By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Copyright © 2003, BC Decker Inc.
Bookshelf ID: NBK12607

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Kufe DW, Pollock RE, Weichselbaum RR, et al., editors. Holland-Frei Cancer Medicine. 6th edition. Hamilton (ON): BC Decker; 2003.

Benign pediatric ocular lesions are very rare. Choroidal nevi, which are present in more than 10% of the adult population, are rare before puberty and are never seen in the infant. Conjunctival and iris nevi are also extremely rare in prepubertal children. Iris nevi detected in children often represent Lisch nodules, a manifestation of neurofibromatosis type 2.
Benign retinal tumors are also rare. When found they are usually astrocytic hamartomas and are frequently part of the tuberous sclerosis syndrome. When viewed with indirect ophthalmoscopy, astrocytic hamartomas usually have a thin, transparent membrane overlying the retina and typically obscure retinal blood vessels. They may enlarge and calcify with time. They may be confused with myelinated nerve fibers which are white, follow the distribution of the nerve fiber layer, and obscure retinal vessels.
Hamartomas of the retinal pigment epithelium are rare in children. They are frequently near the optic disc and are pigmented, with distortion of retinal vessels and a slightly opaque appearance. They have no malignant potential.
The most common primary ocular malignancy of childhood is retinoblastoma.1 Even more common, however, is secondary ocular involvement from leukemia. Retinoblastoma arises from an unidentified retinal progenitor cell. Although retinoblastoma is relatively rare, it has been the subject of great interest because of its well-studied genetic inheritance pattern and molecular biology.2
Retinoblastoma occurs in 1 in 18,000 to 30,000 live births worldwide. Surveys suggest a relatively constant occurrence in this century.3 The incidence in the United States is relatively low, at 3.58 cases for each million children younger than age 15 years, and is closely correlated with age. For ages 1 to 4 years, the incidence is 10.6 per million; for 5 to 9 years, 1.53 per million; and for 10 to 14 years, 0.27 per million. It is the seventh most common pediatric cancer, and in some countries (eg, Mexico), it is the most common solid tumor seen in children.
There are no differences in incidence by sex, race, or right versus left eye.4 Some data suggest geographic clustering, but convincing evidence is lacking. Retinoblastoma does appear to occur more commonly in poor patients worldwide. Preliminary evidence suggests that the reason for this predilection is that the presence of human papillomavirus (HPV) sequences in retinoblastoma tumor tissue may play a role in the development of sporadic retinoblastoma.5
Retinoblastoma occurs in two forms, germinal and nongerminal. In germinal cases, both eyes are usually affected and the mean number of tumors distributed between the two eyes is five. All patients with bilateral retinoblastoma are believed to have a germinal mutation of the rb1 gene on chromosome 13, although only 8% have an antecedent family history of the disease. Approximately 15% of patients with germinal retinoblastoma have only one eye involved. When patients with germinal retinoblastoma have unilateral disease, it is almost always multifocal.
Nongerminal retinoblastoma is always unilateral and unifocal, although lack of tumor cohesiveness may cause the tumor to break apart, resulting in hundreds of tiny intraocular seeds. Table 85-2 schematically presents genetic counseling based on these factors.
Genetic Counseling for Retinoblastoma.
Retinoblastoma was one of the prototypical models demonstrating the genetic etiology of cancer. Hereditary retinoblastoma presents in a mendelian autosomal dominant pattern with 90% penetrance, but only 10% of patients typically have a family history of the disease. Knudson proposed the now classic two-hit model, in 1971, after noting that the timing of tumor development (earlier diagnosis of bilateral tumors than of unilateral tumors) suggested a mechanism in which at least two events would be responsible for the development of the tumor.6 Patients with multifocal bilateral disease are germ line carriers for the first hit, with only one second hit necessary to develop retinoblastoma. Unilateral unifocal patients usually have a normal germline genome, but develop both hits in the progenitor tumor cell.
The nature of these hits was suggested by karyotypic abnormalities on the long arm of chromosome 13 in tumors. In 1986, Friend and colleagues isolated the rb1 gene, located on the long arm of chromosome 13, band 14.2.7 Further characterization of the gene revealed that it spans 200 kilobases (kb) and is composed of 27 exons. The gene encodes a 4.7-kb messenger ribonucleic acid (mRNA) transcript which is expressed in all adult tissues. The 110-kilodalton (kDa) nuclear phosphoprotein consists of 928 amino acids.
The protein is a regulator at the cell-cycle checkpoint between G1 and entry into the S-phase. The phosphorylation pattern of p110 RB varies during the cell cycle and the current model suggests that the unphosphorylated normal RB1 protein binds transcriptional regulators that promote entry into the S-phase (Figure 85-1). When the normal RB1 protein is phosphorylated, it dissociates from E2F (one of these transcription factors) freeing it to bind to deoxyribonucleic acid (DNA) and stimulate transcription of downstream genes that promote progression through the cell cycle. Loss of normal rb1 function, as in the case of the tumors, presumably allows for uncontrolled entry into the S-phase, more rapid cell cycling, and rapid cell division.

Artistic rendition of molecular mechanism of retinoblastoma gene action.
Because it is the loss of function of the rb1 gene that provides the impetus for malignant transformation, the gene falls into the category of tumor-suppressor genes. Retinoblastoma research has provided much of the initial understanding of this class of oncogenes. Whether replacing nonfunctional RB1 protein in an already fully transformed tumor cell with normal RB1 protein is sufficient to reverse the malignant state is still controversial.8 Much evidence does indicate that although rb1 gene mutations are a necessary first step in tumorigenesis, additional steps are also required for the development of retinoblastoma to occur, in which case, such a protein replacement strategy should fail to reverse the malignant state.
The rb1 gene is expressed in all adult tissues, and the fact that the RB1 protein plays such a vital role in controlling cell proliferation is evidenced by its frequently mutated state in human tumors of all types. Specific cell types initiate expression of the protein at different times during development. In developing retinal progenitor cells, it appears that expression of the RB1 protein may occur as the cells undergo terminal differentiation, and it is at that time that the retinal tissue is at risk for tumor development if the RB1 protein is nonfunctional.9 Cells in other tissues of patients who carry the germline mutation are at risk for uncontrolled growth when patients are significantly older, as evidenced by the development of second nonocular cancers as late as 40 years after the initial development of retinoblastoma.
Additional studies designed to better understand the function of the normal RB1 protein have included the creation of knockout mice that have been genetically engineered to lack the normal rb1 gene in their germ line.10 Heterozygotes are normal at birth but have a propensity to develop pituitary tumors. Notably, these animals do not develop retinoblastomas. Animals homozygously lacking rb1 do not survive to birth, dying by embryonic day 16 because of hematopoietic and nervous system abnormalities.
Several groups of investigators have begun to use single-stranded conformational polymorphism analysis and DNA sequencing to identify specific mutations found in the rb1 gene in retinoblastoma tumors and in the germ line of patients with the hereditary form. Although only limited amounts of data have been published to date, it seems that the majority of first hits are point mutations, although the remainder are small deletions.11 Only approximately 2% to 3% of tumors demonstrate karyotypically visible larger deletions of material in the 13q14 band. Some of the children with this cytogenetic abnormality in their germ line have a constitutional disease with severe developmental delay and dysmorphic features, although others are phenotypically normal other than their retinoblastoma. The second hit consists of loss of the normal allele (loss of heterozygosity) in 60% of cases, although the remainder of the patients may have a second distinct point mutation, and an even smaller fraction of patients have a second distinct small deletion.12
Mutations in the rb1 gene seem to occur throughout the gene. The majority of the mutations are nonsense, introducing a premature stop codon and therefore producing a truncated and presumably nonfunctional protein.
Evidence that retinoblastoma, like other cancers such as colon carcinomas and glial brain tumors, requires other genetic abnormalities to occur prior to transformation into malignancy has stimulated much work focused on genetic abnormalities other than mutations in the rb1 gene. A rare benign clinical entity called a retinoma is thought to be the result of the loss of the rb1 gene without the subsequent acquisition of other mutations that are required for progression to the malignant state. It is unclear what these subsequent steps in the development of the retinoblastoma tumor are, but cytogenetic studies have revealed some consistent abnormalities that provide clues. Squire and colleagues noted that gross aneuploidy of chromosome 1q (1q1) was present in 21 of 27 tumors studied.13 Additionally, a cytogenetic abnormality specific to the retinoblastoma tumor was noted, isochromosome 6p, or 6p1, in 15 of 27 cases. Comparative genomic hybridization studies further demonstrated that tumors that contain one or both of these two cytogenetic abnormalities are more likely to contain additional genetic alterations than tumors that do not contain either abnormality.14 This finding suggests that chromosomal instability may be associated with the acquisition of these changes. Which genes at these two loci are responsible for malignant transformation is at present unclear.
Other known oncogenes and tumor-suppressor genes have been investigated in retinoblastoma tumors. Kato studied 25 tumors and found no evidence of p53 mutations in any of the primary tumors.15 The histologic similarity between retinoblastoma and neuroblastoma prompted Doz and colleagues to investigate whether two genetic abnormalities common in stage 4 neuroblastoma, n-myc amplification and loss of material from the short arm of chromosome 1, were also common in retinoblastoma.16 They determined that only 1 of 45 retinoblastoma tumors had amplification of the n-myc protooncogene. They also found that only 9 of 43 primary retinoblastoma tumors had loss of heterozygosity of 1p. However, a higher proportion of distant metastases had this mutation, suggesting that a tumor-suppressor gene on this arm may contribute to the metastatic potential of retinoblastoma.
Although our understanding of the molecular biology of retinoblastoma is impressive, none of our progress to date helps explain the reason that an rb1 mutation occurs in the 90% of patients who have no antecedent family history. It is therefore difficult in the clinical setting to explain to most patients' families why their children have developed this disease. Future discoveries that may explain the mechanisms responsible for the gene mutations are eagerly awaited.
The presenting signs and symptoms of retinoblastoma vary depending on the geographic location in which the child presents. In developing countries, children often develop extraocular disease with proptosis and an orbital mass before they are diagnosed. In these cases, the retinoblastoma often extends directly into the orbit, causing rupture of the globe (Figure 85-2). Regional nodal metastasis may be found in the preauricular or submandibular regions. These children are older at diagnosis (age 4 to 6 years) than patients in the United States, and few survive. In the United States, most children present with intraocular disease by age 2 years and are diagnosed with signs rather than symptoms.
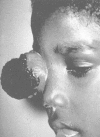
Advanced orbital presentation of retinoblastoma. Courtesy of A. Wachtel, MD, Lima, Peru. (Four-color version of figure on CD-ROM)
In the United States, the most common presenting sign (60% of cases) is leukocoria, a white pupillary reflex or cat's-eye reflex (Figure 85-3).17 The reflex is caused by tumor itself in the vitreous or by the retinal detachment caused by the underlying tumor.
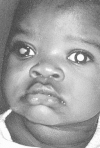
Leukocoria (white pupillary reflex) caused by retinoblastoma. The tumor can be seen in the vitreous. There are seeds in the anterior chamber, anterior to the iris. (Four-color version of figure on CD-ROM)
The second most common sign is strabismus, misalignment of the two eyes. Of the 20% to 25% of patients that present with this sign, two-thirds have eyes crossed in (esotropia) and one-third have eyes crossed out (exotropia).17 Esotropia in children in general is more common than exotropia, such that an infant with exotropia must be suspected to have retinoblastoma until proven otherwise. The crossed eyes are caused by tumor or retinal detachment in the area of central vision, the macula. Although patient survival is independent of whether patients present with strabismus or leukocoria, ocular survival rates are significantly lower for patients who present with leukocoria.18
The third most common sign (6% to 10% of cases) is painful glaucoma with inflammatory signs. These children may present to the pediatric emergency room with a presentation that resembles orbital cellulitis. The patients appear systemically ill with irritability, failure to eat, and, rarely, low-grade fever. Although the clinical exam and diagnostic imaging may suggest extraocular disease, the patients usually have only intraocular disease, frequently with massive necrosis and glaucoma.
Presenting signs in the United States that occur in fewer than 5% of cases include anisocoria (different-sized pupils), heterochromia (different- colored irides), hyphema (blood in the anterior chamber), tumor hypopyon (tumor in the anterior chamber), and nystagmus. Only 7% of patients are detected on routine pediatric screening by a pediatrician, whereas 72% of cases are detected by a family member or friend.18
When retinoblastoma is detected in patients 6 months old or younger, patterns of presentation are different.19 Sixty percent of such patients have no signs or symptoms, but are instead examined because of a family history of retinoblastoma. These examinations have a significant impact on ocular outcome. Patients who began undergoing screening examinations at our center as newborns because of a family history had a 68% ocular 5-year survival rate, whereas nonscreened patients with a family history had only a 38% ocular survival.18
The anatomic location of these tumors within the retina and the age at which the lesions develop have been well studied (Figure 85-4). Retinoblastoma may be present anywhere in the retina at birth, but as age at tumor development increases, tumors develop progressively farther into the periphery. Thus macular tumors present earliest and peripheral anterior tumors present later. In our center, the average macular tumor is diagnosed at 5.6 months and never presents after 15.5 months. In contrast, peripheral tumors are diagnosed at an average of 16.4 months and can present as late as 8 years of age.
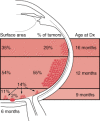
Artistic rendition of relationship between age at diagnosis, surface area of involved retina, and location of intraocular foci of retinoblastoma.
The diagnosis of retinoblastoma is usually made on examination of a child with the indirect ophthalmoscope with scleral indentation under anesthesia if necessary.20 Early in development, tumors appear on exam as glassy hemispheres in one or multiple sites bulging from the retina. With time tumors become pink and vascularized. As the tumors grow, they may detach the retina and/or break apart into pieces, causing characteristic seeds within the vitreous and beneath the retina. At certain stages of disease progression, retinoblastoma may resemble other nonmalignant ophthalmic conditions. Extensive lists of these simulating lesions, called pseudogliomas in ophthalmic texts, are presented elsewhere.21
The Reese- Ellsworth Classification scheme is the most commonly used classification system for describing intraocular tumors.22 It is not a true staging scheme, for untreated patients do not progress from group I to higher groups, but it has served as an excellent ocular reference for comparison of different series and treatment schemes. Because that scheme does not include patients with extraocular disease, we have created a classification scheme for these patients. Table 85-3 outlines both schemes.
Intraocular and Extraocular Classification of Retinoblastoma.
Table 85-4 presents the most common and challenging lesions to differentiate from retinoblastoma. Astrocytic hamartomas may be seen in children with the syndrome of tuberous sclerosis or as incidental findings. If they have the syndrome, the children may have intracranial calcifications, seizures, delayed development, mental retardation, dermal adenoma sebaceum, and characteristic ash-leaf skin findings. With time astrocytic hamartomas may become more defined, increase in number, and calcify in part or completely. They require no treatment.
Lesions Simulating Retinoblastoma.
The lesions of Toxocara canis represent the retinal eosinophilic abscess(es) of presumably dead, migrated second stage larvae of Toxocara canis, the same round worm that causes the clinical syndrome of visceral larval migrans (VLM). Curiously, intraocular lesions are usually not found in the patients who have VLM. Children are diagnosed at an average age of 6 years (later than retinoblastoma) and get the disease by ingesting feces from young puppies. Toxicara canis is a congenital infection of dogs and is more prevalent in warmer parts of the United States.
Coats disease is the ocular condition that most commonly mimics retinoblastoma. It is a unilateral (90%) retinal vascular anomaly of boys (80%) characterized in early stages by localized telangiectasias of the retinal vasculature. With time these vessels leak a macrophage-laden exudate rich in cholesterol crystals, which causes a retinal detachment simulating retinoblastoma. Although it is not a malignancy, Coats disease usually blinds the affected eye and sometimes causes a painful glaucoma for which patients often require enucleation.
Retinopathy of prematurity (ROP), formerly called retrolental fibroplasia (RLF), is a cicatricial disease of the vitreous and retina most commonly seen in low-birth-weight children who received oxygen at birth. It has also been seen in term babies who required no oxygen. It is usually bilateral, with bilateral microphthalmos (small eyes) and myopia.
Persistent hyperplastic primary vitreous (PHPV) is a congenital anomaly that includes combinations of the following findings: unilateral microphthalmos, a vascularized membrane behind the lens simulating a cataract, abnormal iris blood vessels, a vascular stalk extending from the optic nerve to the back of the retrolental membrane, and anomalous formation of some of the layers of the retina. This condition is not heritable.
Patients suspected to have retinoblastoma should undergo indirect ophthalmoscopy and fundus photography, and in some cases, ophthalmic ultrasound. In young children, these examinations are typically done under general anesthesia. Ultrasonography can be useful for this disease, as it demonstrates masses with high reflectivity that block sound, causing characteristic shadowing behind the tumor. False-positive results on ultrasonography are not uncommon, however. Needle biopsies are rarely, if ever, indicated in retinoblastoma, as puncturing the eye can lead to tumor seeding and orbital invasion. Aqueous taps, previously explored as a possible diagnostic tool, are inappropriate for the same reason.23
Computed tomography (CT) scans may no longer be appropriate for retinoblastoma patients, as a recent analysis has demonstrated an increased lifetime risk of other cancers in pediatric patients subjected to this imaging modality.24 Instead, magnetic resonance imaging (MRI) should be routinely performed. In addition to its excellent resolution in the diagnosis of extraocular soft-tissue disease, MRI can readily distinguish between retinoblastoma and Coats disease, as Coats appears brighter on T2-weighted images because of proteinaceous exudates. One disadvantage of MRI is that calcification, a key feature of retinoblastoma, is more readily demonstrated with CT than MRI.
The treatment of retinoblastoma can be divided into treatment of intraocular disease and treatment of extraocular disease. The following discussion focuses on the treatment of intraocular disease.
Exenteration refers to the surgical removal of the eye and lids, orbital portion of the optic nerve, and all orbital tissue including extraocular muscles, fat, nerves, and muscles. It is rarely used for retinoblastoma currently. Extensive orbital disease will not be cured by this technique and excellent local control in such cases can be obtained via external beam radiation and/or systemic chemotherapy. In cases of superimposed life-threatening orbital infection or bleeding, however, it may still be appropriate.
Enucleation is surgical removal of the eye without resecting the lids or extraocular muscles. Patients considered for enucleation include those with advanced retinoblastoma in one or both eyes, active tumor in a blind eye, and painful glaucoma from tumor invasion. More than 99% of patients with unilateral retinoblastoma without microscopic or macroscopic extraocular disease are cured with this procedure.25
In children, the procedure is done under general anesthesia, although the children do not require overnight hospitalization. A dilated funduscopic exam is performed on both eyes prior to surgery. Critical elements of the surgery include obtaining a long stump of optic nerve and avoiding any perforation of the globe.26 A silicone or plastic ball is inserted in place of the eye, and 3 weeks later a thin contact-like prosthesis is molded and painted by an ocularist to match the fellow eye.
Radiation therapy for retinoblastoma is prescribed to the entire retina and at least 1 cm of optic nerve while avoiding as much normal tissue as possible, including the lens and lacrimal gland. For children with bilateral disease, the conventional parallel opposing fields from left and right are designed with a “D” shape.27 The field is designed using a CT simulator or plain films taken in the conventional radiation therapy simulator unit. Information about globe size and lens position obtained from a head CT or MRI is also used in designing the field. The central ray of the beam is aimed at the posterior pole of the lens. The curved portion of the “D” is shaped to encompass the eye as described above, with a margin of approximately 1 cm in all directions. For children with bilateral disease in whom one eye was previously enucleated, a similar field arrangement involving a single field is used with the empty orbit receiving some exit dose of radiation. For patients with unilateral disease, a technique is employed that involves two lateral oblique photon fields with wedges and one lateral electron field to spare the contralateral globe. This technique is described in detail elsewhere.28
The dose prescribed to the retinal target volume ranges from 4,200 to 4,600 cGy with the lower doses reserved for children younger than age 6 months and the higher doses used for children with advanced disease. Children are treated 5 days per week with daily fractions of 180 cGy per day. Although occasionally sedation is necessary, a bivalved plaster of paris cast of the child's head and shoulders molded to a restraining board typically suffices for immobilization.
Following radiation, frequent follow-up visits for examinations under anesthesia are scheduled with the ophthalmologist to assess tumor regression. Regression patterns have been described elsewhere in detail.26
Of note, even after treatment some scarring and residual calcification are the rule rather than the exception in these patients' eyes. After 10 years, 90% of treated tumors still have visible intraocular material when viewed with the ophthalmoscope, ultrasound, or CT scan.
Photocoagulation is employed as primary treatment for selected small retinoblastomas. Patients are treated under general anesthesia with their pupils dilated. The light is aimed through the pupil at the tumor's feeding vessels. Within 1 week of treatment, the tumor begins to show involution and is permanently cured. More than 70% of tumors treated with this modality are cured after 1 to 2 treatment sessions.3 Xenon arc was used in the past but has largely been replaced by the diode laser (transpupillary thermotherapy).
Transpupillary thermotherapy (TTT) is a newer treatment modality for retinoblastoma that is delivered using modifications to the hardware and software of the infrared laser. TTT delivers hyperthermia to the tumor focus with precision by aiming through the pupil transsclerally via diopexy probe. Like photocoagulation, TTT is used to treat selected small retinoblastomas. A preliminary study of 58 patients demonstrated that 86% of tumors treated with TTT and chemotherapy demonstrated regression.29 Ophthalmic complications were significant, however, and included focal iris atrophy, focal paraxial lens opacity, and optic disc atrophy.
Cryotherapy is used as a primary treatment for small peripheral retinoblastomas, or as secondary treatment for recurrent tumors treated previously with external beam radiation. Patients are treated under general anesthesia. A blunt probe is placed on the outside of the sclera directly behind an intraocular focus of retinoblastoma. Rapid freezing (-90°C per minute) results in intracellular ice crystal formation, protein denaturation, pH changes, and finally cell membrane rupture. Ninety percent of tumors that are less than 3 mm in diameter are cured with this technique.30 Complications are rare.
Brachytherapy for retinoblastoma has been in use since the 1930s when 60Co plaques were first employed. Currently, 125Iodine plaques are used, allowing for custom-built plaques that account for the size of each individual lesion. The gold shields of the 125Iodine plaques are also advantageous to both patient and staff because excess radiation exposure is minimized. Ruthenium, palladium, and strontium plaques have also been used with success. After localization of the lesion in the operating room, the plaque is sutured on the outside of the sclera overlying the intraocular tumor. The prescribed dose is 4,000 to 4,500 cGy at a dose rate of approximately 1,000 cGy per day. The plaque is removed in a second operation. The tumor response most commonly seen is a type IV regression pattern.31 When plaques are used after failure of other conservative measures, success rates range from 50% to 92%.32,33
Plaque therapy is currently used increasingly as primary therapy because of increasing awareness of the tumorigenic effects of external beam radiation in children younger than age 1 year and the increasing use of systemic chemotherapy to reduce initial tumor volume. Plaque therapy is thus replacing external beam radiation treatment in appropriate patients.
Systemic chemotherapy for intraocular disease was introduced in the 1950s. Over the years, there have been waves of enthusiasm for different agents. It has been recognized that single or multiple agents cause dramatic reduction in the size of intraocular tumors but rare permanent responses. As a result of this, chemotherapy is presently being investigated to cause reduction of tumors, and when the tumors shrink, they are then treated with an additional modality, such as photocoagulation, hyperthermia, cryotherapy or radioactive plaques which appear to cause permanent inactivation of the tumor. This is an area of active clinical and research interest in the hope of replacing external beam radiation, and with the goal of saving eyes that might previously have been enucleated.
Most recent work has explored the use of carboplatin, vincristine, and an epipodophyllotoxin, either etoposide or teniposide.34–36 Investigators from Toronto, Canada, have added cyclosporine as a P-glycoprotein inhibitor and have suggested that they have better outcome, but that remains controversial.37 It is important to recognize several limitations. First, the use of chemotherapy as an alternative to radiation therapy is based largely on the hypothesis that the chemotherapy will have less long-term morbidity and mortality in retinoblastoma survivors. This cannot be known until many years of follow-up have been performed. However, it is important to recognize that alkylating agents have been demonstrated to increase the risk of secondary bone tumors in survivors of childhood cancer.38 Etoposide is well known to induce secondary leukemia and platinum-based chemotherapy has been demonstrated to increase the risk of secondary leukemia in survivors of ovarian cancer.39 Consequently, the use of chemotherapy may prove to have significant long-term toxicities that will have to be introduced into the risk:benefit ratios that will guide the decision whether to use chemotherapy or radiation therapy for these patients. Second, although it is clear that retinoblastomas are chemotherapy-sensitive tumors, which patients should receive chemotherapy and what the optimal regimens are remain to be defined.
Chemotherapy combined with external beam radiation has been employed in many centers worldwide as the preferred treatment for extraocular retinoblastoma. Precisely which patients deserve treatment is still controversial, however, and the optimal agents and regimens to use are not well-established (Table 85-5). In the most industrialized nations, extraocular disease occurs only in a small minority of patients. It is seen in both unilateral and bilateral cases, but presents sooner after diagnosis in patients with unilateral disease (2.7 to 3.2 months) than in patients with bilateral disease (11.4 to 11.7 months).40
Summary of Recent Reports of Chemotherapy for Extraocular Retinoblastoma a.
The literature regarding efficacy of various chemotherapeutic agents against retinoblastoma is relatively sparse. Alkylating agents are believed to be the most active agents.41 Nitrogen mustard and triethylenemelamine were frequently used in the past; cyclophosphamide and ifosfamide are currently thought to be active agents. Other reportedly active single agents include vincristine, doxorubicin, and cytarabine; a Phase II study recently determined idarubicin to be highly effective.42 Trials using single-agent carboplatin have not been reported, but our unpublished experience clearly shows this to be an active agent in the context of intraocular disease. Most reports of multiagent regimens have used cyclophosphamide and vincristine. Other agents used in multiple-drug regimens include cisplatin, carboplatin, and etoposide.
Extraocular disease can be divided according to the sites of involvement (see Table 85-3). The natural course of untreated retinoblastoma is one of progressive, localized, ocular involvement with eventual extension into the brain along the optic nerve or extension into the overlying conjunctiva with spread to regional nodes. This pattern is still common in less-industrialized countries, but treatment of the intraocular disease significantly alters this pattern. Because unilateral patients and bilateral patients before and after intraocular treatments have different patterns and timing of spread, a useful staging system for extraocular disease has not been developed; consequently, we have used a classification scheme as a guide for treatment (see Table 85-3). Retinoblastoma can spread via several routes. It may grow contiguously through the choroid and into the sclera (stage I) and then into the orbit (stage II), or may grow back through the optic nerve and then invade the brain (stage III). It may enter the subarachnoid space and then spread throughout the leptomeninges via the cerebrospinal fluid (stage IV). It may also spread hematogenously, causing metastatic disease in the bone marrow, bone, and organs such as the liver (stage V). Rarely, retinoblastoma may spread through the lymphatics to produce cervical node disease (stage IIb), but of note, the eye only has lymphatic drainage through the conjunctiva. Figure 85-5 shows an algorithm for the management of extraocular retinoblastoma.
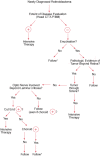
Algorithm for management of extraocular retinoblastoma.
When eyes containing retinoblastoma are enucleated, the extent of disease present in the pathologic specimen has been used to determine the need for chemotherapy.43,44 There are a number of such analyses in the literature and some lack of agreement regarding which criteria indicate a high risk of micrometastases and the need for presumptive treatment with chemotherapy. Two factors extensively discussed are whether optic nerve invasion is present and whether choroidal invasion is present. If optic nerve disease is present at the cut end (positive margin), then there is little controversy that central nervous system spread is likely and that treatment is indicated. More commonly, however, invasion of the nerve is noted with a surgical margin free of tumor. An important landmark used to define the extent of optic nerve invasion is the lamina cribrosa, which is the extension of sclera at the site of the optic nerve. Data suggest that 30% to 40% of patients with optic nerve invasion beyond the lamina cribrosa but with a negative surgical margin will later develop metastatic disease.43–45 Invasion of the choroid only, without associated optic nerve disease, is questionable as a prognostic factor for metastatic disease. It has been suggested that patients with both invasion of the optic nerve beyond the lamina cribrosa and with massive choroidal involvement are those that deserve to be treated with chemotherapy, and that this issue be investigated on a multiinstitutional basis.
Whether a 30% to 40% risk of metastasis justifies treating the 60% to 70% of patients who would not develop metastases is a difficult question. The toxicity, expense, and success of treating presumptively needs to be considered in conjunction with the efficacy of treatment designed to retrieve patients once overt metastatic disease is detected. If a significant proportion of patients with recurrent or metastatic disease can be salvaged with contemporary therapies, then it may be reasonable to closely follow patients with optic nerve invasion beyond the lamina cribrosa but with a negative surgical margin and to reserve aggressive chemotherapy for the minority whose disease recurs.
In the past, disease extending into the orbit was uniformly fatal despite aggressive surgery and radiation therapy. More recently, however, reports of the successful use of aggressive systemic chemotherapy in conjunction with radiation therapy have appeared in the literature.40,42,45–53 Doz and colleagues reported on 33 patients with histologically proven orbital disease treated between 1977 and 1991. Twenty patients had isolated orbital disease, whereas seven also had central nervous system (CNS) metastases and 6 had metastatic disease outside of the CNS. Most patients received both intensive chemotherapy and orbital radiation. Several chemotherapeutic regimens were used, and the agents included cyclophosphamide, platinum compounds, etoposide, doxorubicin, and vincristine. An overall survival of 34% was noted, with most recurrences occurring within the first year following diagnosis of the orbital disease. Patients without CNS disease fared better, and there was a trend toward improvement in the outcome of the more recently treated patients (post-1985). Although survival is clearly possible, orbital disease still carries a high risk of mortality, and aggressive multimodality therapy is warranted. It is also important to note that several groups have reported good outcomes for patients with local nodal disease extension, and it appears that these patients should not be analyzed together with patients with CNS or hematogenously spread distant metastases.
Metastatic disease to the central nervous system or to distant sites was likewise considered incurable until fairly recently. Grabowski and Abramson described 16 children with CNS or hematogenous metastases and showed that aggressive treatment with chemotherapy and radiation could cure a substantial proportion of these patients.40 Of the patients who received treatment at New York Hospital, all 5 with intracranial disease were disease-free survivors, as were 4 of 6 patients with bone and bone marrow disease. Treatment included systemic chemotherapy with cyclophosphamide, doxorubicin, and vincristine, intrathecal methotrexate, and cytarabine, and, in patients at least 1 year-old, whole-brain radiation (1,800 cGy) plus involved field boosts to a total of 4,500 to 5,500 cGy. In contrast, however, Schvartzman and colleagues50 had poor results using a similar approach in children with distant metastases and Doz and colleagues46 reported that all 7 of their patients with CNS involvement in addition to orbital disease died despite aggressive therapy.
Even if the promising results of Grabowski and Abramson40 are replicated, the young age of most of these patients makes the use of craniospinal radiation extremely toxic to normal development and severe developmental delay and intellectual impairment are frequent sequelae. This same problem has been addressed by pediatric oncologists for the treatment of young children with primary brain tumors, and a strategy of using high-dose chemotherapeutic agents that penetrate the blood-brain barrier has been employed to delay or avoid the use of radiation. We and others have used high-dose systemic chemotherapy in conjunction with autologous stem cell rescue for patients with metastatic retinoblastoma with promising results. Namouni and colleagues treated 25 patients with high-risk retinoblastoma with high-dose carboplatin, etoposide and cyclophosphamide, followed by autologous stem cell rescue.51 Five of 11 patients with metastatic disease not involving the central nervous system were event-free survivors. Our group treated 4 children with metastatic retinoblastoma with intensive multimodality therapy that included high-dose carboplatin, thiotepa, and etoposide chemotherapy with autologous stem cell rescue. All 4 survived and were event-free at 46 to 80 months.53 Patients with leptomeningeal metastases should be considered for trials of experimental intrathecal agents such as 131-I-3F8 (an anti-GD2 monoclonal antibody),54 but the role of conventional intrathecal therapy must be questioned because of in vitro data that suggest that retinoblastoma cell lines are invariably resistant to methotrexate, although some are sensitive to cytarabine.55
In 1949, it was first recognized that some retinoblastoma patients developed second nonocular neoplasms years after the successful treatment of the eye cancer.56 Since then, the incidence of additional nonocular cancers in survivors of retinoblastoma who carry the rb1 mutation has been reviewed extensively.57 Previous analyses have also shown that the nonocular cancers are the leading cause of death in survivors of germinal retinoblastoma in the United States. These patients are not at an increased risk of dying of any other causes (eg, pneumonia, heart disease) when compared to patients who have never had retinoblastoma.58
Nearly 95% of all children in the United States who develop retinoblastoma survive the disease. Of the survivors of germinal retinoblastoma, cumulative incidence reports of second cancers vary, but it is estimated that a rate of 1% per year of life approximates the rate.59 Patients who develop a second cancer and then survive that cancer have an increased risk for the development of additional nonocular tumors of approximately 2% per year from the time of second tumor diagnosis.57 The average latency period between subsequent tumor diagnoses becomes progressively shorter with each additional cancer that develops. Because females have a higher overall risk of dying of second tumors than males, more males are at risk for developing third tumors.57,58
Table 85-6outlines several risk factors that increase a patient's likelihood of developing a second nonocular cancer.60–64 A necessary risk factor for the development of additional nonocular tumors in retinoblastoma survivors is the presence of the germinal rb1 mutation. All patients with bilateral retinoblastoma and some patients with unilateral retinoblastoma are carriers of the germinal mutation and are at risk for additional nonocular cancers.57 Patients with unilateral disease who should be considered high risk for carrying the germinal mutation include patients with a family history of retinoblastoma (including a family history of retinoma), patients diagnosed younger than age 6 months, and patients who present with multifocal disease.
Risk Factors for Second Nonocular Cancer Development in Retinoblastoma Survivors.
External beam radiation is also a contributing factor to the development of additional cancers in retinoblastoma survivors. Children radiated during the first year of life are between 2 and 8 times as likely to develop second cancers as those radiated after the age of 1 year.62,63 The dose-response curve for the development of soft-tissue sarcomas in bilateral retinoblastoma patients has been described. The odds ratio at 0 to 4.9 Gy was 1.0, at 5 to 9.9 Gy was 1.91, at 10 to 29.9 Gy was 4.6, at 30 to 59.9 Gy was 6.4, and at greater than 60 Gy was 11.7.61 The radiation-induced risk for additional cancers applies only to patients with germinal retinoblastoma. Unilateral retinoblastoma patients who do not carry the constitutional mutation are at no increased risk for developing other cancers, even if they received external beam radiation.60
Additional nonocular cancers develop both within and outside the field of radiation. Our group has reported that in radiated patients who develop second malignancies, the tumors are within the radiation field two-thirds of the time and outside the field one-third of the time. In nonradiated patients who develop second tumors, the tumors are outside the hypothetical field two-thirds of the time and within the hypothetical field one-third of the time.61,62
Second nonocular neoplasms observed in survivors of germinal retinoblastoma include, in order of most common to least common, softtissue sarcomas; osteogenic sarcomas of the skull and long bones; pinealoblastomas; cutaneous melanomas; brain tumors; Hodgkin's disease; lung cancer; and breast cancer.61,65 Survivors of hereditary retinoblastoma are also at increased risk for the development of lipomas throughout the body (relative risk [RR] = 8.2). Patients who develop lipomas are at an even higher risk of developing second cancers.64
Pinealoblastoma, a tumor of the pineal gland, is often referred to as trilateral retinoblastoma because the pineal gland is considered the primitive third eye. This type of second nonocular cancer typically occurs in survivors of bilateral retinoblastoma who have a family history of the disease, have been diagnosed within the first 6 months of life, and have been treated with radiation. Virtually all such children have died from their pinealoblastoma with leptomeningeal spread despite a variety of attempts to cure the tumor.66 Of the children treated with external beam radiation at our center, half of those who died were diagnosed with a pinealoblastoma.67
Our group recently published a report on the largest series of retinoblastoma survivors who developed a second cancer, survived, and went on to develop third, fourth, or fifth nonocular tumors.57 Survivors of retinoblastoma who develop second malignancies and survive are at an even higher risk for the development of additional cancers than they were for the development of a second tumor. The sites and expected ages at which soft-tissue sarcomas of the head and osteogenic sarcomas of the long bones develop as third, fourth, and fifth cancers are similar to the patterns observed when they develop as second tumors.
Childhood acute lymphocytic leukemia (ALL) is the most common malignant tumor that involves the eyes of children. Leukemia primarily involves the uveal tract: the iris, ciliary body, and/or choroid. It can also involve the retina, optic nerve, and orbit.
Leukemic iris infiltrates can appear as creamy clusters of cells floating on the surface of the iris. When iris infiltrates are present, they can manifest as heterochromia (different color irides), cells in the anterior chamber (tumor hypopyon mimicking idiopathic iritis), or bleeding in the anterior chamber (hyphema). Hyphema can be associated with glaucoma and a painful, photophobic, red, sensitive eye. In contrast to iris involvement, leukemic infiltration is virtually impossible to detect ophthalmoscopically in the ciliary body or choroid. Tumor in the choroid is not seen because ALL diffusely invades and extravasates out of the choroidal blood vessels, causing only a subtle thickening of the choroid. Fortunately B-scan ultrasonogram examination by an ophthalmologist can detect the thickening.68 In fact, leukemic infiltration in the choroid has been identified in 90% of eyes at autopsy after death from leukemia. Retinal involvement is rare.
Leukemic infiltration of the eye generally presents in one of three ways. Most commonly, the infiltration presents simultaneously with the initial presentation of the disease. The majority of patients with ALL demonstrate ultrasonic ocular findings at presentation. When the leukemia is treated, the choroidal involvement usually disappears within days.
Leukemic infiltration of the eye can also present as an isolated site of relapse following induction treatment and CNS radiation prophylaxis. In these children, the CNS has been treated with radiation but the eye has been spared treatment, creating a sanctuary site. Treatment of the eye alone in such cases may be justified.
Finally, leukemic infiltration can present as a sign of CNS recurrence with or without evidence of a gross mass in the CNS. These patients frequently have leukemic cells near the posterior pole and in the vitreous. In these cases, the tumor cells enter the eye via the optic nerve, which has been seeded directly from the CNS.69 In these cases, some centers treat the brain whenever optic nerve recurrences are detected. Even in cases without apparent systemic or CNS recurrence, treatment to the brain should probably be considered. The traditional treatment for ocular recurrence of leukemia has been 800 to 1,800 cGy of radiation, but we have found that treating the eye via a CNS route (ventricular methotrexate through an Ommaya reservoir) effectively eliminates ocular recurrence.
By agreement with the publisher, this book is accessible by the search feature, but cannot be browsed.
Your browsing activity is empty.
Activity recording is turned off.
See more...