CHST3-Related Skeletal Dysplasia
Synonyms: Chondrodysplasia with Congenital Joint Dislocations, CHST3 Type; CHST3 Deficiency; CHST3-Related Dysplasia; Recessive Larsen Syndrome
Andrea Superti-Furga, MD and Sheila Unger, MD.
Author Information and AffiliationsInitial Posting: September 1, 2011; Last Update: January 31, 2019.
Estimated reading time: 13 minutes
Summary
Clinical characteristics.
CHST3-related skeletal dysplasia is characterized by short stature of prenatal onset, joint dislocations (knees, hips, radial heads), clubfeet, and limitation of range of motion that can involve all large joints. Kyphosis and occasionally scoliosis with slight shortening of the trunk develop in childhood. Minor heart valve dysplasia has been described in several persons. Intellect and vision are normal.
Diagnosis/testing.
The diagnosis is based on the radiographic features of progressive spondyloepiphyseal dysplasia with joint anomalies, spinal abnormalities, normal thumbs (not spatulate), and normal bone age. CHST3 is the only gene in which pathogenic variants are known to cause CHST3-related skeletal dysplasia.
Management.
Treatment of manifestations: Surgical correction of the abnormal joints is the only treatment modality; however, surgical correction is often only partially successful and multiple procedures are needed. Physical therapy has not been effective.
Surveillance: If normal at the time of diagnosis, echocardiogram should probably be repeated every five years.
Agents/circumstances to avoid: Activities with a high impact on joints (e.g., jogging) and obesity.
Genetic counseling.
CHST3-related skeletal dysplasia is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk family members and prenatal testing for a prengancy at increased risk are possible if the pathogenic variants in the family have been identified.
Diagnosis
The diagnosis of CHST3-related skeletal dysplasia is based on the combination of characteristic clinical and radiographic signs and confirmation by molecular genetic testing.
Suggestive Findings
CHST3-related skeletal dysplasia should be suspected in individuals with the following clinical and radiographic features.
Clinical features
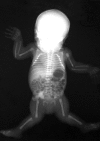
Joint dislocations at birth (knees, hips, radial heads) with short stature ()
Clubfeet
Limitation of range of motion that can involve all large joints
Development of kyphosis and occasionally scoliosis with slight shortening of the trunk in childhood
A newborn with molecularly confirmed CHST3-related skeletal dysplasia. Note the bilateral dislocation of the knees and radial heads.
Radiographic features
Progressive spondyloepiphyseal dysplasia with joint anomalies
Generalized mild epiphyseal dysplasia (small epiphyses)
Delayed ossification of the capital femoral epiphyses and femoral necks
Coxa valga (increase in the angle formed between the head and neck of the femur and the shaft of the femur)
Spinal abnormalities
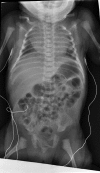
Conspicuous increase in interpediculate distance from T12 to L1 or L2 ()
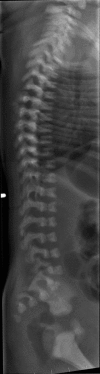
Notching of the vertebral bodies, similar in appearance to coronal clefts ()
Normal thumbs (not spatulate)
Normal or (more rarely) slightly advanced bone age (especially carpal)
Note the conspicuous increase in interpediculate distance from T12 to L1. Also appreciable is the bilateral hip subluxation.
Mild platyspondyly is observed. Note also the coronal clefts throughout the lumbar region.
Establishing the Diagnosis
The diagnosis of CHST3-related skeletal dysplasia is established in a proband by identification of biallelic pathogenic variants in CHST3 by molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include single-gene testing, use of a multigene panel, and more comprehensive genomic testing.
Single-gene testing. Sequence analysis of
CHST3 is performed first. If no pathogenic variants or only a single
pathogenic variant have been identified and the clinical-radiographic index of suspicion is high for a
CHST3-related skeletal dysplasia, consider doing gene-targeted
deletion/duplication analysis. However, no individuals with a deletion/duplication have been reported.
A multigene panel that includes
CHST3 and other genes of interest (see
Differential Diagnosis) may also be considered. Laboratories may offer a general "skeletal dysplasia" panel or a more targeted "skeletal dysplasia with multiple dislocations" panel. Note: (1) The genes included in the panel and the diagnostic
sensitivity of the testing used for each
gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of
uncertain significance and pathogenic variants in genes that do not explain the underlying
phenotype. (3) Methods used in a panel may include
sequence analysis,
deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
More comprehensive genomic testing (when available) including
exome sequencing and
genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different
gene or genes that results in a similar clinical presentation).
For an introduction to comprehensive
genomic testing click
here. More detailed information for clinicians ordering genomic testing can be found
here.
Table 1.
Molecular Genetic Testing Used in CHST3-Related Skeletal Dysplasia
View in own window
| Gene 1 | Method | Proportion of Probands with Pathogenic Variants 2 Detectable by Method |
|---|
|
CHST3
| Sequence analysis 3 | >90% 4, 5 |
| Gene-targeted deletion/duplication analysis 6 | 1 reported 7 |
- 1.
- 2.
- 3.
- 4.
- 5.
The high detection rate applies only to those individuals with clear clinical and radiographic changes consistent with CHST3 deficiency and not to an unselected population of children with joint dislocations.
- 6.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 7.
Biochemical testing. Cultured fibroblasts can be used to determine proteoglycan sulfation (i.e., sulfotransferase activity). The fibroblasts are incubated with radioactive sulfate [35S] and chondroitin. In all patients with CHST3 deficiency studied thus far, the incorporation of sulfate at the C6 position was dramatically decreased while incorporation at the C4 position was within normal levels [Hermanns et al 2008, van Roij et al 2008]. This test requires a skin biopsy and thus is more invasive and less widely available than molecular genetic testing. Biochemical testing can be useful in those cases in which pathogenic variants are not identified or variants of uncertain significance have been detected. The diminished sulfation at carbon 6 of proteoglycans offers unambiguous evidence of CHST3 deficiency.
Clinical Characteristics
Clinical Description
Most children with CHST3-related skeletal dysplasia are identified at birth as having a generalized skeletal disorder. The features of this disorder are generally limited to the skeleton and joints and are progressive in nature.
Occasionally, short stature and knee dislocations are seen on prenatal ultrasound examination [Unger et al 2010]. The prenatal presentation may be that of arthrogryposis [Muys et al 2017].
At birth, affected infants are noted to have short stature (birth length: 39 to 44 cm) and joint dislocations: the large majority have bilateral knee luxation or subluxation. The radial heads and hips are the next most commonly affected joints. Clubfeet are also frequently seen. Despite the congenital joint dislocations, the overall phenotype is one of restricted movement and many children undergo multiple corrective procedures with only limited success [Rajab et al 2004, Unger et al 2010, Searle et al 2014].
In the large family reported from Oman, the adult heights ranged from 110 cm to 130 cm [Rajab et al 2004], while in a large Pakistani family the mean adult height was 84 cm [Waryah et al 2016]. A review article included information on three adults with heights of 117 cm, 121 cm, and 134.5 cm [Unger et al 2010]. Adult height appears to be severely affected in all individuals with CHST3-related skeletal dysplasia but with some intrafamilial variability and large interfamilial variability.
Many adults develop arthritic-type changes. They also develop spinal kyphosis, frequently in the cervical spine, and (rarely) scoliosis.
Other findings include:
Nomenclature
In 1950, Dr LJ Larsen described autosomal dominant Larsen syndrome, now known to be caused by pathogenic variants in FLNB, the gene encoding filamin B [Bicknell et al 2007]. Larsen syndrome is characterized by multiple joint dislocations, dysmorphic facial features, spatulate thumbs, and accelerated carpal ossification (see FLNB-Related Disorders).
Following the delineation of autosomal dominant Larsen syndrome, several reports of "autosomal recessive Larsen syndrome" and other similar disorders were published. By careful reevaluation of patients and through recruitment of cases with autosomal recessive Larsen syndrome, several investigators showed that, in fact, all the various reports of autosomal recessive Larsen syndrome, humerospinal dysostosis, and spondyloepiphyseal dysplasia (SED), Omani type could be attributed to CHST3 deficiency and that the different names had arisen from the part of the phenotype the various authors had emphasized [Hermanns et al 2008, Unger et al 2010]; that is, they were describing the same condition from different viewpoints:
Humerospinal dysostosis was described by
Kozlowski et al [1974] in two brothers with joint dislocations and radiographic abnormalities (bifid humeri and coronal clefts). Because the brothers were reported to be half-sibs,
autosomal dominant inheritance was suspected and no link was made to
autosomal recessive Larsen syndrome.
Mégarbané and Ghanem [2004] described "a newly recognized chondrodysplasia with joint dislocations." Although they made the link to humero-spinal dysostosis, they rejected that diagnosis because the evidence strongly suggested
autosomal recessive inheritance.
Rajab et al [2004] described a large family originating from Oman with what they termed "a new recessive type of SED with progressive spinal involvement." The same group went on to demonstrate that the disorder was caused by CHST3 deficiency and renamed the disorder SED, Omani type [
Thiele et al 2004].
The name "CHST3-related skeletal dysplasia" has been proposed as an unbiased and inclusive designation for this disorder. However, an argument could also be made for retaining the name "autosomal recessive Larsen syndrome," as the joint dislocations are the presenting feature and "Larsen syndrome" is usually the first diagnosis considered; thus, the continued use of this designation is open to debate. The term "recessive Larsen syndrome" is more appropriate for CHST3-related skeletal dysplasia than for the B4GALT7-associated linkeropathy, as bilateral knee dislocation at birth is much more common in CHST3 deficiency than in B4GALT7 deficiency.
Prevalence
No firm data regarding the prevalence of CHST3-related skeletal dysplasia are available. More than 40 cases (including familial recurrences) have been reported.
Differential Diagnosis
A summary of key differentiating clinical and radiographic features for chondrodysplasias with multiple dislocations is available in Ranza et al [2017].
Table 2.
Disorders to Consider in the Differential Diagnosis of CHST3-Related Skeletal Dysplasia
View in own window
| Disorder | Gene(s) | MOI | Clinical Features of This Disorder |
|---|
| Overlapping w/CHST3 Skeletal Dysplasia | Distinguishing from CHST3 Skeletal Dysplasia |
|---|
| Larsen syndrome (See FLNB-Related Disorders.) |
FLNB
| AD | Multiple dislocations | Normal birth length in Larsen syndrome; ↓ in CHST3 skeletal dysplasia Distinctive facial features in Larsen syndrome, w/↑ incidence of cleft palate; normal facies in CHST3 skeletal dysplasia Advanced bone age in Larsen syndrome; normal or delayed bone age in CHST3 skeletal dysplasia
|
|
Diastrophic dysplasia
|
SLC26A2
| AR |
|
|
Desbuquois dysplasia
(OMIM 251450, 615777) |
CANT1
XYLT1
| AR |
|
|
B4GALT7 deficiency 1
(OMIM 604327) |
B4GALT7
| AR |
|
|
B3GALT6 linkeropathy
(OMIM 61521) |
B3GALT6
| AR |
|
|
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease in an individual diagnosed with CHST3-related skeletal dysplasia, the following evaluations are recommended if they have not already been completed:
Treatment of Manifestations
The focus of treatment has thus far been surgical correction of the abnormal joints. Presumably because of the basic defect present in CHST3-related skeletal dysplasia, surgical correction is often only partially successful and most patients have had multiple procedures by adulthood [Unger et al 2010].
Of note, physical therapy has not been demonstrated to be effective in this disorder.
Surveillance
Thus far, heart valve dysplasia has not required correction; thus, no firm guidelines for appropriate surveillance have been developed. Echocardiogram is suggested at time of diagnosis and, if normal, should probably be repeated at intervals of five years.
Agents/Circumstances to Avoid
Activities with a high impact on joints (e.g., jogging) should be avoided.
Obesity, which places an excessive load on the large weight-bearing joints, should be avoided.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
CHST3-related skeletal dysplasia is inherited in an autosomal recessive manner.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the CHST3 pathogenic variants in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the CHST3 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for CHST3-related skeletal dysplasia are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
CHST3-Related Skeletal Dysplasia: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Gene structure.
CHST3 is a relatively small gene, comprising three exons. See Table A, Gene for a detailed summary of gene and protein information.
Pathogenic variants. Several recurrent pathogenic variants have been identified in families of similar ethnic background and, thus, may represent founder variants [Unger et al 2010]. No hot spots have been identified, but the majority of known pathogenic variants are clustered in the sulfotransferase domain.
Normal gene product. Carbohydrate sulfotransferase 3 is the enzyme responsible for the transfer of sulfate from PAPS to position 6 of N-acetyl galactosamine. Proper sulfation of the chondroitin sulfate proteoglycans is essential for normal cartilage structure.
Abnormal gene product. Sulfation studies as well as the nature of the known pathogenic variants and the mode of inheritance suggest that the pathogenesis of the disorder results from decreased/absent catalytic activity of the enzyme.
References
Literature Cited
Bicknell LS, Farrington-Rock C, Shafeghati Y, Rump P, Alanay Y, Alembik Y, Al-Madani N, Firth H, Karimi-Nejad MH, Kim CA, Leask K, Maisenbacher M, Moran E, Pappas JG, Prontera P, de Ravel T, Fryns JP, Sweeney E, Fryer A, Unger S, Wilson LC, Lachman RS, Rimoin DL, Cohn DH, Krakow D, Robertson SP. A molecular and clinical study of Larsen syndrome caused by mutations in FLNB.
J Med Genet. 2007;44:89–98. [
PMC free article: PMC2598053] [
PubMed: 16801345]
Cartault F, Munier P, Jacquemont M-L, Vellayoudom J, Doray B, Payet C, Randrianaivo H, Laville J-M, Munnich A, Cormier-Daire V. Expanding the clinical spectrum of
B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome.
Eur J Hum Genet. 2015;23:49–53. [
PMC free article: PMC4266744] [
PubMed: 24755949]
Hall BD. Humero-spinal dysostosis: report of the fourth case with emphasis on generalized skeletal involvement, abnormal craniofacial features, and mitral valve thickening.
J Pediatr Orthop B. 1997;6:11–4. [
PubMed: 9039660]
Hermanns P, Unger S, Rossi A, Perez-Aytes A, Cortina H, Bonafé L, Boccone L, Setzu V, Dutoit M, Sangiorgi L, Pecora F, Reicherter K, Nishimura G, Spranger J, Zabel B, Superti-Furga A. Congenital joint dislocations caused by carbohydrate sulfotransferase 3 deficiency in recessive Larsen syndrome and humero-spinal dysostosis.
Am J Hum Genet. 2008;82:1368–74. [
PMC free article: PMC2427316] [
PubMed: 18513679]
Kozlowski KS, Celermajer JM, Tink AR. Humero-spinal dysostosis with congenital heart disease.
Am J Dis Child. 1974;127:407–10. [
PubMed: 4814886]
Mégarbané A, Ghanem I. A newly recognized chondrodysplasia with multiple dislocations.
Am J Med Genet A. 2004;130A:107–9. [
PubMed: 15368507]
Muys J, Blaumeiser B, Jacquemyn Y, Janssens K. Prenatal homozygosity mapping detects a novel mutation in CHST3 in a fetus with skeletal dysplasia and joint dislocations.
Clin Case Rep. 2017;5:440–5. [
PMC free article: PMC5378824] [
PubMed: 28396765]
Rajab A, Kunze J, Mundlos S. Spondyloepiphyseal dysplasia Omani type: a new recessive type of SED with progressive spinal involvement.
Am J Med Genet A. 2004;126A:413–9. [
PubMed: 15098240]
Ranza E, Huber C, Levin N, Baujat G, Boyle-Feysot C, Nitschke P, Masson C, Alanay Y, Al-Gazali L, Bitoun P, Boute O, Campeau P, Coubes C, McEntagart M, Elcioglu N, Faivre L, Gezdirici A, Johnson D, Mihci E, Nur BG, Perrin L, Quelin C, Terhal P, Tuysuz B, Cormier-Daire V. Chondrodysplasia with multiple dislocations: comprehensive study of a series of 30 cases.
Clin Genet. 2017;91:868–80. [
PubMed: 28229453]
Searle C, Jewell R, Kraft J, Stoebe P, Chumas P, Titheradge H, Kraus A, Gencik M, Hobson E. Craniosynostosis: a previously unreported association with CHST3-related skeletal dysplasia (autosomal recessive Larsen syndrome).
Clin Dysmorphol. 2014;23:12–5. [
PubMed: 24300290]
Srivastava P, Pandey H, Agarwal D, Mandal K, Phadke SR. Spondyloepiphyseal dysplasia Omani type: CHST3 mutation spectrum and phenotypes in three Indian families.
Am J Med Genet A. 2017;173:163–8. [
PubMed: 27753269]
Thiele H, Sakano M, Kitagawa H, Sugahara K, Rajab A, Höhne W, Ritter H, Leschik G, Nürnberg P, Mundlos S. Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement.
Proc Natl Acad Sci U S A. 2004;101:10155–60. [
PMC free article: PMC454181] [
PubMed: 15215498]
Tuysuz B, Mizumoto S, Sugahara K, Celebi A, Mundlos S, Turkmen S. Omani-type spondyloepiphyseal dysplasia with cardiac involvement caused by a missense mutation in CHST3.
Clin Genet. 2009;75:375–83. [
PubMed: 19320654]
Unger S, Lausch E, Rossi A, Mégarbané A, Sillence D, Alcausin M, Aytes A, Mendoza-Londono R, Nampoothiri S, Afroze B, Hall B, Lo IF, Lam ST, Hoefele J, Rost I, Wakeling E, Mangold E, Godbole K, Vatanavicharn N, Franco LM, Chandler K, Hollander S, Velten T, Reicherter K, Spranger J, Robertson S, Bonafé L, Zabel B, Superti-Furga A. Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: congenital dislocations and vertebral changes as principal diagnostic features.
Am J Med Genet A. 2010;152A:2543–9. [
PubMed: 20830804]
van Roij MH, Mizumoto S, Yamada S, Morgan T, Tan-Sindhunata MB, Meijers-Heijboer H, Verbeke JI, Markie D, Sugahara K, Robertson SP. Spondyloepiphyseal dysplasia, Omani type: further definition of the phenotype.
Am J Med Genet A. 2008;146A:2376–84. [
PubMed: 18698629]
Waryah AM, Shahzad M, Shaikh H, Sheikh SA, Channa NA, Hufnagel RB, Makhdoom A, Riazuddin S, Ahmed ZM. A novel CHST3 allele associated with spondyloepiphyseal dysplasia and hearing loss in Pakistani kindred.
Clin Genet. 2016;90:90–5. [
PMC free article: PMC4870159] [
PubMed: 26572954]
Chapter Notes
Revision History
31 January 2019 (sw) Comprehensive update posted live
1 September 2011 (me) Review posted live
28 March 2011 (asf) Original submission