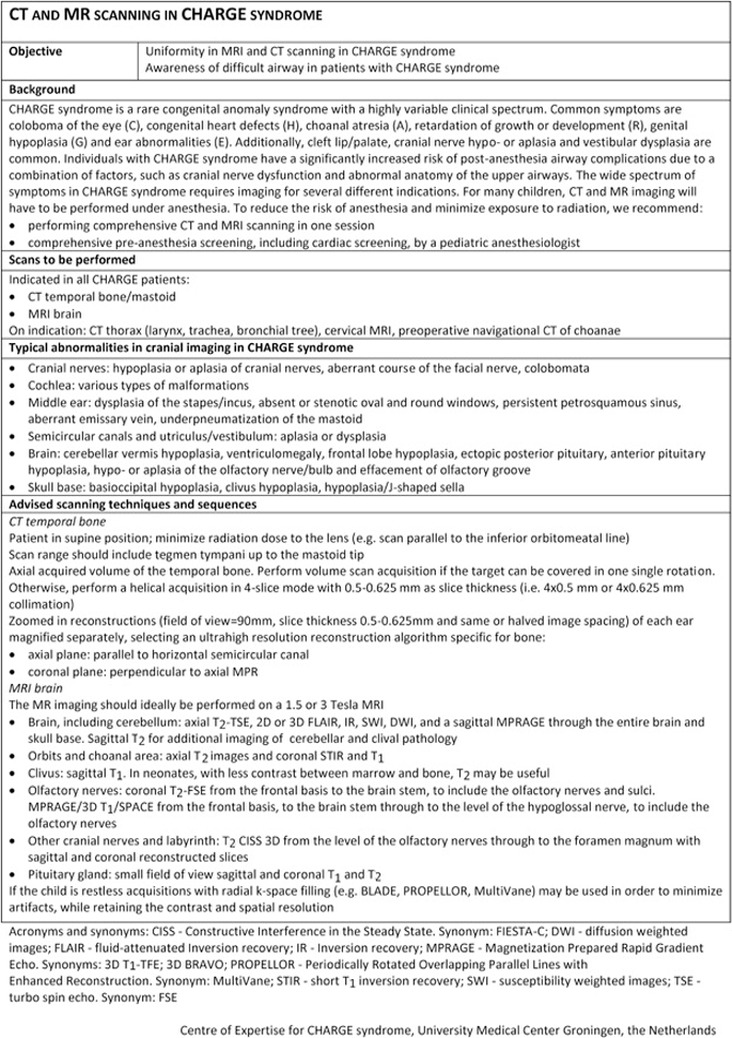
Summary
Clinical characteristics.
CHD7 disorder encompasses the entire phenotypic spectrum of heterozygous CHD7 pathogenic variants that includes CHARGE syndrome as well as subsets of features that comprise the CHARGE syndrome phenotype. The mnemonic CHARGE syndrome, introduced in the premolecular era, stands for coloboma, heart defect, choanal atresia, retarded growth and development, genital hypoplasia, ear anomalies (including deafness). Following the identification of the genetic cause of CHD7 disorder, the phenotypic spectrum expanded to include cranial nerve anomalies, vestibular defects, cleft lip and/or palate, hypothyroidism, tracheoesophageal anomalies, brain anomalies, seizures, and renal anomalies. Life expectancy highly depends on the severity of manifestations; mortality can be high in the first few years when severe birth defects (particularly complex heart defects) are present and often complicated by airway and feeding issues. In childhood, adolescence, and adulthood, decreased life expectancy is likely related to a combination of residual heart defects, infections, aspiration or choking, respiratory issues including obstructive and central apnea, and possibly seizures. Despite these complications, the life expectancy for many individuals can be normal.
Diagnosis/testing.
The diagnosis of CHD7 disorder is established in a proband with suggestive clinical and imaging findings and a heterozygous pathogenic variant in or deletion of CHD7 identified by molecular genetic testing.
Management.
Treatment of manifestations: Management of the manifestations of CHD7 disorder can be complex and require a multidisciplinary approach involving clinicians, therapists, and educators.
Surveillance: Requires routine follow up of manifestations identified in infancy/childhood, as well as ongoing monitoring of growth, development, educational progress, behavior, and possible endocrine issues.
Agents/circumstances to avoid: Because of the increased risk of post-anesthesia airway complications, procedures requiring anesthesia should be minimized and combined whenever possible.
Genetic counseling.
CHD7 disorder is an autosomal dominant disorder typically caused by a de novo pathogenic variant. In rare instances, an individual with CHD7 disorder inherits a pathogenic variant from a heterozygous parent. The risk to the sibs of the proband depends on the genetic status of the proband's parents: (1) If a parent of the proband has a CHD7 pathogenic variant, the risk to the sibs of inheriting the pathogenic variant is 50%; (2) If the CHD7 pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the empiric recurrence risk to sibs of a proband is approximately 1%-2% because of the possibility of parental germline mosaicism. Although many individuals with CHD7 disorder are not able to reproduce, each child of an individual with CHD7 disorder has a 50% chance of inheriting the pathogenic variant. Once the CHD7 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
GeneReview Scope
With the current widespread use of multigene panels and comprehensive genomic testing, it has become apparent that the phenotypic spectrum of heterozygous CHD7 pathogenic variants has broadened to encompass CHARGE syndrome as well as subsets of features that comprise the CHARGE syndrome phenotype. The title of this chapter, CHD7 disorder, refers to the entire phenotypic spectrum that can be associated with heterozygous CHD7 pathogenic variants and emphasizes both the need to evaluate an individual found to have a CHD7 pathogenic variant for medically actionable manifestations in the entire phenotypic spectrum (regardless of clinical findings that prompted molecular genetic testing) and the importance of counseling families that the finding of a CHD7 pathogenic variant is not equivalent to a diagnosis of CHARGE syndrome.
Diagnosis
Suggestive Findings
CHD7 disorder should be suspected in individuals with combinations of the following findings and family history.
Clinical and imaging findings
- Coloboma of the iris, retina, choroid, and/or disc, and/or anophthalmos or microphthalmos
- Choanal atresia or stenosis: unilateral or bilateral, bony or membranous, confirmed by axial sections of non-enhanced axial CT scan
- Cleft palate with or without cleft lip (Note: Choanal atresia is rare in the presence of a cleft palate.)
- Cranial nerve dysfunction or anomaly
- Cranial nerve I. Hyposmia or anosmia
- Cranial nerve VII. Facial palsy (unilateral or bilateral)
- Cranial nerve VIII. Sensorineural hearing loss and/or balance problems, hypoplasia or aplasia on imaging
- Cranial nerve IX/X. Difficulty with sucking/swallowing and aspiration, gut motility problems
- Ear malformations (most characteristic of CHD7 disorder)
- Auricle. Short, wide ear with little or no lobe, "snipped-off" helix, prominent antihelix that is often discontinuous with tragus, triangular concha, decreased cartilage; often protruding and usually asymmetric (See Figure 1.)
- Middle ear. Ossicular malformations (resulting in a typical wedge-shaped audiogram due to mixed sensorineural and conductive hearing loss)
- Temporal bone abnormalities (most commonly determined by temporal bone CT scan). Mondini defect of the cochlea (cochlear hypoplasia), absent or hypoplastic semicircular canals
- Tracheoesophageal fistula or esophageal atresia
- Cardiovascular malformation, including conotruncal defects (e.g., tetralogy of Fallot), AV canal defects, and aortic arch anomalies [Corsten-Janssen & Scambler 2017]
- Hypogonadotropic hypogonadism
- Males at birth. Micropenis and cryptorchidism
- Females at birth. Hypoplastic labia, abnormal or (rarely) absent uterus
- Males and females. Delayed or absent puberty, often in combination with anosmia [Bergman et al 2011a]
- Developmental delay / intellectual disability, delayed motor milestones, often secondary to sensory and balance deficits
- Growth deficiency. Short stature, usually postnatal with or without growth hormone deficiency
- Other clinical features
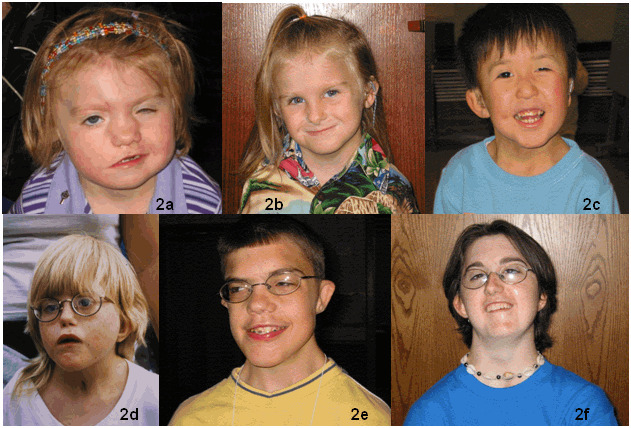
- Face. Square-shaped with broad forehead, broad nasal bridge, prominent nasal columella, flattened malar area, facial palsy or other asymmetry, cleft lip, and small chin (gets larger and broader with age) (See Figure 2.)
- Neck. Short and wide with sloping shoulders [O'Grady et al 2016] (See Figure 2.)
- Hands. Typically, short, wide palm with hockey-stick crease, short fingers, and finger-like thumb (see Figure 3); polydactyly and reduction defects in a small percentage [Van de Laar et al 2007]
- Brain MRI. Clivus hypoplasia [de Geus et al 2018], hypoplasia of cerebellar vermis [Donovan et al 2017]
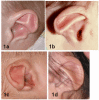
Figure 1.
Ears 1a. Clipped-off helix, prominent antihelix that extends to the outer helical rim, antihelix discontinuous with the antitragus; absent lobe
Figure 2.
Face 2a. Female age 2.5 years; square face, round eye, straight nose with broad nasal root, unilateral facial palsy
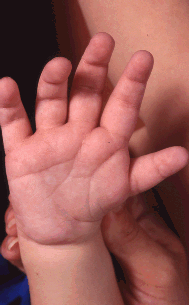
Figure 3.
Hand Typical CHARGE hand: square hand, short fingers, finger-like thumb, hockey-stick palmar crease
Family history is consistent with autosomal dominant inheritance. While the majority of individuals with CHD7 disorder are simplex cases (i.e., a single occurrence in a family resulting from a de novo CHD7 pathogenic variant), familial occurrences consistent with autosomal dominant inheritance and germline mosaicism have been reported [Bergman et al 2011b, Legendre et al 2017]. Note: Absence of a family history of features consistent with CHD7 disorder does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of CHD7 disorder is established in a proband with suggestive clinical and imaging findings and a heterozygous pathogenic (or likely pathogenic) variant in CHD7 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of a heterozygous CHD7 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (CHD7 single-gene testing, multigene panel) and comprehensive genomic testing (chromosomal microarray, exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determines which gene(s) are likely involved, whereas genomic testing does not. Individuals with suggestive findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with atypical findings are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of CHD7 is performed to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications and/or chromosomal microarray (CMA) to detect whole-gene deletions.
A multigene panel (e.g., for developmental delay, coloboma, deafness, heart defects, Kallmann syndrome, normosmic hypogonadotropic hypogonadism) that includes CHD7 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
Because CHD7 disorder typically includes multiple congenital anomalies, it is also reasonable to pursue chromosomal microarray testing first, unless classic features of CHD7 disorder (e.g., the CHARGE syndrome phenotype) are apparent.
Alternatively, if exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Epigenetic signature analysis / methylation array. A distinctive epigenetic signature (disorder-specific genome-wide changes in DNA methylation profiles) in peripheral blood leukocytes has been identified in individuals with CHD7 disorder [Aref-Eshghi et al 2020, Levy et al 2021]. Epigenetic signature analysis of a peripheral blood sample or DNA banked from a blood sample can therefore be considered to clarify the diagnosis in individuals with: (1) suggestive findings of CHD7 disorder but in whom no pathogenic variant in CHD7 has been identified via sequence analysis or genomic testing; or (2) suggestive findings of CHD7 disorder and a variant of uncertain clinical significance identified by molecular genetic testing. For an introduction to epigenetic signature analysis click here.
Table 1.
Molecular Genetic Testing Used in CHD7 Disorder
Clinical Characteristics
Clinical Description
In the premolecular era, the acronym CHARGE was proposed for the combination of the clinical features coloboma, heart defect, choanal atresia, retarded growth and development, genital hypoplasia, ear anomalies (including deafness) of unknown cause [Pagon et al 1981]. Clinical diagnostic criteria were refined for what became called CHARGE association [Blake et al 1998, Verloes 2005]. Following the discovery that heterozygous CHD7 variants and deletions cause CHARGE syndrome [Vissers et al 2004], molecular genetic testing of family members of probands with CHARGE syndrome expanded the phenotypic spectrum to include phenotypes that do not fulfill the previously proposed CHARGE syndrome clinical diagnostic criteria [Lalani et al 2006, Delahaye et al 2007, Jongmans et al 2009, Bergman et al 2011b, Hale et al 2016]. Thus, CHD7 disorder exhibits a high degree of clinical variability even among individuals in the same family and among individuals from different families with the same pathogenic variant [Jongmans et al 2008].
This section discusses only those reports in which a CHD7 pathogenic variant has been confirmed in affected individuals. To date reports of isolated manifestations of CHD7 disorder have been rare – many of which did not document a clinical workup sufficient to identify other features in the CHD7 disorder phenotypic spectrum. Thus, the percentages in Table 2 (based on persons with molecularly confirmed CHARGE syndrome [van Ravenswaaij-Arts & Martin 2017]) are likely to change over time as individuals with a CHD7 pathogenic variant ascertained through use of a multigene panel or genomic testing undergo a complete clinical evaluation (see Table 5).
Table 2.
Features of CHD7 Disorder in Individuals Ascertained for CHARGE Syndrome
Because the majority of individuals with a pathogenic CHD7 variant have a typical CHARGE syndrome or CHARGE syndrome-like phenotype, the clinical features described below are relevant for most individuals with CHD7 disorder. In contrast, isolated hypogonadotropic hypogonadism with or without anosmia due to a pathogenic CHD7 missense variant appears to be rare [Xu et al 2018].
Development
Motor delay is invariably present due to vestibular anomalies and presents as poor head control, five-point crawl, delayed motor milestones, and reduced fine motor skills.
Language delay is caused by hearing loss, vision loss, vestibular anomalies, hospitalizations and illness, and/or cognitive impairment.
Assessment of cognitive abilities is difficult because of the multiple sensory deficits (vision, hearing, balance, smell), and much of the delay observed in motor and speech/language abilities is secondary to these deficits. Nonetheless, intellectual outcome is within the normal range in 50% of the individuals with clinical features consistent with CHARGE syndrome [Vesseur et al 2016b].
Children with better walking skills and fewer medical problems exhibit better adaptive behavior than children with less mobility and more medical problems [Salem-Hartshorne & Jacob 2005].
Behavioral features often reported are attention-deficit/hyperactivity disorder, repetitive behavior, and obsessive-compulsive behaviors. Self-abuse is occasionally seen. An increased pain threshold may predispose children to behaviors that are incorrectly interpreted by others as aggressive [Hartshorne et al 2005].
Many adults with clinical features consistent with CHARGE syndrome live independently, including many who have college or even advanced degrees. However, the level of independence comprises a broad spectrum [Blake et al 2005, Hartshorne et al 2016], depending, for each individual, on the combination of clinical features, educational program designed to address specific needs, and resources available.
Other Features
Gastrointestinal problems are frequently seen, mainly GI-related motility issues such as gastroesophageal reflux disease, constipation, and abdominal pain. Feeding challenges often result in tube feeding and problems with aspiration.
Late-onset issues can include malrotation of intestines, intussusception, and choking due to mouth overstuffing [Hudson et al 2015, Blake & Hudson 2017].
Immunodeficiency due to absent thymus (rarely) or decreased number or function of T-cells may occur [Wong et al 2015b]. Recurrent upper airway infections are common.
Skeletal involvement can include craniosynostosis, vertebral anomalies, scoliosis (in the majority of affected individuals) [Doyle & Blake 2005], extra or missing ribs, absent long bones (rare), ectrodactyly, polydactyly, finger-like thumb, and (more commonly) brachydactyly [Van de Laar et al 2007].
Hypermobility and contractures can be part of the syndrome.
Neuromuscular problems are common in CHARGE syndrome, mostly hypotonia (often resulting in scoliosis) and abnormal shoulder girdle muscles [O'Grady et al 2016]. Proprioception is diminished and, when in combination with balance problems, often results in a preference for pressure-building postures (upside-down position, legs twisted around one another) [Brown 2005].
Dental problems may include overbite, hypodontia, and poor mineralization of teeth [Chetty et al 2020].
Life expectancy highly depends on the severity of manifestations, since the phenotypic spectrum of CHD7 disorder is substantial. Mortality can be high in the first few years, when severe birth defects (particularly complex heart defects) are present, and are often complicated by airway and feeding issues. Feeding difficulties are usually due to cranial nerve abnormalities and improve gradually.
Multiple complex surgeries, along with the breathing problems or difficulty with anesthesia reported in CHARGE syndrome [Blake et al 2009], increase the risks associated with procedures.
After the first two or three years, mortality (and certainly morbidity and medical fragility) remains increased, with parents reporting frequent illnesses, infections, and hospitalizations [Bergman et al 2010].
In childhood, adolescence, and adulthood, increased mortality is likely related to a combination of residual heart defects, infections, aspiration or choking [Corsten-Janssen et al 2016], respiratory issues including obstructive and central apnea, and possibly seizures.
A number of families have reported serious (and in some instances lethal) intestinal issues such as volvulus [Lai & Feng 2006] and intussusception.
Despite these complications, the life span for many individuals can be normal. Individuals with clinical features consistent with CHARGE syndrome in their 60s who are in good health have been observed.
Genotype-Phenotype Correlations
While no clear genotype-phenotype correlations exist for CHD7-related CHARGE syndrome [Legendre et al 2017], in general, but not as a rule, missense variants are associated with a less severe phenotype [Bergman et al 2012].
CHD7-related hypogonadotropic hypogonadism with or without anosmia is more likely to be due to missense variants than nonsense variants.
Prevalence
Because of the more widespread use of genomic testing, it is currently difficult to assess the prevalence of CHD7 disorder.
In the past, when the diagnosis of CHARGE syndrome was based on clinical features or gene-specific molecular testing, its estimated prevalence ranged from one in 15,000 newborns in the Netherlands [Janssen et al 2012] to one in 8,500 in Canada [Issekutz et al 2005].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in CHD7.
Differential Diagnosis
Genetic disorders with multiple features overlapping those associated with CHD7 disorder are summarized in Table 3 and Table 4.
Table 3.
Genes to Consider in the Differential Diagnosis of CHD7 Disorder
Table 4 lists the chromosomal syndromes that overlap significantly with CHD7 disorder. Many other chromosome deletions have at least a few features that overlap with CHD7 disorder.
Table 4.
Chromosomal Syndromes that Significantly Overlap with CHD7 Disorder
VACTERL association (OMIM 192350). CHD7 disorder and VACTERL association can both include vertebral anomalies, cardiac anomalies, tracheoesophageal fistula (or esophageal atresia), renal anomalies, and limb anomalies. Anal atresia, common in VACTERL, is rare in CHD7 disorder. Typical CHD7 disorder findings of temporal bone anomaly, choanal atresia, characteristic ear findings (outer and inner), and cranial nerve anomalies are rarely reported in VACTERL. The genetic basis of VACTERL association is unknown.
CHARGE syndrome-like features secondary to prenatal teratogen exposure. Exposure to Accutane™ at any time during the first trimester may result in malformations associated with abnormal migration of neural crest cells. These may include microtia/anotia, micrognathia, cleft palate, conotruncal heart defects, and aortic-arch abnormalities, thymic defects, retinal or optic nerve abnormalities, and central nervous system malformations [Lammer et al 1985].
Exposure to antithyroid agents, especially methimazole, has been reported to result in a variety of congenital anomalies including choanal and esophageal atresia, iris and retinal coloboma, hearing loss, and delayed neurodevelopment. The risk of birth defects in fetuses exposed during the first trimester of pregnancy has been estimated at 2%-3% [Andersen et al 2013, Komoike et al 2013, Andersen et al 2017].
Management
The management of the manifestations of CHD7 disorder can be complex and require a multidisciplinary approach involving clinicians, therapists, and educators. Published CHARGE syndrome guidelines (including one-page summaries) for clinical management [Trider et al 2017] (see Figure 4) and cranial imaging guidelines [de Geus et al 2017] (full text) are available.

Figure 4.
CHARGE syndrome checklist Reproduced from Trider et al [2017]
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with CHD7 disorder, the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in an Individual with a CHD7 Disorder
Treatment of Manifestations
Management of children with a CHD7 disorder requires coordinated multidisciplinary care (Table 6).
Table 6.
Treatment of Manifestations in Individuals with a CHD7 Disorder
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states and provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services may be provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
- State deaf-blind services:
- In addition to the educational services in the US discussed above, state-level federally funded programs are mandated to provide services for individuals from birth to age 21 years with combined hearing and vision issues (nationaldb.org/). Of note, the designation "deaf-blind," used to qualify individuals with combined vision and hearing loss for these services, does not imply total hearing loss or total vision loss.
- State deaf-blind services typically provide information and training to families, technical assistance to schools and early intervention programs, and assistance with IEPs and transitions.
Communication
A growing body of evidence indicates that normal language development can occur if hearing habilitation is started prior to age six months for hearing-impaired children, whether or not they are visually impaired. (See Hereditary Hearing Loss and Deafness Overview, which provides details about management.)
Depending on the degrees of hearing and vision loss, communication may start with touch cues, followed by object cues and proceeding to auditory/oral and/or sign language. Communication training initiated by age three years is critical to the eventual development of symbolic communication [Thelin & Fussner 2005]. Use of all available means of communication (visual, oral, touch, and incorporating gestures, sign and oral languages) is advisable early on. Most affected children are able to migrate to oral communication as they get older, even if they start with sign and/or total communication.
Surveillance
Table 7.
Recommended Surveillance for Individuals with CHD7 Disorder
Agents/Circumstances to Avoid
Anesthesia. Airway problems associated with anesthesia are common in individuals with CHARGE syndrome. They may be attributed to choanal atresia, cleft lip and palate, and other upper-airway structural anomalies and associated cranial nerve abnormalities. Soft cartilage and resultant floppy trachea add to potential anesthesia risk. Neurogenic incoordination of swallow and closure of the epiglottis may complicate the postoperative course, especially with repeated general anesthetics [Blake et al 2009]. Because of the increased risk of post-anesthesia airway complications, procedures requiring anesthesia should be minimized and combined whenever possible [de Geus et al 2017].
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
CHD7 disorder is an autosomal dominant disorder typically caused by a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
- Most individuals diagnosed with a CHD7 disorder have the disorder as the result of a de novo pathogenic variant [Janssen et al 2012].
- In rare instances, an individual with CHD7 disorder has the disorder as the result of a pathogenic variant inherited from a heterozygous parent. A parent who is heterozygous for a CHD7 pathogenic variant may have one or more features associated with CHD7 disorder [Mitchell et al 1985, Lalani et al 2006, Delahaye et al 2007, Bergman et al 2011b].
- Molecular genetic testing is recommended for the parents of a proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If the CHD7 pathogenic variant identified in the proband is not identified in either parent, the following possibilities should be considered:
- The proband has a de novo pathogenic variant. Note: A pathogenic variant is reported as "de novo" if: (1) the pathogenic variant found in the proband is not detected in parental DNA; and (2) parental identity testing has confirmed biological maternity and paternity. If parental identity testing is not performed, the variant is reported as "assumed de novo" [Richards et al 2015].
- The proband inherited a pathogenic variant from a parent with somatic/germline mosaicism. Somatic mosaicism for a CHD7 pathogenic variant has been documented [Jongmans et al 2008]. (Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism.)
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband has a CHD7 pathogenic variant, the risk to the sibs of inheriting the pathogenic variant is 50%.
- If the CHD7 pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the empiric recurrence risk to sibs of a proband is approximately 1%-2% because of the possibility of parental germline mosaicism [Jongmans et al 2008, Pauli et al 2009].
- The severity of CHD7 disorder in a proband does not predict the severity of the disorder in sibs who inherit a pathogenic variant. (A high degree of clinical variability is observed CHD7 disorder; see Clinical Characteristics.)
Offspring of a proband
- Many individuals with CHD7 disorder are not able to reproduce.
- Each child of an individual with CHD7 disorder has a 50% chance of inheriting the pathogenic variant.
- The severity of CHD7 disorder in the proband does not predict the severity of the disorder in heterozygous offspring (a high degree of clinical variability is observed in CHD7 disorder; see Clinical Characteristics).
Other family members. The risk to other family members depends on the genetic status of the proband's parents; if a parent has the CHD7 pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
For a review of genetic counseling issues in families in which a child has been diagnosed with CHARGE syndrome, see Hefner & Fassi [2017].
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to the parents of affected children and to young adults who are mildly affected.
Prenatal Testing and Preimplantation Genetic Testing
Once the CHD7 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Association CHARGE Enfant SoleilFrance
- CHARGE Family Support GroupLondonUnited KingdomPhone: 020 8265 3604Email: si_howard@hotmail.com
- CHARGE Information PackSense
- CHARGE Syndrome Association of AustralasiaAustraliaPhone: 61 480 121 345Email: admin@chargesyndrome.org.au
- CHARGE Syndrome e.V.GermanyPhone: 0049 – (0)9104 – 826 345Email: info@charge-syndrom.de
- CHARGE Syndrome Foundation18 Half Day Road #305Buffalo Grove IL 60089Phone: 800-442-7604 (toll-free); 516-684-4720Fax: 516-883-9060Email: info@chargesyndrome.org
- Perkins School for the Blind
- Face Equality InternationalUnited Kingdom
- National Consortium on Deaf-Blindness (NCDB)Bibliography and links to state deafblind project resources345 North Monmouth AvenueMonmouth OR 97361Phone: 800-438-9376 (toll-free); 800-854-7013 (TTY)Fax: 503-838-8150Email: info@nationaldb.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
CHD7 Disorder: Genes and Databases
Table B.
OMIM Entries for CHD7 Disorder (View All in OMIM)
Molecular Pathogenesis
CHD7 encodes a chromodomain protein that is involved in the ATP-dependent remodeling of chromatin. CHD7 binds to more than 10,000 sites in the mammalian genome and interacts with dozens of other genes. Features of CHD7-related disorders may be due to loss of ribosomal biogenesis or other mechanisms.
CHD7 functions in a multiprotein complex and uses the energy of ATP to remodel nucleosomes. CHD7 is considered an epigenetic regulator because it modifies the degree to which chromatin is "open" or "closed," making it more or less accessible, respectively, to other proteins that activate or repress gene expression. The broad variability in phenotypes observed in individuals with CHD7 disorder is not fully understood, but is likely related to its unique roles in specific cells and tissues at various times during development.
Mechanism of disease causation. Decreased function or loss of function of CHD7 leads to the clinical manifestations of CHD7 disorder.
Chapter Notes
Author History
John W Belmont, MD, PhD; Baylor College of Medicine (2006-2020)
Kim Blake, MD, MRCP, FRCPC (2020-present)
Sandra LH Davenport, MD, CM; Sensory Genetics/Neurodevelopment, Bloomington, Minnesota (2006-2020)
Meg Hefner, MS (2006-present)
Seema R Lalani, MD; Baylor College of Medicine (2006-2020)
Donna M Martin, MD, PhD (2020-present)
Conny M van Ravenswaaij-Arts, MD, PhD (2020-present)
Revision History
- 29 September 2022 (sw) Revision: epigenetic signature analysis (Establishing the Diagnosis, Option 2)
- 17 September 2020 (bp) Comprehensive update posted live
- 2 February 2012 (me) Comprehensive update posted live
- 22 September 2009 (me) Comprehensive update posted live
- 2 October 2006 (me) Review posted live
- 14 April 2005 (jwb) Original submission
References
Literature Cited
- Andersen SL, Lönn S, Vestergaard P, Törring O. Birth defects after use of antithyroid drugs in early pregnancy: a Swedish nationwide study. Eur J Endocrinol 2017;177:369-78. [PubMed: 28780518]
- Andersen SL, Olsen J, Wu CS, Laurberg P. Birth defects after early pregnancy use of antithyroid drugs: a Danish nationwide study. J Clin Endocrinol Metab. 2013;98:4373-81. [PubMed: 24151287]
- Aref-Eshghi E, Kerkhof J, Pedro VP, Groupe DI. France, Barat-Houari M, Ruiz-Pallares N, Andrau JC, Lacombe D, Van-Gils J, Fergelot P, Dubourg C, Cormier-Daire V, Rondeau S, Lecoquierre F, Saugier-Veber P, Nicolas G, Lesca G, Chatron N, Sanlaville D, Vitobello A, Faivre L, Thauvin-Robinet C, Laumonnier F, Raynaud M, Alders M, Mannens M, Henneman P, Hennekam RC, Velasco G, Francastel C, Ulveling D, Ciolfi A, Pizzi S, Tartaglia M, Heide S, Héron D, Mignot C, Keren B, Whalen S, Afenjar A, Bienvenu T, Campeau PM, Rousseau J, Levy MA, Brick L, Kozenko M, Balci TB, Siu VM, Stuart A, Kadour M, Masters J, Takano K, Kleefstra T, de Leeuw N, Field M, Shaw M, Gecz J, Ainsworth PJ, Lin H, Rodenhiser DI, Friez MJ, Tedder M, Lee JA, DuPont BR, Stevenson RE, Skinner SA, Schwartz CE, Genevieve D, Sadikovic B. Evaluation of DNA methylation episignatures for diagnosis and phenotype correlations in 42 mendelian neurodevelopmental disorders. Am J Hum Genet. 2020;106:356-70. [PMC free article: PMC7058829] [PubMed: 32109418]
- Balasubramanian R, Crowley WF Jr. Reproductive endocrine phenotypes relating to CHD7 mutations in humans. Am J Med Genet Part C Semin Med Genet. 2017;175:507-15. [PMC free article: PMC5790312] [PubMed: 29152903]
- Bergman JE, Blake KD, Bakker MK, du Marchie Sarvaas GJ, Free RH, van Ravenswaaij-Arts CM. Death in CHARGE syndrome after the neonatal period. Clin Genet. 2010;77:232-40. [PubMed: 20447140]
- Bergman JE, Bocca G, Hoefsloot LH, Meiners LC, van Ravenswaaij-Arts CM. Anosmia predicts hypogonadotropichypogonadism in CHARGE syndrome. J Pediatr. 2011a:158:474-9. [PubMed: 20884005]
- Bergman JE, de Wijs I, Jongmans MC, Admiraal RJ, Hoefsloot LH, van Ravenswaaij-Arts CM. Exon copy number alterations of the CHD7 gene are not a major cause of CHARGE and CHARGE-like syndrome. Eur J Med Genet. 2008;51:417-25. [PubMed: 18472328]
- Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2011b;48:334‐42. [PubMed: 21378379]
- Bergman JE, Janssen N, van der Sloot AM, de Walle HEK, Schoots J, Rendtorff ND, Tranebjaerg L, Hoefsloot LH,van Ravenswaaij-Arts CM, Hofstra RM. A novel classification system to predict the pathogenic effects of CHD7 missense variants in CHARGE syndrome. Hum Mutat. 2012;33:1251-60. [PubMed: 22539353]
- Blake K, MacCuspie J, Hartshorne TS, Roy M, Davenport SL, Corsten G. Postoperative airway events of individuals with CHARGE syndrome. Int J Pediatr Otorhinolaryngol. 2009;73:219-26. [PubMed: 19058860]
- Blake KD, Davenport SL, Hall BD, Hefner MA, Pagon RA, Williams MS, Lin AE, Graham JM Jr. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila). 1998;37:159-73. [PubMed: 9545604]
- Blake KD, Hudson AS. Gastrointestinal and feeding difficulties in CHARGE syndrome: a review from head to toe. Am J Med Genet C Semin Med Genet. 2017;175:496-506. [PubMed: 29082627]
- Blake KD, Salem-Hartshorne N, Abi Daoud M, Gradstein J. Adolescent and adult issues in CHARGE syndrome. Clin Pediatr (Phila) 2005;44:151-9. [PubMed: 15735833]
- Brown D. CHARGE syndrome "behaviors": challenges or adaptations. Am J Med Genet A. 2005;133A:268-72. [PubMed: 15666302]
- Chetty M, Roberts TS, Elmubarak M, Bezuidenhout H, Smit L, Urban M. CHARGE syndrome: genetic aspects and dental challenges, a review and case presentation. Head Face Med. 2020;16:10. [PMC free article: PMC7206710] [PubMed: 32384900]
- Corsten-Janssen N, Kerstjens-Frederikse WS, du Marchie Sarvaas GJ, Baardman ME, Bakker MK, Bergman JE, Hove HD, Heimdal KR, Rustad CF, Hennekam RC, Hofstra RM, Hoefsloot LH, van Ravenswaaij-Arts CM. The cardiac phenotype in patients with a CHD7 mutation. Circ Cardiovasc Genet. 2013;6:248-54. [PubMed: 23677905]
- Corsten-Janssen N, Scambler PJ. Clinical and molecular effects of CHD7 in the heart. Am J Med Genet C Semin Med Genet. 2017;175:487-95. [PubMed: 29088513]
- Corsten-Janssen N, van Ravenswaaij-Arts CM, Kaputsa L. Congenital arch vessel anomalies in CHARGE syndrome: a frequent feature with risk for co-morbidity. Int J Cardiol Heart Vasc. 2016;12:21-5. [PMC free article: PMC5454153] [PubMed: 28616537]
- de Geus CM, Bergman JE, van Ravenswaaij-Arts CM, Meiners LC. Imaging of clival hypoplasia in CHARGE syndrome and hypothesis for development: a case-control study. AJNR Am J Neuroradiol. 2018;39:1938-42. [PMC free article: PMC7410728] [PubMed: 30237300]
- de Geus CM, Free RH, Verbist BM, Sival DA, Blake KD, Meiners LC, van Ravenswaaij-Arts CM. Guidelines in CHARGE syndrome and the missing link: cranial imaging. Am J Med Genet Part C Semin Med Genet. 2017;175:450-64. [PMC free article: PMC5765497] [PubMed: 29168326]
- Delahaye A, Sznajer Y, Lyonnet S, Elmaleh-Berges M, Delpierre I, Audollent S, Wiener-Vacher S, Mansbach A-L, Amiel J, Baumann C, Bremond-Cignac D, Attié-Bitach T, Verloes A, Sanlaville D. Familial CHARGE syndrome because of CHD7 mutation: clinical intra- and interfamilial variability. Clin Genet. 2007;72:112-21. [PubMed: 17661815]
- Donovan AP, Yu T, Ellegood J, Riegman KL, de Geus C, van Ravenswaaij-Arts C, Fernandes C, Lerch JP, Basson MA. Cerebellar vermis and midbrain hypoplasia upon conditional deletion of Chd7 from the embryonic mid-hindbrain region. Front Neuroanat. 2017;11:86. [PMC free article: PMC5632662] [PubMed: 29046629]
- Doyle C, Blake KD. Scoliosis in CHARGE: a prospective survey and two case reports. Am J Med Genet A. 2005;133A:340-3. [PubMed: 15688383]
- Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016;170A:344-54. [PMC free article: PMC5102387] [PubMed: 26590800]
- Hartshorne N, Hudson A, MacCuspie J, Kennert B, NacaratoT, Hartshorne T, Blake K. Quality of life in adolescents and adults with CHARGE syndrome. Am J Med Genet Part A. 2016;170:2012-21. [PubMed: 27273681]
- Hartshorne TS, Hefner MA, Davenport SL. Behavior in CHARGE syndrome: introduction to the special topic. Am J Med Genet A. 2005;133A:228-31. [PubMed: 15637707]
- Hefner MA, Fassi E. Genetic counseling in CHARGE syndrome: diagnostic evaluation through follow up. Am J Med Genet C Semin Med Genet. 2017;175:407-16. [PubMed: 29088501]
- Hudson A, Colp M, Blake K. Pocketing of food in cheeks during eating in an adolescent with CHARGE syndrome. J Paediatr Child Health. 2015;51:1143-4. [PubMed: 26541624]
- Issekutz KA, Graham JM, Prasad C, Smith IM, Blake KD. An epidemiological analysis of CHARGE syndrome: preliminary results from a Canadian study. Am J Med Genet A. 2005;133A:309-17. [PubMed: 15637722]
- Janssen N, Bergman JE, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, Hofstra RM, van Ravenswaaij-Arts CM, Hoefsloot LH. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33:1149-60. [PubMed: 22461308]
- Jongmans MC, Hoefsloot LH, van der Donk KP, Admiraal RJ, Magee A, van de Laar I, Hendriks Y, Verheij JB, Walpole I, Brunner HG, van Ravenswaaij CM. Familial CHARGE syndrome and the CHD7 gene: a recurrent missense mutation, intrafamilial recurrence and variability. Am J Med Genet. 2008;146A:43-50. [PubMed: 18074359]
- Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL, van der Donk K, Seminara S, Bergman JE, Brunner HG, Crowley WF Jr, Hoefsloot LH. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin Genet. 2009;75:65-71. [PMC free article: PMC2854009] [PubMed: 19021638]
- Komoike Y, Matsuoka M, Kosaki K. Potential teratogenicity of methimazole: exposure of zebrafish embryos to methimazole causes similar developmental anomalies to human methimazole embryopathy. Birth Defects Res B Dev Reprod Toxicol. 2013;98:222-9. [PubMed: 23630110]
- Lai H-S, Feng C-Y. Cecal volvulus in a child with CHARGE syndrome. Am Surg. 2006;72:356-8. [PubMed: 16676864]
- Lalani SR, Safiullah AM, Fernbach SD, Harutyunyan KG, Thaller C, Peterson LE, McPherson JD, Gibbs RA, White LD, Hefner M, Davenport SL, Graham JM, Bacino CA, Glass NL, Towbin JA, Craigen WJ, Neish SR, Lin AE, Belmont JW. Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am J Hum Genet. 2006;78:303-14. [PMC free article: PMC1380237] [PubMed: 16400610]
- Lammer EJ, Chen DT, Hoar RM, Agnish ND, Benke PJ, Braun JT, Curry CJ, Fernhoff PM, Grix Jr AW, Lott IT. Retionic acid embryopathy. N Engl J Med. 1985;313:837-41. [PubMed: 3162101]
- Legendre M, Abadie V, Attié-Bitach T, Philip N, Busa T, Bonneau D, Colin E, Dollfus H, Lacombe D, Toutain A, Blesson S, Julia S, Martin-Coignard D, Geneviève D, Leheup B, Odent S, Jouk PS, Mercier S, Faivre L, Vincent-Delorme C, Francannet C, Naudion S, Mathieu-Dramard M, Delrue MA, Goldenberg A, Héron D, Parent P, Touraine R, Layet V, Sanlaville D, Quélin C, Moutton S, Fradin M, Jacquette A, Sigaudy S, Pinson L, Sarda P, Guerrot AM, Rossi M, Masurel-Paulet A, El Chehadeh S, Piguel X, Rodriguez-Ballesteros M, Ragot S, Lyonnet S, Bilan F, Gilbert-Dussardier B. Phenotype and genotype analysis of a French cohort of 119 patients with CHARGE syndrome. Am J Med Genet C Semin Med Genet. 2017;175:417-30. [PubMed: 29178447]
- Levy MA, McConkey H, Kerkhof J, Barat-Houari M, Bargiacchi S, Biamino E, Bralo MP, Cappuccio G, Ciolfi A, Clarke A, DuPont BR, Elting MW, Faivre L, Fee T, Fletcher RS, Cherik F, Foroutan A, Friez MJ, Gervasini C, Haghshenas S, Hilton BA, Jenkins Z, Kaur S, Lewis S, Louie RJ, Maitz S, Milani D, Morgan AT, Oegema R, Østergaard E, Pallares NR, Piccione M, Pizzi S, Plomp AS, Poulton C, Reilly J, Relator R, Rius R, Robertson S, Rooney K, Rousseau J, Santen GWE, Santos-Simarro F, Schijns J, Squeo GM, St John M, Thauvin-Robinet C, Traficante G, van der Sluijs PJ, Vergano SA, Vos N, Walden KK, Azmanov D, Balci T, Banka S, Gecz J, Henneman P, Lee JA, Mannens MMAM, Roscioli T, Siu V, Amor DJ, Baynam G, Bend EG, Boycott K, Brunetti-Pierri N, Campeau PM, Christodoulou J, Dyment D, Esber N, Fahrner JA, Fleming MD, Genevieve D, Kerrnohan KD, McNeill A, Menke LA, Merla G, Prontera P, Rockman-Greenberg C, Schwartz C, Skinner SA, Stevenson RE, Vitobello A, Tartaglia M, Alders M, Tedder ML, Sadikovic B. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of mendelian disorders. HGG Adv. 2021;3:100075. [PMC free article: PMC8756545] [PubMed: 35047860]
- Mitchell JA, Giangiacomo J, Hefner MA, Thelin JW, Pickens JM. Dominant CHARGE association. Ophthalmic Paediatr Genet. 1985;6:271-6. [PubMed: 3934623]
- Morgan A, Hudson A, Arra-Robar A, Blake K. Late dumping syndrome in a 17-year-old female with Charge syndrome. J Paediatr Child Health. 2017;53:1244-5. [PubMed: 29205658]
- O'Grady GL, Ma A, Sival D, Wong MT, Peduto T, Menezes MP, Young H, Waddell L, Ghaoui R, Needham M, Lek M, North KN, MacArthur DG, van Ravenswaaij-Arts CM, Clarke NF. Prominent scapulae mimicking an inherited myopathy expands the phenotype of CHD7-related disease. Eur J Hum Genet. 2016;24:1216-9. [PMC free article: PMC4970689] [PubMed: 26813943]
- Pagon RA, Graham JM Jr, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99:223-7. [PubMed: 6166737]
- Pauli S, Pieper L, Häberle J, Grzmil P, Burfeind P, Steckel M, Lenz U, Michelmann HW. Proven germline mosaicism in a father of two children with CHARGE syndrome. Clin Genet. 2009;75:473-9. [PubMed: 19475719]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Salem-Hartshorne N, Jacob S. Adaptive behavior in children with CHARGE syndrome. Am J Med Genet A. 2005;133A:262-7. [PubMed: 15641025]
- Thelin JW, Fussner JC. Factors related to the development of communication in CHARGE syndrome. Am J Med Genet A. 2005;133A:282-90. [PubMed: 15669098]
- Trider C-L, Arra-Robar A, van Ravenswaaij-Arts C, Blake K. Developing a CHARGE syndrome checklist: health supervision across the lifespan (from head to toe). Am J Med Genet A. 2017;173:684-91. [PubMed: 28160409]
- Van de Laar I, Dooijes D, Hoefsloot L, Simon M, Hoogeboom J, Devriendt K. Limb anomalies in patients with CHARGE syndrome: an expansion of the phenotype. Am J Med Genet A. 2007;143A:2712-5. [PubMed: 17937444]
- van Ravenswaaij-Arts C, Martin DM. New insights and advances in CHARGE syndrome: diagnosis, etiologies, treatments, and research discoveries. Am J Med Genet Part C Semin Med Genet. 2017;175:397-406. [PMC free article: PMC6591023] [PubMed: 29171162]
- Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005;133A:306-8. [PubMed: 15666308]
- Vesseur A, Free R, Langereis M, Snels C, Snik A, van Ravenswaaij-Arts C, Mylanus E. Suggestions for a guideline for cochlear implantation in CHARGE syndrome. Otol Neurotol. 2016a;37:1275-83. [PubMed: 27636388]
- Vesseur A, Langereis M, Free R, Snik A, van Ravenswaaij-Arts C, Mylanus E. Influence of hearing loss and cognitive abilities on language development in CHARGE syndrome. Am J Med Genet A. 2016b;170:2022-30. [PubMed: 27145116]
- Vesseur AC, Verbist BM, Westerlaan HE, Kloostra FJ, Admiraal RJ, van Ravenswaaij-Arts CM, Free RH, Mylanus EA. CT findings of the temporal bone in CHARGE syndrome: aspects of importance in cochlear implant surgery. Eur Arch Otorhinolaryngol. 2016c;273:4225‐40. [PMC free article: PMC5104824] [PubMed: 27324890]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BC, Schoenmakers EF, Brunner HG, Veltman JA, Geurts van Kessel A. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955-7. [PubMed: 15300250]
- Vuorela P, Ala-Mello S, Saloranta C, Penttinen M, Pöyhönen M, Huoponen K, Borozdin W, Bausch B, Botzenhart EM, Wilhelm C. Kääriäinen H, Kohlhase J. Molecular analysis of the CHD7 gene in CHARGE syndrome: identification of 22 novel mutations and evidence for a low contribution of large CHD7 deletions. Genet Med. 2007;9:690-4. [PubMed: 18073582]
- Wong MT, Lambeck AJ, van der Burg M, la Bastide-van Gemert S, Hogendorf LA, van Ravenswaaij-Arts CM, Scholvinck EH. Immune dysfunction in children with CHARGE syndrome: a cross-sectional study. PLoS One. 2015a;10:e0142350. [PMC free article: PMC4636349] [PubMed: 26544072]
- Wong MT, Schölvinck EH, Lambeck AJ, van Ravenswaaij-Arts CM. CHARGE syndrome: a review of the immunological phenotype. Eur J Hum Genet. 2015b;23:1451-9. [PMC free article: PMC4613462] [PubMed: 25689927]
- Xu C, Cassatella D, van der Sloot AM, Quinton R, Hauschild M, de Geyter C, Flück C, Feller K, Bartholdi D, Nemeth A, Halperin I, Djurdjevic SP, Maeder P, Papadakis G, Dwyer AA, Marino L, Favre L, Pignatelli D, Niederländer NJ, Acierno J, Pitteloud N. Evaluating CHARGE syndrome in congenital hypogonadotropic hypogonadism patients harboring CHD7 variants. Genet Med. 2018;20:872-81. [PubMed: 29144511]
Suggested Reading
- Hartshorne T, Hefner M, Davenport SLH, Thelin J. CHARGE Syndrome, 1 ed. Plural Publishing. 2011.
- Issue devoted entirely to CHARGE syndrome. Am J Med Genet C Semin Med Genet 2017;175(4).
Publication Details
Author Information and Affiliations
Groningen, the Netherlands
St Louis, Missouri
Halifax, Nova Scotia, Canada
Ann Arbor, Michigan
Publication History
Initial Posting: October 2, 2006; Last Revision: September 29, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
van Ravenswaaij-Arts CM, Hefner M, Blake K, et al. CHD7 Disorder. 2006 Oct 2 [Updated 2022 Sep 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.