By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2000, Geoffrey M Cooper.
Bookshelf ID: NBK9953

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Cooper GM. The Cell: A Molecular Approach. 2nd edition. Sunderland (MA): Sinauer Associates; 2000.

Lysosomes are membrane-enclosed organelles that contain an array of enzymes capable of breaking down all types of biological polymers—proteins, nucleic acids, carbohydrates, and lipids. Lysosomes function as the digestive system of the cell, serving both to degrade material taken up from outside the cell and to digest obsolete components of the cell itself. In their simplest form, lysosomes are visualized as dense spherical vacuoles, but they can display considerable variation in size and shape as a result of differences in the materials that have been taken up for digestion (Figure 9.34). Lysosomes thus represent morphologically diverse organelles defined by the common function of degrading intracellular material.

Electron micrograph of lysosomes and mitochondria in a mammalian cell. Lysosomes are indicated by arrows. (Visuals Unlimited/K. G. Murti.)
Lysosomes contain about 50 different degradative enzymes that can hydrolyze proteins, DNA, RNA, polysaccharides, and lipids. Mutations in the genes that encode these enzymes are responsible for more than 30 different human genetic diseases, which are called lysosomal storage diseases because undegraded material accumulates within the lysosomes of affected individuals. Most of these diseases result from deficiencies in single lysosomal enzymes. For example, Gaucher’s disease (the most common of these disorders) results from a mutation in the gene that encodes a lysosomal enzyme required for the breakdown of glycolipids. An intriguing exception is I-cell disease, which is caused by a deficiency in the enzyme that catalyzes the first step in the tagging of lysosomal enzymes with mannose-6-phosphate in the Golgi apparatus (see Figure 9.25). The result is a general failure of lysosomal enzymes to be incorporated into lysosomes.
All of the lysosomal enzymes are acid hydrolases, which are active at the acidic pH (about 5) that is maintained within lysosomes but not at the neutral pH (about 7.2) characteristic of the rest of the cytoplasm (Figure 9.35). The requirement of these lysosomal hydrolases for acidic pH provides double protection against uncontrolled digestion of the contents of the cytosol; even if the lysosomal membrane were to break down, the released acid hydrolases would be inactive at the neutral pH of the cytosol. To maintain their acidic internal pH, lysosomes must actively concentrate H+ ions (protons). This is accomplished by a proton pump in the lysosomal membrane, which actively transports protons into the lysosome from the cytosol. This pumping requires expenditure of energy in the form of ATP hydrolysis, since it maintains approximately a hundredfold higher H+ concentration inside the lysosome.
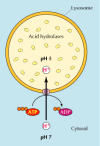
Organization of the lysosome. Lysosomes contain a variety of acid hydrolases that are active at the acidic pH maintained within the lysosome, but not at the neutral pH of the cytosol. The acidic internal pH of lysosomes results from the action of a proton (more...)
One of the major functions of lysosomes is the digestion of material taken up from outside the cell by endocytosis, which is discussed in detail in Chapter 12. However, the role of lysosomes in the digestion of material taken up by endocytosis relates not only to the function of lysosomes but also to their formation. In particular, lysosomes are formed by the fusion of transport vesicles budded from the trans Golgi network with endosomes, which contain molecules taken up by endocytosis at the plasma membrane.
The formation of lysosomes thus represents an intersection between the secretory pathway, through which lysosomal proteins are processed, and the endocytic pathway, through which extracellular molecules are taken up at the cell surface (Figure 9.36). Material from outside the cell is taken up in clathrin-coated endocytic vesicles, which bud from the plasma membrane and then fuse with early endosomes. Membrane components are then recycled to the plasma membrane (discussed in detail in Chapter 12) and the early endosomes gradually mature into late endosomes, which are the precursors to lysosomes. One of the important changes during endosome maturation is the lowering of the internal pH to about 5.5, which plays a key role in the delivery of lysosomal acid hydrolases from the trans Golgi network.
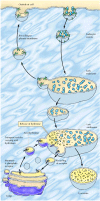
Endocytosis and lysosome formation. Molecules are taken up from outside the cell in endocytic vesicles, which fuse with early endosomes. Membrane components are recycled as the early endosomes mature into late endosomes. Transport vesicles carrying acid (more...)
As discussed earlier, acid hydrolases are targeted to lysosomes by mannose-6-phosphate residues, which are recognized by mannose-6-phosphate receptors in the trans Golgi network and packaged into clathrin-coated vesicles. Following removal of the clathrin coat, these transport vesicles fuse with late endosomes, and the acidic internal pH causes the hydrolases to dissociate from the mannose-6-phosphate receptor (see Figure 9.36). The hydrolases are thus released into the lumen of the endosome, while the receptors remain in the membrane and are eventually recycled to the Golgi. Late endosomes then mature into lysosomes as they acquire a full complement of acid hydrolases, which digest the molecules originally taken up by endocytosis.
In addition to degrading molecules taken up by endocytosis, lysosomes digest material derived from two other routes: phagocytosis and autophagy (Figure 9.37). In phagocytosis, specialized cells, such as macrophages, take up and degrade large particles, including bacteria, cell debris, and aged cells that need to be eliminated from the body. Such large particles are taken up in phagocytic vacuoles (phagosomes), which then fuse with lysosomes, resulting in digestion of their contents. The lysosomes formed in this way (phagolysosomes) can be quite large and heterogeneous, since their size and shape is determined by the content of material that is being digested.
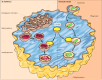
Lysosomes in phagocytosis and autophagy. In phagocytosis, large particles (such as bacteria) are taken up into phagocytic vacuoles or phagosomes. In autophagy, internal organelles (such as mitochondria) are enclosed by membrane fragments from the ER, (more...)
Lysosomes are also responsible for autophagy, the gradual turnover of the cell’s own components. The first step of autophagy appears to be the enclosure of an organelle (e.g., a mitochondrion) in membrane derived from the ER. The resulting vesicle (an autophagosome) then fuses with a lysosome, and its contents are digested (see Figure 9.37). As discussed in Chapter 7, autophagy is responsible for the gradual turnover of cytoplasmic organelles.
Molecular Medicine: Gaucher's Disease.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...