

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Space Occupying Lesions in the hypothalamic/pituitary region include tumours derived from endocrine or neural tissues, as well as a wide spectrum of more uncommon conditions such us inflammatory processes or metastases. The clinical expression of these lesions depends not only of their nature, but also on the size and position of the lesion, with some common patterns that can lead the clinician to the diagnosis, being some of them clinically silent. There is an increasing interest in the study of the molecular abnormalities in these uncommon conditions, and new drugs have started to be used as a result of the understanding of the pathways involved in the tumourigenesis. A literature search in Pubmed was performed, focused on articles referred to craniopharyngioma, non-functioning pituitary adenomas, pituitary carcinoma, hypophysitis, germ-cell tumours, chordoma, cysts and empty sella syndrome. Articles published from 2010 to 2015 in high impact factor journals were selected. Selected articles were mainly referred to new signaling pathways involved in tumourigenesis, somatic mutations, expressed receptors, new treatments and guidelines. Review articles were also used. This chapter reviews the pathogenesis, clinical manifestations and treatments of the main hypothalamic/pituitary space occupying lesions. It also provides a general and practical approach for the evaluation of these patients.
INTRODUCTION
Space Occupying Lesions in the hypothalamic/pituitary region include tumours derived from endocrine or neural tissues, metastatic tumours, chronic inflammatory processes, cystic lesions or vascular aneurysms (Table 1). These lesions may be clinically silent and discovered accidentally when skull X-ray, cranial CT, or MRI is performed for other reasons. These tumours are called pituitary incidentalomas and the management depends on the nature and position of the lesion (1).
More frequently, a variety of endocrine, ophthalmological or neurological symptoms lead the clinician to the diagnosis of a space occupying lesion within the sellar or supra-sellar region, i.e. a pituitary tumour with intra-, supra-, and parasellar extension or a primary hypothalamic tumour which also may extend into the sellar cavity (Fig. 1).
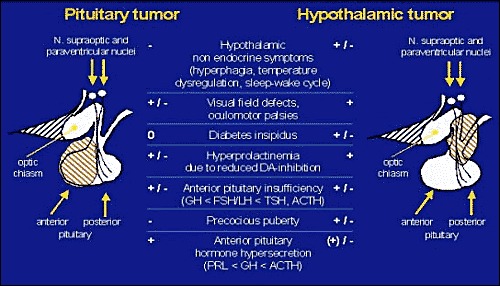
Figure 1Schematic presentation of endocrine, visual and neurological symptoms as they may occur in patients with pituitary (left) and suprasellar, hypothalamic lesions (right).
When there are no typical endocrine features suggesting hormonally active pituitary tumours, the differential diagnosis of the various causes of space occupying lesions in the pituitary-hypothalamic area can be difficult and often impossible without biopsy, which may be hazardous (2).
SPACE OCCUPYING LESIONS OF THE PITUITARY
The increasing use of cranial CT and MRI scanning has increased the number of previously unsuspected pituitary lesions. These incidentalomas have been found in approximately 10% of autopsy studies in adults. Since their natural history, particularly what proportion of them remain stable or increase in size is not known, their management remains a challenge. Once they are discovered, hormonal evaluation as well as radiological and endocrinological follow-up are required.
The most common cause of an expanding lesion within the sella turcica is a pituitary adenoma (3, 4). The mean annual incidence of pituitary adenomas of all sizes and secretory activities is about 2 cases per 100,000 population, prolactinomas being the most common pituitary tumour.
Table 1Space occupying lesions in the pituitary-hypothalamic region
| Pituitary lesionsEndocrine active pituitary adenoma. Prolactinoma. Somatotropinoma . Corticotropinoma . Thyrotropinoma . Gonadotropinoma . Somatomammotropinoma Other mixed endocrine active adenomas Non-Functioning Pituitary Adenomas (NFPA) Null cell adenoma . Non-secreting adenoma (silent adenoma) Biologically inactive sub-unit-secreting adenoma (α-subunits, β-subunits) Malignant pituitary tumours Corticotrophin secreting carcinoma Prolactin-secreting carcinoma GH-secreting carcinoma Endocrine inactive pituitary carcinoma .Sarcoma Metastases in the pituitary (breast, lung, stomach, kidney) Pituitary cysts .Rathke's cleft cyst .Mucocoeles .Arachnoid cysts Empty sella syndrome Primary empty sella Secondary empty sella Hypophysitis Abscess | Hypothalamic lesionsDevelopmental abnormalities cystsCraniopharyngioma (occasionally intrasellar location).Germinoma .Hamartoma Chordoma Epidermoid and Dermoid Primary Tumours of the central nervous system Perisellarmeningioma Optic glioma Ependymoma Vascular tumours Haemangioblastoma Cavernous haemangioma Haemangiopericytoma Malignant systemic diseases of the central nervous system Hodgkin's disease Non-Hodgkin lymphoma Leukaemic infiltration Histiocystosis X Eosinophilic granuloma Giant cell granuloma (tumour) Granulomatous diseases Neurosarcoidosis Wegner's granulomatosis Tuberculoma Syphilis Vascular aneurysms (also intrasellar location) 2Increased intracranial pressure (brain tumours outside the sellar/suprasellar region) |
| The separation into pituitary and hypothalamic lesions is based on the observation that these lesions occur predominantly but not exclusively in the respective region. Thus, metastases are also found in the hypothalamic area and hypophysitis can take place in the pituitary stalk. On the other hand, a craniopharyngioma may present with a purely intrasellar location. | |
NON-FUNCTIONING PITUITARY ADENOMAS
The most common macroadenomas are non functioning pituitary adenomas (NFPA) followed by macroprolactinomas (4). GH-, and ACTH-secreting adenomas (the latter are usually microadenomas) occur more rarely and TSH- and gonadotropin-secreting adenomas are even more uncommon. Prolactinomas, somatotropinomas and corticotropinomas all lead to characteristic clinical features. The detailed pathophysiology, clinical picture, and therapy of patients with pituitary adenomas and hormone hypersecretion are discussed elsewhere and will not be discussed in this chapter. This pertains also for tumours secreting TSH and gonadotropins in vivo.
Subtypes of NFPAs
All tumours in patients in which there is no clinical evidence of enhanced activity of anterior pituitary hormones, are called non-functioning pituitary adenomas (NFPAs), representing a heterogeneous group of tumours (4). A minority of the NFPAs are called null cell adenomas, as no evidence of hormone production can be found using immunostaining or measuring secretory products in the medium when the tumour has been taken into culture after surgery. Most are positive for glycoproteins by in situ hybridisation. However, 80% of the NFPAs show at least positive immunostaining for one glycoprotein hormone, of which 80 to 90% are gonadotroph adenomas. There are also clinically silent somatotroph, lactotroph, and corticotroph adenomas, although these are less common. Non-functioning adenomas which are clinically endocrine inactive may still secrete α or β subunits in vivo (5).
It is of interest that even the null cell adenomas show on electron microscopy a few secretory granules with a diameter up to 250 nm. As noted above, it does seem likely that all NFPAs and null cell adenomas are therefore of gonadotrophin lineage.
Molecular Pathology of NFPAs-- Pathogenesis of pituitary adenomas
The pathogenesis of pituitary tumours, including NFPAs, is a multi-step process with genetic and epigenetic events conferring a growth advantage to a particular cell type which then increase by clonal expansion. These tumours are generally monoclonal, that is, they derive from a single abnormal cell, although there is some evidence to suggest that recurrent tumours may have a different clonal origin from the original tumour (5.1).
There is rapidly increasing evidence for the roles of molecular abnormalities in pituitary adenoma tumorigenesis, including dysregulation of the cell cycle, signal transduction pathways, oncogenes and tumour suppressor genes. The oncogene pituitary tumour transforming gene (PTTG) encodes a multifunctional protein that is implicated in the initiation and perpetuation of pituitary adenoma growth by its regulatory functions in controlling DNA repair and mitosis and gene. Its expression is increased in the majority of pituitary adenomas, and is positively correlated with tumour invasiveness and cell proliferation index Ki-67. One of the tumour suppressor genes which have been related to the pathogenesis of pituitary adenomas is GADD45γ, a P53-regulated human gene involved in growth suppression and apoptosis. While expressed in normal pituitary gland, GADD45γ is absent in the majority of pituitary adenomas (5.2)
There is increasing interest in the study of microRNA (miRNAs), which dysregulation has also been revealed in pituitary adenomas. miRNAs are a class of short noncoding RNAs that post-transcriptionally regulate the target RNAs translation and degradation, and an increasing literature has demonstrated a fundamental role of miRNAs in the pathogenesis of pituitary tumorigenesis (5.3)
Stimulatory factors either from the hypothalamus (releasing factors) or locally produced by the tumour cells themselves (growth factors) may act as progression factors rather than initiators of tumorigenesis.
Two signalling pathways have been implicated in NFPAs tumourigenesis: PI3K/Akt/mTOR/Zac1 and EGFR/Eps8/Raf/MEK/ERK. In all pituitary tumours, Akt has been found to be overexpressed as well as overactivated, but the cause and outcome of this activation requires further investigations. Related to this pathway in NFPAs, Zac1 (a zinc finger transcription factor inducing apoptosis and cell-cycle arrest) RNA and protein levels have been found to be reduced or absent, and a significant correlation has been found between low Zac1 expression and recurrence of NFPAs. Related to EGFR/Eps8/Raf/MEK/ERK pathway, overexpression of Eps8 (Epidermal growth factor receptor substrate), and overactivation of Raf, MEK and ERK, may promote proliferation and cell survival in NFPAs, and may sensitise cells to growth factor activation independent of receptor up-regulation. (6)
Clinical signs and symptoms
In the absence of hormone hypersecretion, NFPAs lead only to clinical symptoms when they cause visual field defects or endocrine disturbances. In very large tumours, frontal headaches as well as ophthalmoplegia due to lateral extension of the tumour and pressure on the 3rd, 4th and 6th nerves in the wall of the cavernous sinus, are common non endocrine symptoms. Concerning anterior pituitary failure, hypogonadism is the most frequently observed clinical endocrine dysfunction in adults (table 2). This hypogonadism may be due to compression of the normal pituitary or the pituitary stalk, which can cause hyperprolactinemia and contribute to the manifestation of hypogonadism by itself. Though menstrual disturbances in females and decline of libido and sexual potency in men are the first obvious clinical events, it is actually the impairment of growth hormone secretion which is the first anterior pituitary hormone deficiency (table 2). However, in contrast to children in whom GH deficiency (GHD) leads to growth failure, the symptoms of GHD in adults are more subtle and not easily recognized by clinical observation alone (7). Often these symptoms become apparent only when these patients have received substitution therapy with growth hormone, which changes their body composition and increases their quality of life (8).
Secondary hypothyroidism and adrenal failure occur only in the more advanced cases. However, the latter may become clinically important in acute stress situations like severe infection, haemorrhage and surgery. In these patients acute adrenal failure may develop though they had previously adequate adrenocortical function under basal conditions.
The diagnosis of NFPAs is made using imaging techniques such as MRI or CT scan, and endocrine evaluation. The latter serves to document the degree of pituitary insufficiency, and possible hypersecretion of biologically inactive hormones such as ɑ- or β-subunits (see chapter 3.3.). In addition, ophthalmological evaluation examining visual fields and function of ocular nerves must be preformed.
Table 2Prevalence of Hypopituitarism (%) in 26 patients with large NFPAs before and 2 - 3 months after transsphenoidal surgery.
| Hormone function | Preoperative (5) | Postoperative*(%) |
| GH-deficiency | 100 | 85 |
| Hypogonadism | 96 | 65 |
| Hypothyroidism | 81 | 35 |
| Adrenal insufficiency | 62 | 46 |
| * The improvement of hormone function after surgery was usually observed in those patients who had hyperprolactinemia and responded with a rise of the anterior pituitary hormone after administration of the respective releasing hormone. Both observations point to pituitary stalk compression as a cause of pituitary hormone deficiency (from Arafah, B. M.: J. Clin. Endoc. Metab., 62: 1173 - 9, 1986(9)). | ||
Therapy
Surgical resection of the tumour is the treatment of choice (10, 11). Tumour removal leads to normalization or at least improvement of visual field defect in those cases in which chiasma compression has occurred only recently and the optic papilla has not become atrophic. Whenever anterior pituitary failure was mainly caused by compression of the pituitary stalk, surgery may lead to improvement of anterior pituitary function (table 2). This is not the case when anterior pituitary failure was caused by compression of the pituitary gland. In this situation endocrine dysfunction is usually irreversible even when surgery has led to resolution of the visual field defects (10). In patients in whom the tumour has grown into the cavernous sinus or in other structures of the brain and could therefore not be totally resected, postoperative radiotherapy should be considered.
Radiotherapy (RDT) is not without its side effects which includes a high frequency of new hypopituitarism, if not already present. In a comparative study between 2 centres the one routinely employing post-operative radiotherapy to NFPA remnants had a much lower incidence of re-operation for recurrence (10.1). There has been concern whether pituitary RDT leads to a higher incidence of new brain tumours and visual impairment but this is minimal if at all (10.1). From epidemiological studies, patients with NFPAs who became hypopituitary do appear to have an increased mortality risk, so this may be a reason to personalise the indication for post-operative RDT. In selected patients with residual tumour laterally in the cavernous sinus away from the optic chiasma highly focused radiotherapy (gamma-knife) may be superior to conventional three field radiotherapy. The advantage of these techniques is the ability to deliver a higher radiation dose to a smaller field increasing the likelihood of tumour control and minimizing damage to residual normal pituitary tissue (12).
In relation to medical treatment, several options have been evaluated. Somatostatin receptors are widely expressed in NFPAs. All subtypes have been found to be present in these tumours, but the level and pattern of expression differ between studies, supporting the theory that NFPAs are a heterogeneous group of neoplasms. The results of somatostatin analogues (SSA) are not as satisfactory as might be expected, with a limited effect on tumour shrinkage, and only some results supporting the stabilisation of tumour remnant. As the EGFR pathway has been related to pathogenesis, EGFR inhibitors have also been investigated, but no study has reported its effects on NFPA cells (13).
The dopamine receptor D2 (D2R) is expressed in the majority of NFPAs, only showing very low expression in silent corticotrophinomas. Primary medical treatment with dopamine agonists has had disappointing outcomes, except for some scattered case reports. However, treatment in postoperative patients with residual tumour has promising results preventing residual tumour enlargement, with its effect depending on the quantity of D2Rs present (14).
PITUITARY CARCINOMA
Though many of the non functioning pituitary adenomas are invasive, they are not considered to be malignant as there are no metastases. However, the rare true pituitary carcinomas usually present as invasive macroadenomas in the early stage and only later develop metastases distant from the pituitary site (15). Occasionally local tumour growth may be massive, leading to destruction of the skull base and other vital centres causing early death of the patient not allowing the development of distant metastases (Fig. 2).

Figure 2
MRI of a 58 years old patient with an invasive, cystic giant prolactinoma, which has destroyed the base of the scull, caused blindness, and infiltrated the lateral ventricle. He did not respond to DA-agonists and died because of the local tumour complications, though he had no distant metastases, in the CNS or elsewhere
Using strict criteria for making the diagnosis of a pituitary carcinoma, i. e. presence of metastases, only about 100 cases have been reported. Particularly likely to become aggressive are the so-called ‘silent’ corticotroph adenomas which clinically present as NFPAs but immunostain positive for ACTH. These tumours may secrete precursors of ACTH in the circulation which cannot bind and activate the ACTH receptor in an effective way. This particular sub-group requires more frequent follow-up scanning than null-cell adenomas. Typically, pituitary cancers develop from invasively growing macroadenomas. They present with anterior pituitary failure or symptoms and signs of hormone hypersecretion, i.e. Cushing's syndrome, rarely acromegaly. In patients with prolactin secreting carcinomas the diagnosis of a macroprolactinoma had usually been made some years before. These tumours were often already partially dopamine agonist resistant and, in the course of the disease they lost responsiveness to dopamine agonists completely, though in some patients dopamine receptors can still be documented in vivo.
Malignancies in the pituitary fossa may also be metastases from breast, bronchial, renal, or gastric cancers leading to similar symptoms to those caused by NFPAs. Although many of these patients with metastases are asymptomatic and often diagnosed at autopsy, they may cause posterior pituitary failure.
Though patients with pituitary carcinomas have been treated aggressively with surgery, conventional and highly focussed radiotherapy, interstitial application of radioisotopes, and medical treatment including cytotoxic chemotherapy, the prognosis of these patients is overall unfavourable (15). Only temozolomide, an alkylating agent routinely used in the treatment of gliobastoma, has been reported to be of benefit in isolated cases (15.1). The outcome of the treatment may depend on the expression of the enzyme MGMT, which interferes with the drug efficacy, but because of the inconsistency of the data, its expression should not be taken as a reason to deny treatment (15.2)
BENIGN CYSTS
The most common non-neoplastic cystic lesions in the sellar/suprasellar region are Rathkes's pouch or cleft cysts, mucocoeles and arachnoid cysts, which all can cause headaches, visual disturbances and pituitary failure (16). Rathke's cleft cysts derive from remnants of Rathke's pouch and are pathologically indistinguishable from mucocoeles which are expansions from the paranasal sinuses. Arachnoid cysts develop from a duplication of the arachnoid and can develop anywhere in the brain, also intrasellarly and in the suprasellar area. Arachnoid cysts represent less than 1% of all space occupying lesions in the brain and occur mainly in children, whereas Rathke's cysts occur more frequently in adult females compared to males.
Typically, Rathke's cleft cysts lead to amenorrhea/galactorrhea by causing hyperprolactinemia due to impingement on the pituitary stalk and visual disturbances and headaches. Though MRI after gadolinium administration may lead to rim enhancement (Fig. 3), the diagnosis of an intrasellar Rathke's cleft cyst can not be made with absolute certainty. Thus, it is not unusual that in a patient operated for a presumed NFPA the diagnosis of Rathke's cleft cyst is made histologically after surgery.

Figure 3
MRI of a 34 years old patient with a surgically confirmed Rathke's cleft cyst. The MRI above suggests a NFPA, the enhanced rim seen after gadolinium administration (below) however, raises the suspicion of a cystic intrasellar, suprasellar extending lesion.
Treatment of choice is surgical total removal of the cyst (17). If this is not possible, drainage of the cyst content should be attempted. Particularly, in large mucocoeles drainage into the nasal sinuses can be achieved with good therapeutic results. Rathke's cleft cysts are related to craniopharyngiomas and epidermoid cysts concerning their origin. The latter two are mainly located in the suprasellar space, though intrasellar manifestations are also observed.
EMPTY SELLA SYNDROME
Empty sella syndrome is a condition in which the sella turcica is partially or completely filled with CSF resulting in a displacement of the normal pituitary gland. Usually the sella is enlarged and the pituitary gland is compressed. MRI scan may reveal CSF within the sella due to herniation of the sellar diaphragm with evagination of the third ventricle.
Empty sella syndrome is divided into primary or secondary. A primary empty sella syndrome can be due to transmission of intracranial pressure in the presence of an incompetent diaphragm and is often discovered accidentally since it is usually not associated with any clinical symptoms (18). A secondary empty sella syndrome may be caused by previous surgery or radiotherapy of a pituitary adenoma leading to shrinkage of the sella content. Auto-apoplexy of an intrasellar pituitary macroadenoma is also a possible cause of the empty sella syndrome (Fig. 4).

Figure 4
Empty sella: MRI of a 40 years old female patient with acromegalic features but a GH-level below 1 μg/l and an IGF-1-level below the normal age matched range. She had hypogonadotropic hypogonadism and her enlarged sellar cavity is filled with CSF with a rim of pituitary tissue, suggesting that she had an apoplexy of a somatotroph tumour.
HYPOPHYSITIS
Inflammatory lesions of the pituitary gland may clinically and radiologically mimic tumours of the sellar region, causing mass effects like headaches and visual impairment and endocrine symptoms such as hyperprolactinemia, diabetes insipidus, and anterior pituitary failure. Since CT- or MRI-scans produce images which are not significantly different from those of other space occupying lesions, the diagnosis of hypophysitis is often made only after transsphenoidal surgery has been performed (19).
We differentiate primary from secondary hypophysitis, the latter being composed of inflammatory lesions induced by defined pathogens or as a manifestation of distinct diseases such as sarcoidosis, Wegner's granulomatosis, etc. (see Chapter 2.5).
The aetiology of primary hypophysitis is not known, though an autoimmune pathology is most likely. The most common form of primary hypophysitis is lymphocytic hypophysitis, which occurs more frequently in females around late pregnancy or early postpartum. It has been speculated that pituitary failure occurring postpartum, which has been attributed to Sheehan's syndrome, may in fact have been caused by lymphocytic hypophysitis (20). IgG4-related disease is a newly recognised clinical entity that was first proposed in 2001. It involves various tissues and is associated with autoimmune pancreatitis, Riedel’s thyroiditis, interstitial pneumonitis, interstitial nephritis and others. It is characterised by IgG4C plasma cell and lymphocyte infiltration and an elevated serum IgG4 concentration, and should be considered in all cases presenting with hypophysitis since it responds well to treatment and also as this is part of a systemic disease. (21)
Several types of secondary hypophysitis have been described. An increasing diagnosis is lymphocytic hypophysitis secondary to ipilimumab theraphy, an immunomodulating drug used for the treatment of unresectable or metastatic malignant melanoma. As a clinical response to ipilimumab results from immunostimulation, it may generate autoimmunity as well, causing immune-related adverse events in many patients (22)
More uncommon are two other forms of hypophysitis. Granulomatous hypophysitis is a rare disorder which cases are diagnosed at autopsy. In addition to the endocrine symptoms, affected patients have clinical symptoms like nausea, vomiting, and meningeal irritation (19). Xanthomatous hypophysitis presents lipid rich foamy histiocytes resembling xanthomatous inflammatory processes, which occur elsewhere in the body.
Anterior pituitary failure occuring in patients with hypophysitis differs from that observed in patients with other space occupying lesions. In patients with hypophysitis secondary adrenal failure due to isolated ACTH deficiency, sometimes combined with TSH deficiency is the hallmark of anterior pituitary dysfunction, while growth hormone and gonadotropin secretion are often only slightly impaired (23). Occasionally, hypophysitis occurs in the area of the pituitary stalk and presents as infundibulitis, which invariably leads to clinically apparent diabetes insipidus (24).
Though conservative management may lead to resolution of clinical symptoms caused by hypophysitis, many patients have been subjected to transsphenoidal surgery because a pituitary tumour had been suspected. Transsphenoidal surgery seems to be indicated also in those patients in whom progression of visual field defects is noted or further derangement of pituitary function is observed. In those cases in which the diagnosis has been made on account of astute clinical observation, conservative management is justified. Spontaneous resolution of the abnormal endocrine function has been observed. In some patients glucocorticoid treatment has been shown to be effective to reduce the inflammatory process (19). However, since the development of the disease is unpredictable, patients with diagnosed or only suspected hypophysitis have to be followed up carefully.
SPACE OCCUPYING LESIONS IN THE HYPOTHALAMIC REGION
An enlarged pituitary fossa may also be caused by primary hypothalamic tumours extending into the pituitary fossa (table 1).
Hypothalamic tumours may extend into the pituitary fossa thereby leading to endocrine, visual, and neurological symptoms similar to those observed in patients with primary pituitary tumours. Furthermore, in contrast to primary pituitary tumours, hypothalamic tumours may cause posterior pituitary failure. Thus, diabetes insipidus is not seen in patients with pituitary tumours before surgery, though it is frequently observed in patients with primary suprasellar tumours in the pituitary stalk- or hypothalamic region (Fig. 1). Furthermore, hypothalamic tumours can cause disturbances of thirst control and dysregulation of osmolality, may lead to massive obesity by abnormal control of satiety, or to dysregulation of body temperature control. Extension of the hypothalamic tumour into upper parts of the brain may lead to blockage of the foramen of Monroe, internal hydrocephalus, and at the end, coma. Many hypothalamic tumours are developmental tumours, the most common is the craniopharyngioma followed by the rare germinomas, chordomas and hamartomas (Table 1). Furthermore, whereas metastases of solid cancers usually develop within the pituitary, systemic malignant diseases such as Hodgkin's disease, leukaemia, histiocytosis X (Langherhan's cell histiocytosis), and Wegner's granulomatosis manifest as space occupying lesions of the basal hypothalamus. In addition, benign granulomatous or infectious diseases such as neurosarcoidosis, tuberculosis, syphilis, and cysticercosis are rare causes of a suprasellar mass.
CRANIOPHARYNGIOMA
Craniopharyngioma is an intracranial tumour derived from remnants of Rathke's pouch. They account for about 1% of all intracranial tumours in adults and 10% of all intracranial tumours in children (25).
Though craniopharyngiomas are benign and grow rather slowly, they frequently infiltrate surrounding structures and are associated with increased mortality as well as physical and cognitive damages, and decreased quality of life, with a worse prognosis when diagnosed in children (26).
According to the histology, two craniopharyngioma subtypes can be differentiated: adamantinomatous (aCPs) and papillary (pCPs), the latter only seen in adults. Mutations have been reported in both subtypes. BRAF p.V600E mutations have been found in pCPs and mutations in CTNNB1, encoding β-catenin, have been found in aCPs. These mutations were thought to be mutually exclusive and specific to tumour subtype, but recently BRAF mutations have been found in aCPs, coexisting with mutations in CTNNB1 (27)
The clinical presentation of craniopharyngiomas in adults depends on the location of the tumour. Endocrine deficits are frequently present, and affect GH secretion (75%), gonadotropins (40%), ACTH (25%), and TSH (25%) (28). At the time of diagnosis, 40 to 87% of patients present with at least one hormonal deficit, and other endocrine symptoms such as neurohormonal diabetes insipidus are present preoperatively in 17 to 27% of patients (28). Patients with suprasellar invasion may also present neurological defects such as cranial nerve palsy, ataxia and convulsions. Raised intracranial pressure is mainly seen in children and has been associated with worse outcomes.
Intense calcifications present in aCPs can be detected by TC: MRI before and after gadolinium is the standard imaging for detection of craniopharingyomas, as it will show the extension of the space occupying lesion. The fluid in the cysts in craniopharyngiomas has been shown to contain high concentrations of immunoreactive b-hCG, which can also be found in the CSF, bringing up the differential diagnosis of germinomas, in which measurable b-hCG levels are often found in the CSF and occasionally also in the general circulation.
The treatment of craniopharyngiomas is with surgery, though particularly in children it is difficult to obtain radical removal of the tumour. After surgery, several radiation techniques have been proposed: stereotatic radiotherapy, radiosurgery and intracavitary instillation of α-emitting isotopes into cystic craniopharingyomas, causing destruction of the secretory epithelial lining, elimination of the fluid production and cyst shrinkage. The presence of BRAF mutations in the majority of pCPs offers the possibility for targeted BRAF-inhibitor therapy for patients with this tumour type.
Endocrine management is not different from those of other hypothalamic pituitary deficiencies. However, non-endocrine hypothalamic disturbances may be life threatening and extremely difficult to control. Among other morbidities, it is worth mentioning that obesity is paramount, and has to its complex pathophysiology and difficult management. The damage of the hypothalamus, either preoperatively by the tumour or surgery-related, seems to lead to a disruption of satiety system and reduced energy expenditure. Data regarding bariatric surgery with different techniques are scarce and contradictory, with modest results reported in some series (29)
GERM CELL TUMOURS
Germ cell tumours are differentiated into germinomas, which represent 65% of intracranial germ cell neoplasms, and non-germinomatous germ cell tumours (NGGCT) such as teratomas, embryonal carcinomas and chorioncarcinomas. The incidence of these neoplasms is rather low in western countries but is much higher in Japan and China
Germinomas and NGGCT differ with regard to their age of onset, location and prognosis. Germ cell tumours may secrete β-hCG and ɑ-fetoprotein which can be found in the CSF and the peripheral circulation (30). NGGCTs occur in children whereas germinomas are more frequently observed in adult males. Germ cell tumours are usually found in the region of third ventricle between the suprasellar space (mainly germinomas) and the pineal gland (mainly NGGCTs).
The clinical features of these tumours, which may spread by direct invasion or via the CSF into the ventricles or suparachnoid pathways down to the spinal cord, are anterior pituitary failure, diabetes insipidus, visual disturbances, and other neurological deficits depending on the size, location and histological tumour type. The clinical presentation in younger patients includes precocious puberty which may occur due to pressure on the median eminence or as a consequence of the elevated β-hCG levels which may directly stimulate Leydig cells of the testes.
The role of neurosurgery consists of biopsy and treatment of hydrocephalus if present. Radiotherapy is the treatment of choice after tumour extension has been delineated carefully by appropriate imaging techniques. Germinomas have a better prognosis than NGGCTs, where radiation therapy alone rarely is curative, and the combination with platinum-based chemotherapy has become the standard treatment in intracranial location. Chemotherapy is also used particularly in patients with wide spread disease whereas surgery is only reserved for those patients with radiotherapy resistant tumours (31).
A new study has found the KIT/RAS signalling pathway mutated in more than 50% of intracranial germ cell tumours as well as novel somatic mutations in the AKT/mTOR pathway, establishing a molecular foundation for understanding the biology of these tumours that suggests potentially promising therapeutic strategies focusing on the inhibition these pathways (32)
Where initially elevated, tumours markers can also be used for response evaluation (30).
CHORDOMA
Chordomas are very rare neoplasms representing less than 0,5 % of primary intracranial tumours. It is thought that the chordoma derives from remnants of the embryonic notocord, the precursor of the intraspinal nucleus pulposus which extends up to the sphenooccipital suture (33). They can present any time in life with symptoms of headaches, diplopia and palsies of the 6th, the 9th, the 10th and the 11th nerves. In addition, when the tumour mass is located in sellar area near the clivus, endocrine symptoms with hypogonadism, hypothyroidism and adrenal failure, hyperprolactinemia and diabetes insipidus may occur. Chordomas do not only occur intracranially. If they occur in the spinal cord they are more likely to give rise to distant metastases, which occur rarely when they are located in the pituitary hypothalamic area.
The diagnosis is made by MRI. A chordoma is suspected whenever the clivus is involved. However, the diagnosis of an isolated intrasellar chordoma is usually made only on account of the histology after surgery has been performed (34).
Surgery is the established treatment but the site of origin of the disease often prevents complete removal. Whereas surgery can be performed in purely intrasellar chordomas and in extra cranial sacral chordoma, larger skull base chordomas are treated with debulking surgery followed by RDT or exclusive highdose RDT. Systemic therapy is needed in patients not amenable to surgery or RDT, but up to the present, only some evidence about antitumour activity has been reported for imatinib (35) and lapatinib (36), with very modest results. The complex molecular profile of chordomas could be better dealt by the combination of different drugs, and some studies with sunitib and imatinib plus mTOR inhibitors are currently ongoing
Hypothalamic hamartomas (HH) are hyperplastic formations consisting of foci, of neurons and glia. HH are rare tumours which have profound influence on endocrine function since they may secret gonadotropin releasing hormone (GnRH) or growth hormone releasing hormone (GHRH). Since they occur exclusively in children they can cause precocious puberty and gigantism in addition to neurological complications such as intractable seizures. Diagnosis and treatment will be discussed in the chapter Pituitary - Hypothalamic Tumour Syndromes: Children.
MALIGNANT SYSTEMIC DISEASES OF THE CENTRAL NERVOUS SYSTEM
Hodgin's disease, lymphomas, leukaemic infiltration, Langerhan's cell histiocytosis (histiocystosis X), eosinophilic granuloma and giant cell tumours can occur in the suprasellar area. They lead to the classical triad of anterior pituitary failure, diabetes insipidus and visual disturbances, which can be accompanied with other neurological deficits and symptoms due to the original disease (37, 38, 39, 40). Treatment depends on the diagnosis, the extent of endocrine dysfunction, the space occupying lesion and staging of the disease. The reversibility of endocrine dysfunctions, i. e. hyperprolactinemia or diabetes insipidus can be used as an indicator for the effectiveness of the respective therapy.
NEUROSARCOIDOSIS AND OTHER GRANULOMATOUS DISEASES
Sarcoidosis of the nervous system can lead to peripheral and central manifestations (37, 39). When granulomatous disease in the CNS leads to local infiltration of brain substance and impedes the flow of CSF giving rise to increased intracranial pressure, diabetes insipidus, hyperprolactinemia and anterior pituitary failure as well as other hypothalamic disturbances like somnolence, disturbance of water balance, food intake, and temperature regulation are observed (41). Hypothalamic involvement is the most common feature of neurosarcoidosis though other neurological symptoms like seizures and psychiatric disturbances are also encountered. The diagnosis can not be made by MRI or CT alone. Determination of angiotensin converting enzyme is usual not helpful though analysis of the CSF may reveal an elevated protein content or an increase of CSF lymphocytes. Lymph node enlargement and pulmonary sarcoid disease are seldom present. Often the diagnosis is made only after a pituitary biopsy reveals the classical granulomas. Treatment of choice is prednisone (1,5 mg/kg body weight) which usually rapidly normalizes endocrine disturbances. However, other diseases should be excluded, before steroid treatment is considered (39).
Other rare differential diagnoses include hypophysitis (see above), infectious diseases at the base of the skull like tuberculosis, syphilis, fungal infections, and cysticercosis, and Wegner's granulomatosis which may all lead to endocrine and ophthalmological symptoms. In patients with HIV, manifestations of opportunistic infections may occur in the pituitary-hypothalamic area (42). CSF-examination may be helpful in making the differential diagnosis.
EVALUATION OF PATIENTS WITH PITUITARY/ HYPOTHALAMIC SPACE OCCUPYING LESIONS
There are 3 reasons why patients need special investigation of their pituitary/hypothalamic axis (Fig. 5).
- Disturbance of pituitary hormone secretion.
- Neurologic symptoms, such as headache or visual field defect
- Incidental finding of a sellar or suprasellar space occupying lesion by MRI or CT

Figure 5Evaluation and treatment of pituitary incidentaloma (43)
Insufficiency of GH secretion followed by secondary hypogonadism, followed by secondary thyroid and adrenal failure is the same regardless whether patients suffer from a primary pituitary or primary hypothalamic lesion. Typical for hypothalamic involvement however is diabetes insipidus and behavioural symptoms such as increased appetite with pathological weight gain, somnolence, disturbances of temperature regulation etc. Furthermore, the clinical manifestation of hypopituitarism depends not only on the type and the degree of hormone deficiency but whether it is an acute or chronic deficiency. Patients with chronic hypopituitarism are often hypogonadal and have partial secondary hypothyroidism and hypoadrenalism, whereas in acute pituitary failure, such as that caused by tumour apoplexy the clinical picture is dominated by the consequence of acute cortisol deficiency with severe headaches, hypotension, prostration and coma.
The diagnostic work-up of pituitary or hypothalamic tumour patients should therefore answer the following questions (Figure 5):
- What is the nature of the pituitary or hypothalamic space-occupying lesion?
- Is there hormone hypersecretion?
- Is there impairment of pituitary function?
- Is there involvement of the optic pathways with visual field defects or other cranial nerve abnormalities?
Imaging studies
The MRI with gadolinium is the imaging technique of choice. Gadolinium does not pass the intact blood-brain barrier, so enhances visualisation of normal tissues such us the pituitary which lack it. Adenomas, as well as other sellar masses such us craniopharyngiomas, usually take up gadolinium to a lesser degree than the normal pituitary. Compared to CT, MRI is clearly superior in showing the extension to neural tissue. Unlike CT however, MRI will not show the calcification associated with craniopharyngioma.
Ophthalmology
Since changes in vision are often early signs of tumour progression, ophthalmological investigations are useful in the primary diagnosis as well as during follow up. All patients presenting with a pituitary mass abutting the optic nerves or chiasm on MRI should undergo a formal visual field examination, computerised perimetry being a reliable tool.
Endocrine evaluation
Full assessment of pituitary function is needed in all patients with a pituitary hypothalamic tumour, evaluating hormone hypersecretion and hypopituitarism. The latter can often not be preformed by measuring basal hormones only, thus stimulation tests may be required (44).
With serum PRL levels over 5000 mU/L, the diagnosis is more likely to a prolactinoma. Moderately elevated serum PRL in the presence of a large space occupying lesion is more often due to stalk compression, but ideally the laboratory should measure levels in diluted serum to ensure that these are not falsely lowered by “hook effect”. If a macroprolactinoma is mistakenly diagnosed as a NFPA, it would have serious therapeutic consequences. In pituitary incidentalomas, IGF-1 should also be measured, with no clear recommendations about screening for glucocorticoid excess in asymptomatic patients. Related to hormone deficiency, basal cortisol, IGF-1, free T4 and TSH, gonadotrophin, oestradiol and testosterone should be measured, and if baseline test suggest a hypopituitarism, further stimulation test will be performed to evaluated IGF-1 or pituitary-adrenal axis.
The insulin stress test (IST), which stimulates ACTH, GH, and PRL secretion, is still the ‘gold standard’ for testing anterior pituitary function (45). However, this test should not usually be performed in patients more than 70 years of age, or those with coronary disease, uncontrolled hypertension or epilepsy, because of the high risk of complications. The short synacthen test with 250 mcg ACTH 1-24 is used in many centres as an alternative to the IST for assessment of the hypothalamic – pituitary – adrenal axis although it cannot assess growth hormone reserve, for which other tests are required. The rationale for this test is that an ACTH deficiency would lead to adrenal atrophy, since they have not been stimulated for a prolonged period. As a result of this atrophy, the adrenals will not be able to secrete cortisol normally in response to a bolus of ACTH. There is still debate about the accuracy of this test, its sensitivity and positive and negative predictive value, and it should not be used soon after surgery.
Posterior pituitary function is evaluated by measuring daily urine output and serum and urine osmolality, and is tested formally by the water deprivation/desmopressin test. Often partial diabetes insipidus becomes manifest only after appropriate hydrocortisone substitution therapy in ACTH deficient patients.
Supplementary test
TRH-test should not be done in a patient with a macroadenoma, because of serious side effects which have been reported, i. e. pituitary apoplexy, sudden visual loss, etc. (46). β-hCG and ɑ-fetoprotein in the peripheral circulation or the CSF may help to identify a suprasellar mass as a germ cell tumour.
Table 6Triad of symptoms in hypothalamic tumour patients
1. Hyperprolactinaemic anterior pituitary failure (at least hypogonadism) 2. Diabetes insipidus 3. Visual disturbances.
Therapy and follow-up
Pituitary hypothalamic space occupying lesions need to be treated, either by surgery, radiotherapy or both. In special situations medical therapy may be indicated.
A more complex problem is presented in pituitary incidentalomas. Clinical judgement is needed not only in determining the extent of evaluation but also in respect to therapy and follow-up. In the absence of visual field abnormalities and endocrine dysfunction, an expectant policy of no intervention and periodic follow-up is indicated. In the case of macroadenomas, a new MRI should be performed 6 months after the initial scan. In case of microadenomas, 12 months later seems to be more adequate. If no change, then MRI should be repeated every year during the next 3 years, and gradually less frequently thereafter. (43) Limited information suggests that this will occur rarely in patients with microadenomas but in up to 25% of larger tumours (1). Timing of surgical intervention depends on several factors including patient’s general medical fitness, development of headache or visual symptoms, or hypopituitarism.
Since patients with pituitary or hypothalamic tumours are rarely cured completely or at least harbour one or several hormonal deficits needing substitution therapy, lifelong follow-up of these patients is warranted.
REFERENCES
- Feldkamp, J., Santen, R., Harms, E., Aulich, A., Mödder, U., Scherbaum, W. A. (1999): Incidentally discovered pituitary lesions: high frequency of macroadenomas and hormone-secreting adenomas - results of a prospective study. Clinical Endocrinology, 51: 109 - 113.
- Thapar, K., Kovacs, K., Scheithauer, B. W., Lloyd, R. V. (eds): Diagnosis and management of pituitary tumors. Humana Press, Totowa; USA,, 2001.
- Kovacs, K. Scheithauer, B. W., Horvath, G., Lloyd, R. V. (1996): The World Health Organization classification of adenohypophysial neoplasms. Cancer, 78: 502 - 10.
- Katznelson, L., Alexander, J. M., Klibanski, A. (1993): Clinically non-functioning pituitary adenomas. J Clin Endocrinol Metab, 76: 1089 - 94.
- Samejima, N., Yamada, S., Takada, K., et. al. (2001): Serum alpha-subunit levels in patients with pituitary adenomas. Clinical Endocrinology, 54:479 - 84
5.1 Clayton RN & Farrell WE – Frontiers in Hormone Research 2004 32:186-204, Pituitary Tumor Clonality Revisited
5.2 Zhang X, Sun H, Danila DC, Johnson SR, Zhou Y, Swearingen B, Klibanski A. (2002): Loss of expression of GADD45 gamma, a growth inhibitory gene, in human pituitary adenomas: implications for tumorigenesis. J Clin Endocrinol Metab. 87(3):1262-7.
5.3 Li XH, Wang EL, Zhou HM, Yoshimoto K, Qian ZR. (2014): MicroRNAs in Human Pituitary Adenomas. International Journal of Endocrinology
- 6.
Rubinfeld H, Shimon I. (2012) PI3K/Akt/mTOR and Raf/MEK/ERK signaling pathways perturbations in non-functioning pituitary adenomas. Endocrine
. 42:285-91.
- 7.
Vance, M. L. (1994): Hypopituitarism. New England Journal of Medicine, 330: 1651 - 1662.
- 8.
Bengtsson B. A., Eden, S., Lonn, L., et al. (1993): Treatment of adults with growth hormone (GH) deficiency with recombinant human GH. J Clin Endocrinol Metab, 76: 309 - 17.
- 9.
Arafah, B. M. (1986): Reversible hypopituitarism in patients with large non-functioning pituitary adenomas. J Clin Endocrinol Metab, 62: 1173 - 9.
- 10.
Oyesiku, N., Tindall, G. T.: Endocrine-inactive adenomas: surgical results and prognosis. In: Landolt, A. M., Vance, M. L., Reilly, P. R. (eds) Pituitary adenomas, Churchill-Livingstone, New York, USA, 458 - 501, 1996 .
10.1 Gittoes NJ, Pituitary Radiotherapy Current Controversies, Trends Endo Metab 2005 16: 407-413
- 11.
Fahlbusch, R., Honegger, J. (1997): Extended trans-sphenoidal approach to the pituitary region and upper clivus. In: Torrens, M., Al-Mefty, O., Kobayashi, S. (eds.) Operative skull base surgery. Churchill Livingstone, New York, 69 - 87, 1997.
- 12.
Swords SM, Monson JP, Besser GM, Chew SL, Drake WM, Grossman AB, Plowman PN (2009). Gamma knifev radiosurgery: a safe and effective salvage treatment for pituitary tumors not controlled despite conventional radiotherapy. Eur. J . Endocrinol. 16:819-28.
- 13.
Fusco A, Giampietro A, Bianchi A, Cimino V, Lugli F, Piacentini S, Lorusso M, Tofani A, Perotti G, Lauriola L, Anile C, Maira G, Pontecorvi A, De Marinis L. (2012) Treatment with octreotide LAR in clinically non-functioning pituitary adenoma: results from a case-control study. Pituitary. 15:571-8.
- 14.
Gabalec F, Beranek M, Netuka D, Masopust V, Nahlovsky J, Cesak T, Marek J, Cap J. (2012) Dopamine 2 receptor expression in various pathological types of clinically non-functioning pituitary adenomas. Pituitary. 15:222-6.
- 15.
Kaltsas, G. A., Grossmann, A. B. (1998): Malignant pituitary tumors. Pituitary 1: 69-81
15.1. Hagen c, Schroeder HD, Hansen S, Hagen C, Andersen M, (2009) Temozolomide treatment of a pituitary carcinoma and two pituitary macroadenomas resistant to conventional therapy. Eur. J. Endocrinol. 16:631-7
15.2. Raverot G, Castinetti F, Jouanneau E, Morange I, Figarella-Branger D, Dufour H, Trouillas J, Brue T. (2012) Pituitary carcinomas and aggressive pituitary tumours: merits and pitfalls of temozolomide treatment. Clin Endocrinol, 76:769-75
- 16.
Pierre-Kahn, A., Capelle, L., Brauner, R. et al. (1990): Presentation and management of suprasellar arachnoid cysts. Review of 20 cases. J. Neurosurg. 73: 355-9.
- 17.
Ross, D. A., Norman, D., Wilson, G. B. (1992): Radiologie characteristics and results of surgical management of Rathke's cysts in 43 patients. Neurosurgery, 30: 173 - 9.
- 18.
Lambert, M., Gaillard, R. C., Vallotton, M. B., Megret, M., Delavelle, J. (1989): Empty sella syndrome associated with diabetes insipidus: case report and review of the literature. J Endocrinol. Invest., 12: 433 - 437.
- 19.
Cheung, C. C., Ezzat, S., Smyth, H. S., Asa, S. L. (2001): The spectrum and significance of primary hypophysitis. J Clin Endocrinol Metab, 86: 1048 - 1053.
- 20.
Asa, S. L., Bilbao, J. M., Kovacs, K., Josse, R. G., Kreines, K. (1981): Lymphocytic hypophysitis of pregnancy resulting in hypopituitarism: a distinct clicicopathologic entity. Ann Intern Med, 95: 166 - 171.
- 21.
Bando H, Iguchi G, Fukuoka H, Taniguchi M, Yamamoto M, Matsumoto R, Suda K, Nishizawa H, Takahashi M, Kohmura E, Takahashi Y. (2013) The prevalence of IgG4-related hypophysitis in 170 consecutive patients with hypopituitarism and/or central diabetes insipidus and review of the literature. Eur J Endocrinol. 170:161-72
- 22.
Juszczak A, Gupta A, Karavitaki N, Middleton MR, Grossman AB. (2012) Ipilimumab: a novel immunomodulating therapy causing autoimmune hypophysitis: a case report and review. Eur J Endocrinol. 167:1-5.
- 23.
Jenkins, P. J., Chew, S. L., Lowe, D. G., Afshar, F., Charlesworth, M., Besser, G. M., Wass, J. A. H. (1995): Lymphocytic hypophysitis: unusual features of a rare disorder. Clin Endocrinol, 42: 529 - 34.
- 24.
Imura, H., Nakao, K., Shimatsu, A., Ogawa, Y., Sando, T., Fujisawa, I., Yamabe, H. (1993): Lymphocytic infundibuloneuro-hypophysitis as a cause of central diabetes insipidus. N Engl J Med, 329: 683 - 689.
- 25.
Braunstein, G. D.: The hypothalamus. In: The pituitary. S. Melmed (Ed.) Blackwell, London, 309 - 340, 1995.
- 26.
Adamson, T. E., Wiestler, O. D., Kleihues, P., Yasargil, M. G. (1990): Correlation of clinical and pathological features in surgically treated craniopharyngiomas. J. Neurosurg., 73: 12 - 17.
- 27.
Larkin SJ, Preda V, Karavitaki N, Grossman A, Ansorge O. (2014) BRAF V600E mutations are characteristic for papillary craniopharyngioma and may coexist with CTNNB1-mutated adamantinomatous craniopharyngioma. Acta Neuropathol. 127:927-9
- 28.
Müller HL. (2014). Craniopharyngioma. Endocr Rev. 35:513-43.
- 29.
Gatta B, Nunes ML, Bailacq-Auder C, Etchechoury L, Collet D, Tabarin A. (2013) Is bariatric surgery really inefficient in hypothalamic obesity? Clin Endocrinol; 78(4):636-8.
- 30.
Arita, N., Ustico, Y., Hayakawa, T. et al. (1987): Role of tumour markers in the management of intracranial germ cell tumours. Prog Exp Tumor Res, 30: 289 - 95.
- 31.
Allen, J. C., Kim, J. H., Packer, R. J. (1987): Neoadjuvant chemotherapy for newly diagnosed germ cell tumours of the central nervous system. J Neurosurg, 67: 65 - 70.
- 32.
Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, Nakamura H, He Z, Doddapaneni H, Lewis L, Wang M, Suzuki T, Nishikawa R, Natsume A, Terasaka S, Dauser R, Whitehead W, Adekunle A, Sun J, Qiao Y, Marth G, Muzny DM, Gibbs RA, Leal SM, Wheeler DA, Lau CC. (2014) Novel somatic and germline mutations in intracranial germ cell tumours. Nature;511:241-5.
- 33.
Rich, T., Schiller, A., Suit, H., Mankin, H. (1985): Clinical and pathological review of 48 cases of chordoma. Cancer, 56: 182 - 7.
- 34.
Imura, H., Kato, Y., Nakai, Y. (1987): Endocrine aspects of tumours arising from suprasellar, third ventricle regions. Prog Exp Tumor Res, 30: 313 - 24.
- 35.
Stacchiotti S, Longhi A, Ferraresi V, Grignani G, Comandone A, Stupp R, Bertuzzi A, Tamborini E, Pilotti S, Messina A, Spreafico C, Gronchi A, Amore P, Vinaccia V, Casali PG. (2012) Phase II study of imatinib in advanced chordoma. J Clin Oncol. 20;30:914-20
- 36.
Stacchiotti S, Tamborini E, Lo Vullo S, Bozzi F, Messina A, Morosi C, Casale A, Crippa F, Conca E, Negri T, Palassini E, Marrari A, Palmerini E, Mariani L, Gronchi A, Pilotti S, Casali PG. (2013) Phase II study on lapatinib in advanced EGFR-positive chordoma. Ann Oncol. 24:1931-6
- 37.
Delany, P. (1977): Neurologixal manifestations in sarcoidosis. Ann Intern Med, 87: 336 - 45.
- 38.
Braunstein, G. D., Kohler, P. O. (1981): Endocrine manifestations of histiocystosis . Am J Pediatr Hematol Oncol, 3: 65 - 75.
- 39.
Chapelon, C., Ziza, J. M., Piette, J. C., Levy, Y., Raguin, G., Wechsler, B., Bitker, M. O., Bletry, O., Laplane, D., Bousser, M. G. (1990): Neurosarcoidosis: signs, course and treatment in 35 confirmed cases. Medicine, 69: 261 - 276.
- 40.
Lohr, K. M., Ryan, L. M., Toohill, R. J., Anderson, T. (1988): Anterior pituitary involvement in Wegener's granulomatosis. J Rheumatol, 15: 855 - 857.
- 41.
Bullmann, C., Faust, M., Hoffmann, A., Heppner, C., Jockenhövel, F., Müller-Wieland, D., Krone, W. (2000): Five cases with central diabetes insipidus and hypogonadism as first presentation of neurosarcoidosis. Eur J Endocrinol, 142: 365 - 372.
- 42.
Sano, T., Kovacs, K., Scheithauer, B. W., Rosenblum, M. K., Petito, G. K., Greco, C. M. (1989): Pituitary pathology in acquired immunodeficiency syndrome. Arch Pathol Lab Med, 113: 1066 - 1070.
- 43.
Freda PU, Beckers AM, Katznelson L, Molitch ME, Montori VM, Post KD, Vance ML; Endocrine Society. (2011) Pituitary incidentaloma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab; 96:894-904
- 44.
von Werder, K. (1996): Endocrine investigation. Pituitary enlargement. Clin. Endocrinol (Oxf) 44: 299 - 303.
- 45.
Erturk, E., Jaffe, C. A., Barkan, A. L. (1998): Evaluation of the integrity of the hypothalamic-pituitary- adrenal axis by insulin hypoglycemia tests. J Clin Endocrinol Metab, 83: 2350 - 4.
- 46.
Otsuka, F., Kageyama, J., Ogura, T., Makino, H. (1998): Pituitary apoplexy induced by a combined anterior pituitary test: case report and literature review. Endocr J, 45: 393 - 821
- ABSTRACT
- INTRODUCTION
- SPACE OCCUPYING LESIONS OF THE PITUITARY
- NON-FUNCTIONING PITUITARY ADENOMAS
- PITUITARY CARCINOMA
- BENIGN CYSTS
- EMPTY SELLA SYNDROME
- HYPOPHYSITIS
- SPACE OCCUPYING LESIONS IN THE HYPOTHALAMIC REGION
- CRANIOPHARYNGIOMA
- GERM CELL TUMOURS
- CHORDOMA
- MALIGNANT SYSTEMIC DISEASES OF THE CENTRAL NERVOUS SYSTEM
- NEUROSARCOIDOSIS AND OTHER GRANULOMATOUS DISEASES
- EVALUATION OF PATIENTS WITH PITUITARY/ HYPOTHALAMIC SPACE OCCUPYING LESIONS
- REFERENCES
- Review [Space occupying processes of the sellar region with emphasis on tumor-like lesions].[Pathologe. 2003]Review [Space occupying processes of the sellar region with emphasis on tumor-like lesions].Saeger W. Pathologe. 2003 Jul; 24(4):247-54. Epub 2003 May 29.
- Review Nonneuroendocrine Neoplasms of the Pituitary Region.[J Clin Endocrinol Metab. 2019]Review Nonneuroendocrine Neoplasms of the Pituitary Region.Kaltsas GA, Kolomodi D, Randeva H, Grossman A. J Clin Endocrinol Metab. 2019 Aug 1; 104(8):3108-3123.
- Review Clinical features and differential diagnosis of pituitary tumours with emphasis on acromegaly.[Baillieres Clin Endocrinol Met...]Review Clinical features and differential diagnosis of pituitary tumours with emphasis on acromegaly.Hennessey JV, Jackson IM. Baillieres Clin Endocrinol Metab. 1995 Apr; 9(2):271-314.
- Review A clinical approach to parasellar lesions in the transition age.[J Neuroendocrinol. 2021]Review A clinical approach to parasellar lesions in the transition age.Sbardella E, Puliani G, Feola T, Pofi R, Pirchio R, Sesti F, Verdecchia F, Gianfrilli D, Moffat D, Isidori AM, et al. J Neuroendocrinol. 2021 Jun; 33(6):e12995.
- Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.[Cochrane Database Syst Rev. 2022]Folic acid supplementation and malaria susceptibility and severity among people taking antifolate antimalarial drugs in endemic areas.Crider K, Williams J, Qi YP, Gutman J, Yeung L, Mai C, Finkelstain J, Mehta S, Pons-Duran C, Menéndez C, et al. Cochrane Database Syst Rev. 2022 Feb 1; 2(2022). Epub 2022 Feb 1.
- Pituitary-Hypothalamic Tumor Syndromes: Adults - EndotextPituitary-Hypothalamic Tumor Syndromes: Adults - Endotext
- Mus musculus galactosamine (N-acetyl)-6-sulfatase (Galns), transcript variant 2,...Mus musculus galactosamine (N-acetyl)-6-sulfatase (Galns), transcript variant 2, mRNAgi|302370950|ref|NM_001193645.1|Nucleotide
- Mus musculus galactosamine (N-acetyl)-6-sulfate sulfatase (Galns), mRNAMus musculus galactosamine (N-acetyl)-6-sulfate sulfatase (Galns), mRNAgi|31980653|ref|NM_016722.2|Nucleotide
- Homo sapiens kinesin family member 5A (KIF5A), transcript variant 1, mRNAHomo sapiens kinesin family member 5A (KIF5A), transcript variant 1, mRNAgi|1519243698|ref|NM_004984.4|Nucleotide
- biotin-dependent carboxyltransferase family protein [Enterocloster bolteae]biotin-dependent carboxyltransferase family protein [Enterocloster bolteae]gi|488630228|ref|WP_002566917.1|Protein
Your browsing activity is empty.
Activity recording is turned off.
See more...