Clinical Description
Hereditary motor and sensory neuropathy with agenesis of the corpus callosum (HMSN/ACC) is both a neurodevelopmental disorder (with variable degrees of dysgenesis of the corpus callosum and mild-to-severe intellectual disability) and a neurodegenerative disorder (severe progressive sensorimotor neuropathy).
The neurologic findings of HMSN/ACC in 64 individuals (ages 2 to 34 years) in the French Canadian population reported by Mathieu et al [1990] are summarized in Table 2, with additional information from Larbrisseau et al [1984], Salin-Cantegrel et al [2007], and Auer et al [2016].
Table 2.
Hereditary Motor and Sensory Neuropathy with Agenesis of the Corpus Callosum: Select Features
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
|
Motor & sensory neuropathy
| 100% | |
|
Cranial nerve involvement
|
Ptosis
| 33%-59% | Symmetric or asymmetric |
|
Gaze palsy
| 13%-30% | |
|
Horizontal nystagmus
| 20% | |
|
Facial weakness
| 34%-100% | Symmetric or asymmetric; may be assoc w/hemifacial atrophy |
|
Cognitive function
|
Normal
| 8% | Based on Taft clinical classification to stratify cognitive function in 53 persons 1 |
|
Mild ID
| 49% |
|
Moderate ID
| 40% |
|
Severe ID
| 4% |
|
Psychotic episodes
| 39% (25/64) | After age 15 yrs 1 |
|
Scoliosis
| 86% | Average onset age 10.4 yrs |
|
Pulmonary restrictive syndrome
| Unknown | Related to scoliosis & axonal loss affecting respiratory muscles 2 |
|
Contractures
| 59% | MCP joint (flexion) contracture |
| 50% | Valgus foot deviation |
| 47% | Early Achilles tendon retraction |
| 31% | Varus foot deviation |
|
Seizures
| 17% | Generalized, absence, or focal seizures 3 |
|
Tremor
| 25% | |
ID = intellectual disability; MCP = metacarpophalangeal
- 1.
- 2.
- 3.
Progressive motor and sensory neuropathy
Reflexes invariably absent from infancy
Hypotonia invariably present in the first year of life
Progressive distal and proximal symmetric limb weakness
Muscle atrophy
Diffuse limb tremor (probably secondary to polyneuropathy)
Contractures
Loss of sensation to touch and pinprick in a glove and stocking distribution; easier to evaluate in older children
The average age of onset for sitting alone is 2.1 years, for standing is more than two years, for walking is 3.8 years, and average age of loss of ability to walk is 13.8 years.
Cognitive function. The Taft clinical classification uses an IQ test to rank individuals in four categories: normal intelligence (IQ >75), mild intellectual disability (IQ = 50-75), moderate intellectual disability (IQ = 25-50), and severe intellectual disability (IQ <25). Individuals with mild intellectual disability can achieve five to six years of elementary school and live independently. Individuals with moderate intellectual disability can help with activities of daily living but require supervision on a daily basis. Individuals with severe intellectual disability require assistance for daily living and supervision but may be able to take care of some routine daily needs. The range of intelligence of individuals with HSMN/ACC is normal IQ to severe intellectual disability.
Psychotic episodes.
Mathieu et al [1990] reported that after age 15 years, 39% (25/64) developed "psychotic episodes" characterized by paranoid delusions, depressive states, visual hallucinations, auditory hallucinations, or "autistic-like" features.
Life expectancy. Average age of death is 33 years and is usually related to respiratory insufficiency [Larbrisseau & Sarnat 2017].
Other
Lumbar puncture usually reveals mild elevation of CSF proteins [
Dupré et al 2003].
Sural nerve biopsy shows an almost total lack of large myelinated fibers, signs of axonal loss (ovoids of Wallerian degeneration), and some enlarged axons that on electron microscopy show decreased density of neurofilaments. Isolated fibers may have disproportionately thin myelin sheaths, suggesting that the axoplasm is swollen. Electron microscopy may show decreased packing density of neurofilaments, without signs of their degradation [
Dupré et al 2003,
Auer et al 2016].
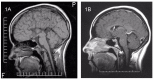
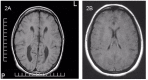
Autopsy examination. The hallmark is swollen axons in cranial nerve samples (especially cranial nerves 3 and 7), as well as in the dorsal and ventral nerve roots. Swollen axons can also be scattered in the white matter. The brain shows either no agenesis of the corpus callosum (ACC), partial ACC, or complete ACC with preservation of Probst bundle [Dupré et al 2003, Auer et al 2016].
Prevalence
In the French Canadian population of the Saguenay and Lac-St-Jean regions of Quebec, Canada, the overall incidence of HMSN/ACC is 1:2,117 live births; the carrier rate is 1:23 inhabitants due to the c.2436+1delG (also known as c.2436delG) founder variant. Otherwise, HMSN/ACC is extremely rare worldwide. Three pairs of sibs (of Italian, Mexican, and Turkish origin) have been reported to have molecularly confirmed HMSN/ACC.