Summary
Clinical characteristics.
Cleidocranial dysplasia (CCD) spectrum disorder is a skeletal dysplasia that represents a clinical continuum ranging from classic CCD (triad of delayed closure of the cranial sutures, hypoplastic or aplastic clavicles, and dental abnormalities), to mild CCD, to isolated dental anomalies without other skeletal features. Individuals with classic CCD spectrum disorder typically have abnormally large, wide-open fontanelles at birth that may remain open throughout life. Clavicular hypoplasia can result in narrow, sloping shoulders that can be opposed at the midline. Moderate short stature may be observed, with most affected individuals being shorter than their unaffected sibs. Dental anomalies may include delayed eruption of secondary dentition, failure to shed the primary teeth, and supernumerary teeth. Individuals with CCD spectrum disorder are at increased risk of developing recurrent sinus infections, recurrent ear infections leading to conductive hearing loss, and upper airway obstruction. Intelligence is typically normal.
Diagnosis/testing.
The diagnosis of CCD spectrum disorder is established in an individual with typical clinical and radiographic findings and/or a heterozygous pathogenic variant in RUNX2 identified by molecular genetic testing.
Management.
Treatment of manifestations: If the cranial vault defect is significant, the head needs protection from blunt trauma; helmets may be used for high-risk activities. Surgical cosmesis for depressed forehead or lengthening of hypoplastic clavicles can be considered. Careful planning of anesthetic management due to craniofacial and dental abnormalities. Consultation with an otolaryngologist to assist in securing the airway. Consideration of alternative anesthetic approaches, including neuraxial block, taking into account possible spine abnormalities. If bone density is below normal, treatment with calcium and vitamin D supplementation. Dental procedures to address retention of primary dentition, presence of supernumerary teeth, and non-eruption of secondary dentition. Such procedures may include prosthetic replacements, removal of the supernumerary teeth followed by surgical repositioning of the secondary teeth, and a combination of surgical and orthodontic measures for actively erupting and aligning the impacted secondary teeth. Speech therapy as needed. Aggressive treatment of sinus and middle ear infections; consideration of tympanostomy tubes for recurrent middle ear infections; regular immunizations including influenza. Sleep study in those with manifestations of obstructive sleep apnea; surgical intervention may be required for upper airway obstruction.
Surveillance: Monitor children for orthopedic complications, dental abnormalities, sinus and ear infections, upper airway obstruction, hearing loss, and speech issues. DXA scan to assess bone mineral density beginning in early adolescence and every five to ten years thereafter.
Agents/circumstances to avoid: Helmets and protective devices should be worn when participating in high-risk activities.
Pregnancy management: Monitor affected women during pregnancy for cephalopelvic disproportion.
Genetic counseling.
CCD spectrum disorder is inherited in an autosomal dominant manner. The proportion of individuals with CCD spectrum disorder caused by a de novo pathogenic variant is high. Each child of an individual with CCD spectrum disorder has a 50% chance of inheriting the RUNX2 pathogenic variant. Once the RUNX2 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing for CCD spectrum disorder are possible.
Diagnosis
Cleidocranial dysplasia (CCD) spectrum disorder is a skeletal dysplasia that represents a continuum of clinical and radiographic findings ranging from classic CCD (triad of delayed closure of the cranial sutures, hypoplastic or aplastic clavicles, and dental abnormalities), to mild CCD, to isolated dental anomalies without other skeletal features. No formal clinical diagnostic criteria for CCD spectrum disorder have been established.
Suggestive Findings
CCD spectrum disorder should be suspected in probands with the following clinical and radiographic findings.
Clinical findings
- Abnormally large, wide-open fontanelles at birth that may remain open throughout life. The wide-open metopic suture results in separation of the frontal bones by a metopic groove. The forehead is otherwise broad and flat; the cranium is brachycephalic.
- Additional craniofacial features. Frontal and parietal bossing, hypertelorism, midface retrusion, and depressed nasal bridge
- Narrow, sloping shoulders that can be opposed at the midline due to clavicular hypoplasia or aplasia (see Figure 1).
- Abnormal dentition including delayed eruption of secondary dentition, failure to shed the primary teeth, variable numbers of supernumerary teeth along with dental crowding, and malocclusion
- Digit abnormalities including brachydactyly, tapering fingers, and short, broad thumbs
- Short stature (typically moderate)
- Normal intellect
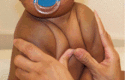
Figure 1.
Shoulders in an individual with clavicular hypoplasia may be brought to the midline.
Radiographic findings
- Cranium
- Wide-open sutures, patent fontanelles, presence of Wormian bones
- Delayed ossification of the skull
- Poor or absent pneumatization of the paranasal, frontal, and mastoid sinuses
- Impacted, crowded teeth; supernumerary teeth
- Thorax
- Cone-shaped thorax with narrow upper thoracic diameter
- Typically bilateral (but not necessarily symmetric) clavicular abnormalities ranging from complete absence to hypoplastic or discontinuous clavicles. The lateral portions are more affected than the medial aspects of the clavicles (see Figure 2).
- Hypoplastic scapulae
- Pelvis
- Delayed ossification of the pubic bone with wide pubic symphysis
- Hypoplasia of the iliac wings
- Widening of the sacroiliac joints
- Elongated femoral head with short femoral neck and elongated epiphyses ("chef-hat" appearance)
- Coxa vara
- Hands
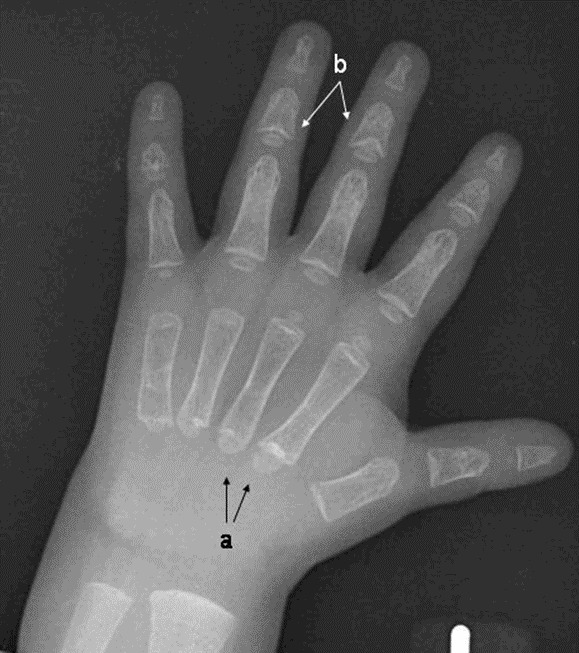
- Pseudoepiphyses of the metacarpal and metatarsal bones, which may result in a characteristic lengthening of the second metacarpal (See Figure 3.)
- Hypoplastic distal phalanges
- Deformed and short middle phalanges of the third, fourth, and fifth digits with cone-shaped epiphyses
- Other. Osteopenia/osteoporosis with evidence of decreased bone mineral density on DXA scan; some affected individuals sustain multiple fractures.
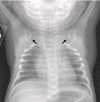
Figure 2.
Chest x-ray demonstrates clavicular hypoplasia.
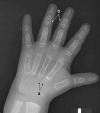
Figure 3.
Hand x-ray of a male age 2.5 years with cleidocranial dysplasia spectrum disorder a. Note pseudoepiphyses at the bases of the second and third metacarpals with accessory physes seen at the base of the fourth and fifth metacarpals.
Establishing the Diagnosis
The clinical diagnosis of a CCD spectrum disorder can be established in a proband with characteristic clinical and radiographic findings, or a molecular diagnosis can be established in a proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in RUNX2 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic, and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variant" in this section is understood to include any likely pathogenic variant. (2) Identification of a heterozygous variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of single-gene testing, multigene panel, and karyotype.
- Single-gene testing. Sequence analysis of RUNX2 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform RUNX2 gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
- A multigene panel that includes RUNX2 and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- Karyotype. If RUNX2 testing is not diagnostic in an individual with features of a CCD spectrum disorder who also has multiple congenital anomalies and/or developmental delay, a karyotype may be considered to evaluate for complex chromosome rearrangements or translocations that involve 6p21.1 (RUNX2 locus) but do not result in RUNX2 copy number changes.
Table 1.
Molecular Genetic Testing Used in Cleidocranial Dysplasia Spectrum Disorder
Clinical Characteristics
Clinical Description
Cleidocranial dysplasia (CCD) spectrum disorder is a skeletal dysplasia representing a clinical continuum ranging from classic CCD (triad of delayed closure of the cranial sutures, hypoplastic or aplastic clavicles, and dental abnormalities), to mild CCD, to isolated dental anomalies without other skeletal features [Golan et al 2000]. Most individuals are diagnosed because they have classic features. CCD spectrum disorder affects most prominently those bones derived from intramembranous ossification, such as the cranium and the clavicles, although bones formed through endochondral ossification can also be affected. Cooper et al [2001] recorded the natural history of 90 probands and 56 first- and second-degree relatives; findings highlight the clinical variability of this condition within affected members of the same family who harbor the same pathogenic variant. Roberts et al [2013] reviewed their experience with more than 100 affected individuals in South Africa.
Classic CCD. The most prominent clinical findings in individuals with classic CCD are listed in Suggestive Findings and include: abnormally large, wide-open fontanelles at birth that may remain open throughout life; clavicular hypoplasia resulting in narrow, sloping shoulders that can be opposed at the midline; and abnormal dentition (see Dental complications in this section).
Further medical problems identified in individuals with CCD spectrum disorder include short stature, skeletal/orthopedic findings, dental complications, ENT complications, endocrine findings, and mild developmental delay.
Height. Individuals with CCD spectrum disorder are often shorter than their unaffected sibs and present with postnatal growth deficiency.
- Males are on average six inches shorter than their unaffected brothers and have an average height of 165 cm (± 8 cm).
- Females are on average three inches shorter than their unaffected sisters and have an average height of 156 cm (± 10 cm) [Cooper et al 2001].
Skeletal/orthopedic findings. Affected individuals are more likely to have other bone-related problems:
- Pes planus (flat feet) in 57%
- Genu valgum (knock-knee deformity) in 28%
- Scoliosis in 18% [Cooper et al 2001]
- Osteoporosis, found in 8/14 (57%) affected individuals; and osteopenia, identified in 3/14 (21%) individuals [Dinçsoy Bir et al 2017]
Other less common orthopedic problems include joint dislocation at the shoulder and elbow [El-Gharbawy et al 2010].
Dental complications. Up to 94% of persons with CCD spectrum disorder have dental findings, including delayed eruption of secondary dentition and failure to shed the primary teeth [Golan et al 2003]. The most consistent dental findings in individuals with a CCD spectrum disorder are the presence of the second permanent molar with the primary dentition (80%), wide spacing in the lower incisor area, supernumerary tooth germs (70%), and parallel-sided ascending rami [Cooper et al 2001, Golan et al 2003, Golan et al 2004, Bufalino et al 2012]. Individuals with a CCD spectrum disorder are more likely to have an underbite and to have gingival cysts that usually form around extra teeth [McNamara et al 1999].
ENT complications. Recurrent sinus infections and other upper airway complications are observed significantly more often in individuals with CCD spectrum disorder than in the general population. When symptoms are suggestive of upper airway obstruction, a sleep study is indicated, and surgical intervention may be required. Conductive hearing loss occurs in 39% of affected individuals. Individuals with CCD spectrum disorder of any age are more likely to have recurrent ear infections.
Endocrinology. Individuals with CCD spectrum disorder can have low insulin-like growth factor 1 levels. Low vitamin D with no consistent association with osteoporosis has also been reported [Dinçsoy Bir et al 2017]. Rarely, individuals with CCD spectrum disorder have low levels of alkaline phosphatase [Morava et al 2002, Unger et al 2002, El-Gharbawy et al 2010].
Development. Intelligence is typically normal. Children younger than age five years may show mild motor delay, particularly in gross motor abilities. This delay may be associated with orthopedic complications such as flat feet and genu valgum. No significant differences are observed among elementary school-age children.
Genotype-Phenotype Correlations
Some genotype-phenotype correlations have been established for the dental manifestations seen in CCD spectrum disorder. No clear correlation has been established between genotype and clavicular involvement [Otto et al 2002, Bufalino et al 2012, Jaruga et al 2016].
- Heterozygous RUNX2 pathogenic variants located in the runt domain (or predicting a premature termination upstream of or within the runt domain) that abolish the transactivation activity of the mutated protein with consequent haploinsufficiency result in classic CCD.
- Short stature and dental anomalies were found to be milder in individuals with a classic CCD phenotype who had an intact runt domain and higher residual RUNX2 activity when compared to individuals with a classic CCD phenotype in whom the pathogenic variant affected the runt domain [Yoshida et al 2002].
- A clinical spectrum ranging from isolated dental anomalies without the skeletal features of CCD, to mild CCD, to classic CCD results from hypomorphic pathogenic variants that result in partial loss of protein function (c.1171C>T [p.Arg391Ter], c.598A>G [p.Thr200Ala], and c.90dupC [p.Ser31LeufsTer130]) (see Molecular Genetics). Intrafamilial variability is significant [Zhou et al 1999].
- Osteoporosis leading to recurrent bone fractures and scoliosis has been associated with a heterozygous pathogenic frameshift variant c.1205dupC, reflecting the role of RUNX2 in the maintenance of adult bone [Quack et al 1999].
Penetrance
Pathogenic variants in RUNX2 have high penetrance. To date, there are no reports of incomplete penetrance.
Nomenclature
Cleidocranial dysplasia spectrum disorder was originally described as dento-osseous dysplasia affecting several individuals in a large pedigree.
While the term "cleidocranial dysostosis" has been used, the disease is more correctly considered a dysplasia given that RUNX2 has important functions both during skeletal formation and in bone maintenance.
Prevalence
Stevenson et al [2012] found the frequency to be 0.12 in 10,000 individuals in the Utah (US) population, suggesting that the frequency may be higher than previously recognized.
Genetically Related (Allelic) Disorders
Partial intragenic duplication of RUNX2 has been associated with metaphyseal dysplasia, maxillary hypoplasia, and brachydactyly (MDMHB) (OMIM 156510). Affected individuals have short stature, long bone and spinal abnormalities, dystrophic teeth, and enlargement of the medial half of the clavicle bones.
Complete duplications of RUNX2 have been described in individuals with craniosynostosis and oligodontia [Mefford et al 2010, Greives et al 2013, Molin et al 2015].
Differential Diagnosis
Other conditions share some characteristics with cleidocranial dysplasia (CCD) spectrum disorder. The fact that similar skeletal elements are affected suggests that some of these conditions may result from mutation of genes (most notably CBFB) that affect the action of RUNX2 on its downstream targets (CBFB forms a heterodimer with RUNX2 to activate transcription of downstream targets).
Table 2.
Genes of Interest in the Differential Diagnosis of Cleidocranial Dysplasia Spectrum Disorder
Other disorders/conditions in the differential diagnosis of CCD spectrum disorder
- Crane-Heise syndrome (OMIM 218090). A lethal condition of unknown genetic cause associated with a poorly mineralized calvarium; macrocephaly; cleft lip and palate; low-set, dysplastic ears; severe vertebral and limb anomalies with absence of cervical vertebrae; hypoplastic clavicles and scapulae; hypoplastic and absent phalanges; multiple joint contractures; intrauterine growth restriction; and genital hypoplasia.
- CDAGS syndrome (OMIM 603116). A disorder caused by biallelic pathogenic variants in small nuclear RNA encoding RNU12 [Xing et al 2021] that combines the apparently opposing pathophysiologic and developmental processes of accelerated suture closure and delayed ossification [Mendoza-Londono et al 2005]. CDAGS syndrome is associated with delayed closure of the fontanelles, clavicular hypoplasia, craniosynostosis, anal anomalies, genitourinary malformations, and skin lesions (porokeratosis).
- Familial supernumerary teeth. Nonsyndromic supernumerary premolar teeth [Bae et al 2017]
- Hypothyroidism. Can be associated with delayed fontanelle closure
Management
No clinical practice guidelines for cleidocranial dysplasia (CCD) spectrum disorder have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with CCD spectrum disorder, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Cleidocranial Dysplasia Spectrum Disorder
Treatment of Manifestations
Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 4).
Table 4.
Treatment of Manifestations in Individuals with Cleidocranial Dysplasia Spectrum Disorder
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 5 are recommended.
Table 5.
Recommended Surveillance for Individuals with Cleidocranial Dysplasia Spectrum Disorder
Agents/Circumstances to Avoid
To avoid head trauma, helmets and protective devices should be worn when participating in high-risk sports and activities.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Pregnant women with CCD spectrum disorder should be monitored closely for cephalopelvic disproportion, which may require delivery by cesarean section. The primary cesarean section rate among women with a CCD spectrum disorder is 69%, which is higher than in controls [Cooper et al 2001].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Cleidocranial dysplasia (CCD) spectrum disorder is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Some individuals diagnosed with CCD spectrum disorder have an affected parent.
- A proband with CCD spectrum disorder may have the disorder as the result of a de novo RUNX2 pathogenic variant. The proportion of individuals with CCD spectrum disorder caused by a de novo pathogenic variant is high.
- If the proband appears to be the only affected family member (i.e., a simplex case), recommendations for the evaluation of the parents of the proband include:
- Detailed clinical examination and consideration of craniofacial and skeletal x-rays if there are signs suggestive of dental or bone abnormalities. (Note: The phenotype may vary between parent and child even though they have the same pathogenic variant.)
- Molecular genetic testing (if a molecular diagnosis has been established in the proband) to confirm the genetic status of the parents and to allow reliable recurrence risk counseling.
- If the proband has a known pathogenic variant that is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism* [Pal et al 2007, Qian et al 2018, Muurinen et al 2022]. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.* A parent with somatic and germline mosaicism for a RUNX2 pathogenic variant may be mildly/minimally affected.
- The family history of some individuals diagnosed with CCD spectrum disorder may appear to be negative because of failure to recognize the disorder in family members. Therefore, an apparently negative family history cannot be confirmed without appropriate clinical examination (with skeletal x-rays) of the parents and/or molecular genetic testing (to establish that neither parent is heterozygous for the pathogenic variant identified in the proband).
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs is 50%.
- The phenotype may vary among sibs who inherit the same RUNX2 pathogenic variant (although the penetrance of RUNX2 pathogenic variants is high, significant clinical variability is observed between affected family members; see Genotype-Phenotype Correlations and Penetrance).
- If the proband has a known RUNX2 pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism. Germline mosaicism has been demonstrated in a family with three affected sibs and an apparently unaffected mother [Pal et al 2007].
- If the parents are clinically unaffected but their genetic status is unknown, the risk to the sibs of a proband appears to be low but increased over that of the general population because of the possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with CCD spectrum disorder has a 50% chance of inheriting the RUNX2 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has features of CCD spectrum disorder and/or is known to be heterozygous for a RUNX2 pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the RUNX2 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing for CCD spectrum disorder are possible.
Ultrasound examination. Classic CCD can be diagnosed by ultrasound examination in the offspring of an affected parent as early as 14 weeks' gestation. The most consistent features are abnormal clavicles, which are either short (<5th centile for gestational age) or partially or totally absent. Other less specific findings include brachycephalic skull with undermineralization, frontal bossing, and generalized immature ossification [Stewart et al 2000, Hermann et al 2009].
Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- About Kids HealthCanada
- CCD Smiles
- Children's Craniofacial AssociationPhone: 800-535-3643Email: contactCCA@ccakids.com
- FACES: National Craniofacial AssociationPhone: 800-332-2373; 423-266-1632Email: info@faces-cranio.org
- Human Growth Foundation
- MAGIC FoundationPhone: 800-362-4423Email: contactus@magicfoundation.org
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Cleidocranial Dysplasia Spectrum Disorder: Genes and Databases
Table B.
OMIM Entries for Cleidocranial Dysplasia Spectrum Disorder (View All in OMIM)
Molecular Pathogenesis
RUNX2 encodes runt-related transcription factor 2 (RUNX2), a transcription factor involved in osteoblast differentiation and skeletal morphogenesis. RUNX2 is essential for osteoblast differentiation during intramembranous ossification as well as chondrocyte maturation during endochondral ossification [Zheng et al 2005]. RUNX2 contains an N-terminal stretch of consecutive polyglutamine and polyalanine repeats known as the Q/A domain, a runt domain, and a C-terminal proline/serine/threonine-rich (PST) activation domain. The runt domain is a 128-amino-acid polypeptide motif originally described in the Drosophila runt gene that has the unique ability to independently mediate DNA binding and protein heterodimerization [Zhou et al 1999].
The majority of RUNX2 pathogenic variants in individuals with classic CCD affect the runt domain and most are predicted to abolish DNA binding [Lee et al 1997, Mundlos et al 1997, Otto et al 2002]. Pathogenic missense variants cluster at arginine 225 (p.Arg225) of RUNX2, a critical residue for RUNX2 function. In vitro studies have shown that pathogenic missense variants at p.Arg225 interfere with nuclear accumulation of RUNX2.
Hypomorphic RUNX2 alleles with partial loss of protein function, c.90dupC and c.598A>G, are associated with mild CCD, isolated dental anomalies, and significant intrafamilial variability.
Mechanism of disease causation. Loss of function
RUNX2-specific laboratory technical considerations. At the genomic level, the longest RUNX2 transcript variant (NM_001024630.4) contains nine exons. Transcript variants that encode different protein isoforms [Geoffroy et al 1998] result from the use of alternate promoters and alternate splicing.
Table 6.
Notable RUNX2 Pathogenic Variants
Chapter Notes
Author Notes
Dr Keren Machol's web pages: Texas Children's Hospital and Baylor College of Medicine
Dr Mendoza-Londono's web page
Dr Lee's web pages: Baylor College of Medicine, People and Baylor College of Medicine, Find a Physician
Revision History
- 13 April 2023 (sw) Comprehensive update posted live
- 16 November 2017 (ma) Comprehensive update posted live
- 29 August 2013 (me) Comprehensive update posted live
- 25 June 2009 (me) Comprehensive update posted live
- 3 January 2006 (me) Review posted live
- 28 June 2005 (rml) Original submission
References
Literature Cited
- Bae DH, Lee JH, Song JS, Jung HS, Choi HJ, Kim JH. Genetic analysis of non-syndromic familial multiple supernumerary premolars. Acta Odontol Scand. 2017;75:350–4. [PubMed: 28393601]
- Becker A, Lustmann J, Shteyer A. Cleidocranial dysplasia: Part 1--General principles of the orthodontic and surgical treatment modality. Am J Orthod Dentofacial Orthop. 1997a;111:28–33. [PubMed: 9009920]
- Becker A, Shteyer A, Bimstein E, Lustmann J. Cleidocranial dysplasia: Part 2--Treatment protocol for the orthodontic and surgical modality. Am J Orthod Dentofacial Orthop. 1997b;111:173–83. [PubMed: 9057617]
- Bufalino A, Paranaíba LM, Gouvêa AF, Gueiros LA, Martelli-Júnior H, Junior JJ, Lopes MA, Graner E, De Almeida OP, Vargas PA, Coletta RD. Cleidocranial dysplasia: oral features and genetic analysis of 11 patients. Oral Dis. 2012;18:184–90. [PubMed: 22023169]
- Cooper SC, Flaitz CM, Johnston DA, Lee B, Hecht JT. A natural history of cleidocranial dysplasia. Am J Med Genet. 2001;104:1–6. [PubMed: 11746020]
- Dinçsoy Bir F, Dinçkan N, Güven Y, Baş F, Altunoğlu U, Kuvvetli SS, Poyrazoğlu Ş, Toksoy G, Kayserili H, Uyguner ZO. Cleidocranial dysplasia: clinical, endocrinologic and molecular findings in 15 patients from 11 families. Eur J Med Genet. 2017;60:163–8. [PubMed: 28027977]
- El-Gharbawy AH, Peeden JN Jr, Lachman RS, Graham JM Jr, Moore SR, Rimoin DL. Severe cleidocranial dysplasia and hypophosphatasia in a child with microdeletion of the C-terminal region of RUNX2. Am J Med Genet A. 2010;152A:169–74. [PMC free article: PMC2799546] [PubMed: 20014132]
- Farrow E, Nicot R, Wiss A, Laborde A, Ferri J. Cleidocranial Dysplasia: A Review of Clinical, Radiological, Genetic Implications and a Guidelines Proposal. J Craniofac Surg. 2018;29:382–9. [PubMed: 29189406]
- Geoffroy V, Corral DA, Zhou L, Lee B, Karsenty G. Genomic organization, expression of the human CBFA1 gene, and evidence for an alternative splicing event affecting protein function. Mamm Genome. 1998;9:54–7. [PubMed: 9434946]
- Golan I, Baumert U, Hrala BP, Mussig D. Dentomaxillofacial variability of cleidocranial dysplasia: clinicoradiological presentation and systematic review. Dentomaxillofac Radiol. 2003;32:347–54. [PubMed: 15070835]
- Golan I, Baumert U, Hrala BP, Mussig D. Early craniofacial signs of cleidocranial dysplasia. Int J Paediatr Dent. 2004;14:49–53. [PubMed: 14706028]
- Golan I, Preising M, Wagener H, Baumert U, Niederdellmann H, Lorenz B, Mussig D. A novel missense mutation of the CBFA1 gene in a family with cleidocranial dysplasia (CCD) and variable expressivity. J Craniofac Genet Dev Biol. 2000;20:113–20. [PubMed: 11321595]
- Goto T, Aramaki M, Yoshihashi H, Nishimura G, Hasegawa Y, Takahashi T, Ishii T, Fukushima Y, Kosaki K. Large fontanels are a shared feature of haploinsufficiency of RUNX2 and its co-activator CBFB. Congenit Anom (Kyoto). 2004;44:225–9. [PubMed: 15566413]
- Greives MR, Odessey EA, Waggoner DJ, Shenaq DS, Aradhya S, Mitchell A, Whitcomb E, Warshawsky N, He TC, Reid RR. RUNX2 quadruplication: additional evidence toward a new form of syndromic craniosynostosis. J Craniofac Surg. 2013;24:126–9. [PubMed: 23348268]
- Hermann NV, Hove HD, Jørgensen C, Larsen P, Darvann TA, Kreiborg S, Sundberg K. Prenatal 3D ultrasound diagnostics in cleidocranial dysplasia. Fetal Diagn Ther. 2009;25:36–9. [PubMed: 19169035]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Ioscovich A, Barth D, Samueloff A, Grisaru-Granovsky S, Halpern S. Anesthetic management of a patient with cleidocranial dysplasia undergoing various obstetric procedures. Int J Obstet Anesth. 2010;19:106–8. [PubMed: 19945847]
- Jaruga A, Hordyjewska E, Kandzierski G, Tylzanowski P. Cleidocranial dysplasia and RUNX2-clinical phenotype-genotype correlation. Clin Genet. 2016;90:393–402. [PubMed: 27272193]
- Kang N, Kim SZ, Jung SN. Correction of depressed forehead with BoneSource in cleidocranial dysplasia. J Craniofac Surg. 2009;20:564–6. [PubMed: 19305258]
- Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, Geoffroy V, Ducy P, Karsenty G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307–10. [PubMed: 9207800]
- McNamara CM, O'Riordan BC, Blake M, Sandy JR. Cleidocranial dysplasia: radiological appearances on dental panoramic radiography. Dentomaxillofac Radiol. 1999;28:89–97. [PubMed: 10522197]
- Mefford HC, Shafer N, Antonacci F, Tsai JM, Park SS, Hing AV, Rieder MJ, Smyth MD, Speltz ML, Eichler EE, Cunningham ML. Copy number variation analysis in single-suture craniosynostosis: multiple rare variants including RUNX2 duplication in two cousins with metopic craniosynostosis. Am J Med Genet A. 2010;152A:2203–10. [PMC free article: PMC3104131] [PubMed: 20683987]
- Mendoza-Londono R, Lammer E, Watson R, Harper J, Hatamochi A, Hatamochi-Hayashi S, Napierala D, Hermanns P, Collins S, Roa BB, Hedge MR, Wakui K, Nguyen D, Stockton DW, Lee B. Characterization of a new syndrome that associates craniosynostosis, delayed fontanel closure, parietal foramina, imperforate anus, and skin eruption: CDAGS. Am J Hum Genet. 2005;77:161–8. [PMC free article: PMC1226190] [PubMed: 15924278]
- Molin A, Lopez-Cazaux S, Pichon O, Vincent M, Isidor B, Le Caignec C. Patients with isolated oligo/hypodontia caused by RUNX2 duplication. Am J Med Genet A. 2015;167:1386–90. [PubMed: 25899668]
- Morava E, Karteszi J, Weisenbach J, Caliebe A, Mundlos S, Mehes K. Cleidocranial dysplasia with decreased bone density and biochemical findings of hypophosphatasia. Eur J Pediatr. 2002;161:619–22. [PubMed: 12424590]
- Motaei J, Salmaninejad A, Jamali E, Khorsand I, Ahmadvand M, Shabani S, Karimi F, Nazari MS, Ketabchi G, Naqipour F. Molecular Genetics of Cleidocranial Dysplasia. Fetal Pediatr Pathol. 2021;40:442–54. [PubMed: 31984822]
- Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JH, Owen MJ, Mertelsmann R, Zabel BU, Olsen BR. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–9. [PubMed: 9182765]
- Northup JK, Matalon R, Lockhart LH, Hawkins JC, Velagaleti GV. A complex chromosome rearrangement, der(6)ins(6)(p21.1q25.3q27)inv(6)(p25.3q27), in a child with cleidocranial dysplasia. Eur J Med Genet. 2011;54:e394–8. [PubMed: 21466863]
- Muurinen M, Taylan F, Tournis S, Eisfeldt J, Balanika A, Vastardis H, Ala-Mello S, Mäkitie O, Costantini A. Mosaic Deletions of Known Genes Explain Skeletal Dysplasias With High and Low Bone Mass. JBMR Plus. 2022;6:e10660. [PMC free article: PMC9382864] [PubMed: 35991531]
- Ott CE, Hein H, Lohan S, Hoogeboom J, Foulds N, Grünhagen J, Stricker S, Villavicencio-Lorini P, Klopocki E, Mundlos S. Microduplications upstream of MSX2 are associated with a phenocopy of cleidocranial dysplasia. J Med Genet. 2012;49:437–41. [PubMed: 22717651]
- Ott CE, Leschik G, Trotier F, Brueton L, Brunner HG, Brussel W, Guillen-Navarro E, Haase C, Kohlhase J, Kotzot D, Lane A, Lee-Kirsch MA, Morlot S, Simon ME, Steichen-Gersdorf E, Tegay DH, Peters H, Mundlos S, Klopocki E. Deletions of the RUNX2 gene are present in about 10% of individuals with cleidocranial dysplasia. Hum Mutat. 2010;31:E1587–93. [PubMed: 20648631]
- Otto F, Kanegane H, Mundlos S. Mutations in the RUNX2 gene in patients with cleidocranial dysplasia. Hum Mutat. 2002;19:209–16. [PubMed: 11857736]
- Pal T, Napierala D, Becker TA, Loscalzo M, Baldridge D, Lee B, Sutphen R. The presence of germ line mosaicism in cleidocranial dysplasia. Clin Genet. 2007;71:589–91. [PubMed: 17539909]
- Purandare SM, Mendoza-Londono R, Yatsenko SA, Napierala D, Scott DA, Sibai T, Casas K, Wilson P, Lee J, Muneer R, Leonard JC, Ramji FG, Lachman R, Li S, Stankiewicz P, Lee B, Mulvihill JJ. De novo three-way chromosome translocation 46,XY,t(4;6;21)(p16;p21.1;q21) in a male with cleidocranial dysplasia. Am J Med Genet A. 2008;146A:453–8. [PMC free article: PMC2663417] [PubMed: 18203189]
- Puvabanditsin S, February M, Mayne J, McConnell J, Mehta R. Cleidocranial Dysplasia with 6p21.1-p12.3 Microdeletion: A Case Report and Literature Review. Cleft Palate Craniofac J. 2018;55:891–4. [PubMed: 27500518]
- Qian Y, Zhang Y, Wei B, Zhang M, Yang J, Leng C, Ge Z, Xu X, Sun M. A novel Alu-mediated microdeletion in the RUNX2 gene in a Chinese patient with cleidocranial dysplasia. J Genet. 2018;97:137–43. [PubMed: 29666333]
- Quack I, Vonderstrass B, Stock M, Aylsworth AS, Becker A, Brueton L, Lee PJ, Majewski F, Mulliken JB, Suri M, Zenker M, Mundlos S, Otto F. Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am J Hum Genet. 1999;65:1268–78. [PMC free article: PMC1288279] [PubMed: 10521292]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Roberts T, Stephen L, Beighton P. Cleidocranial dysplasia: a review of the dental, historical, and practical implications with an overview of the South African experience. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;115:46–55. [PubMed: 23102800]
- Sewell MD, Higgs DS, Lambert SM. Clavicle lengthening by distraction osteogenesis for congenital clavicular hypoplasia: case series and description of technique. J Pediatr Orthop. 2013;33:314–20. [PubMed: 23482270]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Stevenson DA, Carey JC, Byrne JL, Srisukhumbowornchai S, Feldkamp ML. Analysis of skeletal dysplasias in the Utah population. Am J Med Genet A. 2012;158A:1046–54. [PubMed: 22461456]
- Stewart PA, Wallerstein R, Moran E, Lee MJ. Early prenatal ultrasound diagnosis of cleidocranial dysplasia. Ultrasound Obstet Gynecol. 2000;15:154–6. [PubMed: 10776001]
- Unger S, Mornet E, Mundlos S, Blaser S, Cole DE. Severe cleidocranial dysplasia can mimic hypophosphatasia. Eur J Pediatr. 2002;161:623–6. [PubMed: 12424591]
- Visosky AM, Johnson J, Bingea B, Gurney T, Lalwani AK. Otolaryngological manifestations of cleidocranial dysplasia, concentrating on audiological findings. Laryngoscope. 2003;113:1508–14. [PubMed: 12972925]
- Xing C, Kanchwala M, Rios JJ, Hyatt T, Wang RC, Tran A, Dougherty I, Tovar-Garza A, Purnadi C, Kumar MG, Berk D, Shinawi M, Irvine AD, Toledo-Bahena M, Agim NG, Glass DA 2nd. Biallelic variants in RNU12 cause CDAGS syndrome. Hum Mutat. 2021;42:1042–52. [PubMed: 34085356]
- Yoshida T, Kanegane H, Osato M, Yanagida M, Miyawaki T, Ito Y, Shigesada K. Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations. Am J Hum Genet. 2002;71:724–38. [PMC free article: PMC378531] [PubMed: 12196916]
- Zheng Q, Sebald E, Zhou G, Chen Y, Wilcox W, Lee B, Krakow D. Dysregulation of chondrogenesis in human cleidocranial dysplasia. Am J Hum Genet. 2005;77:305–12. [PMC free article: PMC1224532] [PubMed: 15952089]
- Zhou G, Chen Y, Zhou L, Thirunavukkarasu K, Hecht J, Chitayat D, Gelb BD, Pirinen S, Berry SA, Greenberg CR, Karsenty G, Lee B. CBFA1 mutation analysis and functional correlation with phenotypic variability in cleidocranial dysplasia. Hum Mol Genet. 1999;8:2311–6. [PubMed: 10545612]
Publication Details
Author Information and Affiliations
Department of Molecular and Human Genetics
Baylor College of Medicine
Houston, Texas
University of Toronto
Toronto, Canada
Division of Clinical and Metabolic Genetics
The Hospital for Sick Children
Toronto, Canada
Chair, Molecular and Human Genetics
Baylor College of Medicine
Houston, Texas
Publication History
Initial Posting: January 3, 2006; Last Update: April 13, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Machol K, Mendoza-Londono R, Lee B. Cleidocranial Dysplasia Spectrum Disorder. 2006 Jan 3 [Updated 2023 Apr 13]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.