hallmark of CADASIL is electron-dense granules in the media of arterioles that can often be identified by electron microscopic evaluation of skin biopsies. Mutation of NOTCH3 causes CADASIL [Joutel et al 1996]. Inheritance is autosomal dominant.
Autosomal dominant retinal vasculopathy with cerebral leukodystrophy (RVCL) (OMIM 192315) is a microvascular endotheliopathy, which variably associates a retinal vasculopathy, migraine, Raynaud phenomenon, stroke, and dementia with onset in middle age [Ophoff et al 2001].
Hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS), a distinctive subtype of RVCL, is characterized by brain disease, kidney disease (hematuria and proteinuria), and Raynaud phenomenon. Ultrastructural alterations affecting the glomerular basement membrane and the basement membrane of capillaries in the brain and other tissues have been observed in HERNS [Jen et al 1997].
C terminus pathogenic variants in TREX1 cause RVCL [Richards et al 2007]. Inheritance is autosomal dominant.
Porencephalic cysts may occur after antenatal or neonatal parenchymal hemorrhagic infarction in the context of neonatal alloimmune thrombocytopenia; a coagulopathy like von Willebrand disease, factor V deficiency (OMIM 227400), or factor X deficiency (OMIM 227600); maternal warfarin use; or thrombophilia (most often heterozygosity for factor V Leiden mutation) (see Factor V Leiden Thrombophilia).
CARASIL (cerebral autosomal recessive arteriopathy with subcortical infarcts and leucoencephalopathy; Maeda-syndrome) is characterized by early-onset changes in the deep white matter of the brain observed on MRI and associated neurologic findings. The most frequent initial symptom is gait disturbance from spasticity beginning between ages 20 and 30 years; 23% of affected individuals have stroke-like episodes before age 40 years. Mood changes (apathy and irritability), pseudobulbar palsy, and cognitive dysfunction begin between ages 20 and 50 years. The disease progresses slowly over the five to 20 years following the onset of neurologic symptoms. This rare disease is caused by pathogenic variants in HTRA1; inheritance is autosomal recessive. CARASIL has been reported mostly in Japanese and Chinese families. See HTRA1 Disorder.
Anophthalmia/microphthalmia (A/M). Microphthalmia refers to a globe with a total axial length that is at least two standard deviations below the mean for age. Anophthalmia refers to complete absence of the globe in the presence of ocular adnexa (eyelids, conjunctiva, and lacrimal apparatus). A/M is a genetically heterogenous disorder that has been associated with pathogenic variants in more than 70 genes. SOX2, OTX2, and FOXE3 are among the genes most commonly associated with A/M [Chassaing et al 2014].
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with COL4A1-related disorders, the following are recommended:
Brain MRI including T1-weighted saggital, T2-weighted axial, and FLAIR axial images
Brain angiographic CT scan
Ophthalmologic examination including fundoscopic examination and slit-lamp examination
Kidney and liver ultrasound examination or CT
Measurement of serum CK concentration
Measurement of serum creatinine concentration and estimation of the glomerular filtration rate
Evaluation for the presence of hematuria
Electrocardiogram (EKG); echocardiography and ambulatory EKG monitoring in individuals presenting with palpitations
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Hypertensive individuals must be treated to reduce the global risk of stroke.
Supportive care including practical help, emotional support, and counseling are appropriate for affected individuals and their families.
No specific support exists for individuals with COL4A1-related disorders.
Seizures are managed using standard protocols.
Cataract surgery may be required for individuals with severe lens opacities.
Glaucoma is initially treated with topical anti-glaucoma medication. Surgery is reserved for eyes that do not respond to medical therapy.
Symptomatic paroxysmal supraventricular arrhythmia should be treated with antiarrhythmic drugs (beta blockers).
Surgical or endovascular treatment should be discussed for asymptomatic intracranial aneurysms >10.0 mm in diameter.
Prevention of Primary Manifestations
Avoidance of anticoagulant exposure and activities that involve an increased risk for head trauma may decrease the risk for intracerebral hemorrhage.
Surveillance
The interval at which individuals with COL4A1-related disorders should be seen for follow up depends on the severity and type of symptoms.
Annual clinical evaluation is reasonable.
Regular brain imaging can be proposed, especially to evaluate the size of asymptomatic cerebral aneurysms.
Agents/Circumstances to Avoid
The following should be avoided:
Pregnancy Management
Cesarean delivery for pregnancies in which the fetus is at risk for a COL4A1-related disorder is recommended to prevent brain vascular injury attributable to birth trauma in newborns [Gould et al 2006].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
COL4A1-related disorders are inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
At least 50% of individuals diagnosed with a
COL4A1-related disorder have an affected parent [
Meuwissen et al 2015].
If the
COL4A1 pathogenic variant found in the
proband cannot be detected in leukocyte DNA of either parent, possible explanations include a
de novo pathogenic variant in the proband or
germline mosaicism in a parent. Though theoretically possible, no instances of germline mosaicism have been reported.
The family history of some individuals diagnosed with a
COL4A1-related disorder may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent. Therefore, an apparently negative family history cannot be confirmed unless appropriate evaluations and
molecular genetic testing have been performed on the parents of the
proband.
Sibs of a proband
The risk to the sibs of the
proband depends on the genetic status of the proband’s parents: if a parent of the proband is affected, the risk to the sibs is 50%.
Although
penetrance of
COL4A1-related disorders is probably close to 100%, age of onset and severity of the clinical symptoms vary, even in the same family.
The sibs of a
proband with clinically unaffected parents are still at increased risk (for the disorder) because of the possibility of reduced
penetrance in a parent.
Offspring of a proband. Each child of an individual with a COL4A1-related disorder has a 50% chance of inheriting the COL4A1 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the COL4A1 pathogenic variant, other members of the parent's family may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the COL4A1 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for a COL4A1-related disorder are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
National Institute of Neurological Disorders and Stroke (NINDS)
Phone: 800-352-9424
CDC - Developmental Disabilities
Phone: 800-CDC-INFO
Email: cdcinfo@cdc.gov
Epilepsy Foundation
Phone: 301-459-3700
Fax: 301-577-2684
Filière Orphan Kidney Diseases
CHRU de Montpellier - Hôpital Arnaud de Villeneuve
371, Avenue du Doyen Gaston Giraud
France
Phone: 33 4 67 33 55 99
Email: contact@filiereorkid.com
Kidney Foundation of Canada
Canada
Phone: 514-369-4806; 800-361-7494
Email: info@kidney.ca
National Eye Institute
Phone: 301-496-5248
Email: 2020@nei.nih.gov
National Kidney Foundation (NFK)
Phone: 855-NKF-CARES; 855-653-2273
Email: nkfcares@kidney.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
COL4A1-Related Disorders: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
View in own window
|
120130 | COLLAGEN, TYPE IV, ALPHA-1; COL4A1 |
|
175780 | BRAIN SMALL VESSEL DISEASE 1 WITH OR WITHOUT OCULAR ANOMALIES; BSVD1 |
|
611773 | ANGIOPATHY, HEREDITARY, WITH NEPHROPATHY, ANEURYSMS, AND MUSCLE CRAMPS; HANAC |
Gene structure.
COL4A1 consists of 52 exons spanning roughly 158 kb. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Most of the sequence alterations in COL4A1 are missense variants (leading to the substitution of a glycine residue in the collagenous domain of the protein) located between exons 24 and 49. One pathogenic variant affects the start codon. One small duplication is located within the non-collagenous domain (NC-1) located at the C-terminal end of the protein.
Table 2.
Selected COL4A1 Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.1A>T | p.Met1Leu |
NM_001845.4
NP_001836.2
|
| c.1493G>T | p.Gly498Val |
| c.1528G>A | p.Gly510Arg |
| c.1555G>A | p.Gly519Arg |
| c.1583G>A | p.Gly528Glu |
| c.1769G>A | p.Gly562Glu |
| c.2122G>A | p.Gly708Arg |
| c.2159G>A | p.Gly720Asp |
| c.2245G>A | p.Gly749Ser |
| c.2317G>A | p.Gly773 |
| c.2345G>C | p.Gly782Ala |
| c.2413G>A | p.Gly805Arg |
| c.3389G>A | p.Gly1130Asp |
| c.3706G>A | p.Gly1236Arg |
| c.3976G>A | p.Gly1326Arg |
| c.4267G>C | p.Gly1423Arg |
| c.4582-4586dupCCCAT | p.Met1529IlefsTer15 |
| c.4611_4612insG | p.Thr1537fs |
| c.4738G>C | p.Gly1580Arg |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
COL4A1 encodes the alpha 1 chain of type IV collagen. Type IV collagen is the main component of basement membranes. The six different type IV collagen alpha chains described all consist of a small amino-terminal 7S domain, a large collagenous domain containing the classic Gly-X-Y repeat, and a carboxy-terminal non-collagenous NC1 domain. Specific interactions between NC1 domains initiate the formation of only three different trimers: α1α1α2, α3α4α5, and α5α5α6. Glycine residues play an important role for the stabilization of collagenous triple helical domain. Isoforms of type IV collagen display a tissue- and developmental-specific distribution that explains the heterogeneity of basement membrane composition. The α1α1α2 (IV) trimer is widely expressed, whereas the α3α4α5 (IV) and α5α5α6 (IV) trimers display a more tissue-restricted expression.
Abnormal gene product. Most of the pathogenic variants reported in COL4A1-related disorders affect highly conserved glycine residues within the collagenous domain of the protein. These amino-acid changes are predicted to result in detrimental effect on collagen α1α1α2 (IV) triple helix formation and stability. Six pathogenic variants are localized in the non-collagenous (NC1) domain that contains recognition sequences involved in the collagen α1α1α2 (IV) triple helix assembly. One pathogenic variant (p.Met1Leu) affects the start codon, resulting in an unknown effect on protein synthesis (see Table 2). Two pathogenic variants affect splice sites, leading to exon deletions as demonstrated by cDNA analysis.
Chapter Notes
Revision History
7 July 2016 (ha) Comprehensive update posted live
8 March 2011 (me) Comprehensive update posted live
25 June 2009 (et) Review posted live
27 February 2009 (ep) Original submission
References
Literature Cited
Alamowitch S, Plaisier E, Favrole P, Prost C, Chen Z, Van Agtmael T, Marro B, Ronco P. Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome.
Neurology. 2009;73:1873–82. [
PMC free article: PMC2881859] [
PubMed: 19949034]
Ayrignac X, Carra-Dalliere C, Menjot de Champfleur N, Denier C, Aubourg P, Bellesme C, Castelnovo G, Pelletier J, Audoin B, Kaphan E, de Seze J, Collongues N, Blanc F, Chanson JB, Magnin E, Berger E, Vukusic S, Durand-Dubief F, Camdessanche JP, Cohen M, Lebrun-Frenay C, Brassat D, Clanet M, Vermersch P, Zephir H, Outteryck O, Wiertlewski S, Laplaud DA, Ouallet JC, Brochet B, Goizet C, Debouverie M, Pittion S, Edan G, Deburghgraeve V, Le Page E, Verny C, Amati-Bonneau P, Bonneau D, Hannequin D, Guyant-Maréchal L, Derache N, Defer GL, Moreau T, Giroud M, Guennoc AM, Clavelou P, Taithe F, Mathis S, Neau JP, Magy L, Devoize JL, Bataillard M, Masliah-Planchon J, Dorboz I, Tournier-Lasserve E, Levade T, Boespflug Tanguy O, Labauge P. Adult-onset genetic leukoencephalopathies: A MRI pattern-based approach in a comprehensive study of 154 patients.
Brain. 2015;138:284–92. [
PubMed: 25527826]
Breedveld G, de Coo RF, Lequin MH, Arts WFM, Heuting P, Gould DB, John SWM, Oostra B, Mancini GMS. Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly.
J Med Genet. 2006;43:490–5. [
PMC free article: PMC2593028] [
PubMed: 16107487]
Chassaing N, Causse A, Vigouroux A, Delahaye A, Alessandri JL, Boespflug-Tanguy O, Boute-Benejean O, Dollfus H, Duban-Bedu B, Gilbert-Dussardier B, Giuliano F, Gonzales M, Holder-Espinasse M, Isidor B, Jacquemont ML, Lacombe D, Martin-Coignard D, Mathieu-Dramard M, Odent S, Picone O, Pinson L, Quelin C, Sigaudy S, Toutain A, Thauvin-Robinet C, Kaplan J, Calvas P. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia.
Clin Genet. 2014;86:326–34. [
PubMed: 24033328]
Coupry I, Sibon I, Mortemousque B, Rouanet F, Mine M, Goizet C. Ophthalmological features associated with COL4A1 mutations.
Arch Ophthalmol. 2010;128:483–9. [
PubMed: 20385946]
Gale DP, Oygar DD, Lin F, Oygar PD, Khan N, Connor TM, Lapsley M, Maxwell PH, Neild GH. A novel COL4A1 frameshift mutation in familial kidney disease: the importance of the C-terminal NC1 domain of type IV collagen.
Nephrol Dial Transplant. 2016;31:1908–14. [
PMC free article: PMC5091614] [
PubMed: 27190376]
Gould DB, Phalan FC, Breedveld GJ. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly.
Science. 2005;308:1167–71. [
PubMed: 15905400]
Gould DB, Phalan FC, van Mil SE, Sundberg JP, Vahedi K, Massin P, Bousser MG, Heuting P, Miner JH, Tournier-Lasserve E, John SWM. Role of COL4A1 in small-vessel disease and hemorrhagic stroke.
N Engl J Med. 2006;354:1489–96. [
PubMed: 16598045]
Gunda B, Mine M, Kovács T, Hornyák C, Bereczki D, Várallyay G, Rudas G, Audrezet MP, Tournier-Lasserve E. COL4A2 mutation causing adult onset recurrent intracerebral hemorrhage and leukoencephalopathy.
J Neurol. 2014;261:500–3. [
PubMed: 24390199]
Jeanne M, Labelle-Dumais C, Jorgensen J, Kauffman WB, Mancini GM, Favor J, Valant V, Greenberg SM, Rosand J, Gould DB. COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke.
Am J Hum Genet. 2012;90:91–101. [
PMC free article: PMC3257894] [
PubMed: 22209247]
Jen J, Cohen AH, Yue Q, Stout JT, Vinters HV, Nelson S, Baloh RW. Hereditary endotheliopathy with retinopathy, nephropathy, and stroke.
Neurology. 1997;49:1322–30. [
PubMed: 9371916]
John S, Jehi L, Manno EM, Conway DS, Uchino K. COL4A1 gene mutation--beyond a vascular syndrome.
Seizure. 2015;31:19–21. [
PubMed: 26362372]
Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cécillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia.
Nature. 1996;383:707–10. [
PubMed: 8878478]
Livingston J, Doherty D, Orcesi S, Tonduti D, Piechiecchio A, La Piana R, Tournier-Lasserve E, Majumdar A, Tomkins S, Rice G, Kneen R, van der Knaap M, Crow Y. COL4A1 mutations associated with a characteristic pattern of intracranial calcification.
Neuropediatrics. 2011;42:227–33. [
PubMed: 22134833]
Meuwissen ME, Halley DJ, Smit LS, Lequin MH, Cobben JM, de Coo R, van Harssel J, Sallevelt S, Woldringh G, van der Knaap MS, de Vries LS, Mancini GM. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature.
Genet Med. 2015;17:843–53. [
PubMed: 25719457]
Ophoff RA, DeYoung J, Service SK. Hereditary vascular retinopathy, cerebrovascular vasculopathy, and hereditary endotheliopathy with retinopathy, nephropathy, and stroke map to a single locus on chromosome 3p21.1-p21.3.
Am J Hum Genet. 2001;69:447–53. [
PMC free article: PMC1235317] [
PubMed: 11438888]
Plaisier E, Chen Z, Gekeler F, Benhassine S, Dahan K, Marro B, Alamowitch S, Paques M, Ronco P. Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain.
Am J Med Genet A. 2010;152A:2550–5. [
PubMed: 20818663]
Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, Verpont MC, Marro B, Desmettre T, Cohen SY, Roullet E, Dracon M, Fardeau M, Van Agtmael T, Kerjaschki D, Antignac C, Ronco P. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps.
N Engl J Med. 2007;357:2687–95. [
PubMed: 18160688]
Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-Labarca ML, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KR, de Vries B, Wan J, Kane MJ, Mamsa H, Schafer R, Stam AH, Haan J, de Jong PT, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP. C-terminal truncations in human 3'-5' DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy.
Nat Genet. 2007;39:1068–70. [
PubMed: 17660820]
Rødahl E, Knappskog PM, Majewski J, Johansson S, Telstad W, Kråkenes J, Boman H. Variants of anterior segment dysgenesis and cerebral involvement in a large family with a novel COL4A1 mutation.
Am J Ophthalmol. 2013;155:946–53. [
PubMed: 23394911]
Shah S, Ellard S, Kneen R, Lim M, Osborne N, Rankin J, Stoodley N, van der Knaap M, Whitney A, Jardine P. Childhood presentation of COL4A1 mutations.
Dev Med Child Neurol. 2012;54:569–74. [
PubMed: 22574627]
Shah S, Kumar Y, McLean B, Churchill A, Stoodley N, Rankin J, Rizzu P, van der Knaap M, Jardine P. A dominantly inherited mutation in collagen IV A1 (COL4A1) causing childhood onset stroke without porencephaly.
Eur J Paediatr Neurol. 2010;14:182–7. [
PubMed: 19477666]
Sibon I, Coupry I, Menegon P, Bouchet JP, Gorry P, Burgelin I, Calvas P, Orignac I, Dousset V, Lacombe D, Orgogozo JM, Arveiler B, Goizet C. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke.
Ann Neurol. 2007;62:177–84. [
PubMed: 17696175]
Vahedi K, Kubi N, Boukobza M, Arnoult M, Massin P, Gould DB, Tournier-Lasserve E, Bousser MG. COL4A1 mutation in patient with sporadic recurrent intracerebral hemorrhage.
Stroke. 2007;38:1461–4. [
PubMed: 17379824]
van der Knaap MS, Smit LM, Barkhof F, Pijnenburg YA, Zweegman S, Niessen HW, Imhof S, Heutink P. Neonatal porencephaly and adult stroke related to mutations in collagen IV A1.
Ann Neurol. 2006;59:504–11. [
PubMed: 16374828]
Verbeek E, Meuwissen ME, Verheijen FW, Govaert PP, Licht DJ, Kuo DS, Poulton CJ, Schot R, Lequin MH, Dudink J, Halley DJ, de Coo RI, den Hollander JC, Oegema R, Gould DB, Mancini GM. COL4A2 mutation associated with familial porencephaly and small-vessel disease.
Eur J Hum Genet. 2012;20:844–51. [
PMC free article: PMC3400734] [
PubMed: 22333902]
Xia XY, Li N, Cao X, Wu QY, Li TF, Zhang C, Li WW, Cui YX, Li XJ, Xue CY. A novel COL4A1 gene mutation results in autosomal dominant non-syndromic congenital cataract in a Chinese family.
BMC Med Genet. 2014;15:97. [
PMC free article: PMC4236509] [
PubMed: 25124159]
Yoneda Y, Haginoya K, Arai H, Yamaoka S, Tsurusaki Y, Doi H, Miyake N, Yokochi K, Osaka H, Kato M, Matsumoto N, Saitsu H. De novo and inherited mutations in COL4A2, encoding the type IV collagen α2 chain cause porencephaly.
Am J Hum Genet. 2012;90:86–90. [
PMC free article: PMC3257897] [
PubMed: 22209246]
Yoneda Y, Haginoya K, Kato M, Osaka H, Yokochi K, Arai H, Kakita A, Yamamoto T, Otsuki Y, Shimizu S, Wada T, Koyama N, Mino Y, Kondo N, Takahashi S, Hirabayashi S, Takanashi J, Okumura A, Kumagai T, Hirai S, Nabetani M, Saitoh S, Hattori A, Yamasaki M, Kumakura A, Sugo Y, Nishiyama K, Miyatake S, Tsurusaki Y, Doi H, Miyake N, Matsumoto N, Saitsu H. Phenotypic spectrum of COL4A1 mutations: porencephaly to schizencephaly.
Ann Neurol. 2013;73:48–57. [
PubMed: 23225343]
Zenteno JC, Crespi J, Buentello-Volante B, Buil JA, Bassaganyas F, Vela-Segarra JI, Diaz-Cascjosa J, Marieges MT. Next generation sequencing uncovers a missense mutation in COL4A1 as the cause of familial retinal arteriolar tortuosity.
Graefes Arch Clin Exp Ophthalmol. 2014;252:1789–94. [
PubMed: 25228067]