All of the proteins that pass through the Golgi apparatus, except those that are retained there as permanent residents, are sorted in the trans Golgi network according to their final destination. The mechanism of sorting is especially well understood for those proteins destined for the lumen of lysosomes, and in this section we consider this selective transport process. We begin with a brief account of lysosome structure and function.
Lysosomes Are the Principal Sites of Intracellular Digestion
Lysosomes are membrane-enclosed compartments filled with hydrolytic enzymes that are used for the controlled intracellular digestion of macromolecules. They contain about 40 types of hydrolytic enzymes, including proteases, nucleases, glycosidases, lipases, phospholipases, phosphatases, and sulfatases. All are acid hydrolases. For optimal activity they require an acid environment, and the lysosome provides this by maintaining a pH of about 5.0 in its interior. In this way, the contents of the cytosol are doubly protected against attack by the cell's own digestive system. The membrane of the lysosome normally keeps the digestive enzymes out of the cytosol, but even if they should leak out, they can do little damage at the cytosolic pH of about 7.2.
Like all other intracellular organelles, the lysosome not only contains a unique collection of enzymes, but also has a unique surrounding membrane. Transport proteins in this membrane allow the final products of the digestion of macromolecules—such as amino acids, sugars, and nucleotides—to be transported to the cytosol, from where they can be either excreted or reutilized by the cell. An H+ pump in the lysosomal membrane uses the energy of ATP hydrolysis to pump H+ into the lysosome, thereby maintaining the lumen at its acidic pH (). A similar or identical vacuolar H
+
ATPase is thought to acidify all endocytic and exocytic organelles, including lysosomes, endosomes, selected compartments of the Golgi apparatus, and many transport and secretory vesicles. Most of the lysosomal membrane proteins are unusually highly glycosylated, which helps to protect them from the lysosomal proteases in the lumen.
Lysosomes. The acid hydrolases are hydrolytic enzymes that are active under acidic conditions. The lumen is maintained at an acidic pH by an H+ ATPase in the membrane that pumps H+ into the lysosome.
Lysosomes Are Heterogeneous
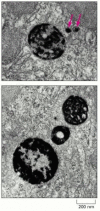
Lysosomes were initially discovered by the biochemical fractionation of cell extracts; only later were they seen clearly in the electron microscope. Although extraordinarily diverse in shape and size, they can be identified as members of a single family of organelles by staining them with specific antibodies. They can also be identified by histochemistry, using the precipitate produced by the action of an acid hydrolase on its substrate to indicate which organelles contain the hydrolase (). By this criterion, lysosomes are found in all eucaryotic cells.
Histochemical visualization of lysosomes. These electron micrographs show two sections of a cell stained to reveal the location of acid phosphatase, a marker enzyme for lysosomes. The larger membrane-enclosed organelles, containing dense precipitates (more...)
The heterogeneity of lysosomal morphology contrasts with the relatively uniform structures of most other cell organelles. The diversity reflects the wide variety of digestive functions mediated by acid hydrolases, including the breakdown of intra- and extracellular debris, the destruction of phagocytosed microorganisms, and the production of nutrients for the cell. For this reason, lysosomes are sometimes viewed as a heterogeneous collection of distinct organelles whose common feature is a high content of hydrolytic enzymes. It is especially hard to apply a narrower definition than this in plant cells, as we see next.
Plant and Fungal Vacuoles Are Remarkably Versatile Lysosomes
Most plant and fungal cells (including yeasts) contain one or several very large, fluid-filled vesicles called vacuoles. They typically occupy more than 30% of the cell volume, and as much as 90% in some cell types (). Vacuoles are related to the lysosomes of animal cells, containing a variety of hydrolytic enzymes, but their functions are remarkably diverse. The plant vacuole can act as a storage organelle for both nutrients and waste products, as a degradative compartment, as an economical way of increasing cell size (), and as a controller of turgor pressure (the osmotic pressure that pushes outward on the cell wall and keeps the plant from wilting). Different vacuoles with distinct functions (e.g., digestion and storage) are often present in the same cell.
The plant cell vacuole. This electron micrograph of cells in a young tobacco leaf shows the cytosol as a thin layer, containing chloroplasts, pressed against the cell wall by the enormous vacuole. The membrane of the vacuole is called the tonoplast. (Courtesy (more...)
The role of the vacuole in controlling the size of plant cells. A large increase in cell volume can be achieved without increasing the volume of the cytosol. Localized weakening of the cell wall orients a turgor-driven cell enlargement that accompanies (more...)
The vacuole is important as a homeostatic device, enabling plant cells to withstand wide variations in their environment. When the pH in the environment drops, for example, the flux of H+ into the cytosol is balanced, at least in part, by an increased transport of H+ into the vacuole to keep the pH in the cytosol constant. Similarly, many plant cells maintain an almost constant turgor pressure in the face of large changes in the tonicity of the fluid in their immediate environment. They do so by changing the osmotic pressure of the cytosol and vacuole—in part by the controlled breakdown and resynthesis of polymers, such as polyphosphate, in the vacuole, and in part by altering the transport rates of sugars, amino acids, and other metabolites across the plasma membrane and the vacuolar membrane. The turgor pressure controls these fluxes by regulating the activities of the distinct sets of transporters in each membrane.
Substances stored in plant vacuoles are often harvested for human use: in different species, these range from rubber to opium to the flavoring of garlic. Many stored products have a metabolic function. Proteins, for example, can be preserved for years in the vacuoles of the storage cells of many seeds, such as those of peas and beans. When the seeds germinate, these proteins are hydrolyzed and the resulting amino acids provide a food supply for the developing embryo. Anthocyanin pigments stored in vacuoles color the petals of many flowers so as to attract pollinating insects, while noxious molecules released from vacuoles when a plant is eaten or damaged provide a defense against predators.
Multiple Pathways Deliver Materials to Lysosomes
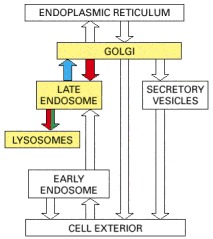
Lysosomes are usually meeting-places where several streams of intracellular traffic converge. Digestive enzymes are delivered to them by a route that leads outward from the ER via the Golgi apparatus, while substances to be digested are fed in by at least three paths, depending on their source.
The best studied of these paths to degradation in lysosomes is that followed by the macromolecules taken up from extracellular fluid by endocytosis. As discussed in detail later, endocytosed molecules are initially delivered in vesicles to small, irregularly shaped intracellular organelles called early endosomes. Some of these ingested molecules are selectively retrieved and recycled to the plasma membrane, while others pass on into late endosomes. It is here that endocytosed materials first meet the lysosomal hydrolases, which are delivered to the endosome from the Golgi apparatus. The interior of the late endosomes is mildly acidic (pH ~6), and it is the site where the hydrolytic digestion of the endocytosed molecules begins. Mature lysosomes form from the late endosomes, accompanied by a further decrease in internal pH. Lysosomes are thought to be produced by a gradual maturation process, during which endosomal membrane proteins are selectively retrieved from the developing lysosome by transport vesicles that deliver these proteins back to endosomes or the trans Golgi network.
A second pathway to degradation in lysosomes is used in all cell types for the disposal of obsolete parts of the cell itself—a process called autophagy. In a liver cell, for example, an average mitochondrion has a lifetime of about 10 days, and electron microscopic images of normal cells reveal lysosomes containing (and presumably digesting) mitochondria, as well as other organelles. The process seems to begin with the enclosure of an organelle by membranes of unknown origin, creating an autophagosome, which then fuses with a lysosome (or a late endosome). The process is highly regulated, and selected cell components can somehow be marked for lysosomal destruction during cell remodeling. The smooth ER that proliferates in a liver cell in response to the drug phenobarbital (discussed in Chapter 12), for example, is selectively removed by autophagy after the drug is withdrawn.
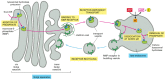
As we discuss later, the third pathway that brings materials to lysosomes for degradation is found mainly in cells specialized for the phagocytosis of large particles and microorganisms. Such professional phagocytes (macrophages and neutrophils in vertebrates) engulf objects to form a phagosome, which is then converted to a lysosome in the manner described for the autophagosome. The three pathways are summarized in .
Three pathways to degradation in lysosomes. (A) Each pathway leads to the intracellular digestion of materials derived from a different source. (B) An electron micrograph of an autophagosome containing a mitochondrion and a peroxisome. (B, courtesy of (more...)
A Mannose 6-Phosphate Receptor Recognizes Lysosomal Proteins in the Trans Golgi Network
We now consider the pathway that delivers lysosomal hydrolases and membrane proteins to lysosomes. Both classes of proteins are synthesized in the rough ER and transported through the Golgi apparatus to the trans Golgi network. The transport vesicles that deliver these proteins to late endosomes (which later form lysosomes) bud from the trans Golgi network. The vesicles incorporate the lysosomal proteins and exclude the many other proteins being packaged into different transport vesicles for delivery elsewhere.
How are lysosomal proteins recognized and selected in the trans Golgi network with the required accuracy? For the lysosomal hydrolases the answer is known. They carry a unique marker in the form of mannose 6-phosphate (M6P) groups, which are added exclusively to the N-linked oligosaccharides of these soluble lysosomal enzymes as they pass through the lumen of the cis Golgi network (). The M6P groups are recognized by transmembrane M6P receptor proteins, which are present in the trans Golgi network. The receptor proteins bind to lysosomal hydrolases on the lumenal side of the membrane and to adaptins in assembling clathrin coats on the cytosolic side. In this way, they help package the hydrolases into clathrin-coated vesicles that bud from the trans Golgi network. The vesicles subsequently deliver their contents to a late endosome.
The structure of mannose 6-phosphate on a lysosomal enzyme.
The M6P Receptor Shuttles Between Specific Membranes
The M6P receptor protein binds its specific oligosaccharide at pH 6.5–6.7 in the trans Golgi network and releases it at pH 6, which is the pH in the interior of late endosomes. Thus, in the late endosomes, the lysosomal hydrolases dissociate from the M6P receptor. As the pH drops further during endosomal maturation, the hydrolases begin to digest the endocytosed material delivered from early endosomes. Having released their bound enzymes, the M6P receptors are retrieved into transport vesicles that bud from late endosomes; the receptors are then returned to the membrane of the trans Golgi network for reuse (). Transport in either direction requires signal peptides in the cytoplasmic tail of the M6P receptor that specify transport of this protein to the late endosome or back to the Golgi apparatus. Thus, the recycling of the M6P receptor closely resembles the recycling of the KDEL receptor, discussed earlier.
The transport of newly synthesized lysosomal hydrolases to lysosomes. The precursors of lysosomal hydrolases are covalently modified by the addition of mannose 6-phosphate (M6P) groups in the cis Golgi network. They then become segregated from all other (more...)
Not all of the hydrolase molecules that are tagged with M6P for delivery to lysosomes get to their proper destination. Some escape the normal packaging process in the trans Golgi network and are transported “by default” to the cell surface, where they are secreted into the extracellular fluid. Some M6P receptors, however, also take a detour to the plasma membrane, where they recapture the escaped lysosomal hydrolases and return them by receptor-mediated endocytosis to lysosomes via early and late endosomes. As lysosomal hydrolases require an acidic milieu to work, they can do little harm in the extracellular fluids, which usually has a neutral pH.
A Signal Patch in the Hydrolyase Polypeptide Chain Provides the Cue for M6P Addition
The sorting system that segregates lysosomal hydrolases and dispatches them to late endosomes works because M6P groups are added only to the appropriate glycoproteins in the Golgi apparatus. This requires specific recognition of the hydrolases by the Golgi enzymes responsible for adding M6P. Since all glycoproteins leave the ER with identical N-linked oligosaccharide chains, the signal for adding the M6P units to oligosaccharides must reside somewhere in the polypeptide chain of each hydrolase. Genetic engineering experiments have revealed that the recognition signal is a cluster of neighboring amino acids on each protein's surface, known as a signal patch.
Two enzymes act sequentially to catalyze the addition of M6P groups to lysosomal hydrolases. The first is a GlcNAc phosphotransferase that specifically binds the hydrolase and adds GlcNAc-phosphate to one or two of the mannose residues on each oligosaccharide chain (). A second enzyme then cleaves off the GlcNAc residue, leaving behind a newly created M6P marker. Since most lysosomal hydrolases contain multiple oligosaccharides, they acquire many M6P residues, providing a high affinity signal for the M6P receptor.
The recognition of a lysosomal hydrolase. The GlcNAc phosphotransferase enzyme that recognizes lysosomal hydrolases in the Golgi apparatus has separate catalytic and recognition sites. The catalytic site binds both high-mannose N-linked oligosaccharides (more...)
Defects in the GlcNAc Phosphotransferase Cause a Lysosomal Storage Disease in Humans
Lysosomal storage diseases are caused by genetic defects that affect one or more of the lysosomal hydrolases. The defect results in the accumulation of the undigested substrates in lysosomes, with severe pathological consequences, most often in the nervous system. In most cases there is a mutation in a structural gene that codes for an individual lysosomal hydrolase. This occurs in Hurler's disease, for example, in which the enzyme required for the breakdown of glycosaminoglycans is defective or missing. The most severe form of lysosomal storage disease, however, is a very rare disorder called inclusion-cell disease (I-cell disease). In this disease almost all of the hydrolytic enzymes are missing from the lysosomes of fibroblasts, and their undigested substrates accumulate in lysosomes, which consequently form large “inclusions” in the patients' cells.
I-cell disease is due to a single gene defect and, like most genetic enzyme deficiencies, it is recessive—that is, it is seen only in individuals in whom both copies of the gene are defective. In I-cell disease patients, all the hydrolases missing from lysosomes are found in the blood. Because they fail to be sorted properly in the Golgi apparatus, the hydrolases are secreted rather than transported to lysosomes. The missorting has been traced to a defective or missing GlcNAc-phosphotransferase. Because lysosomal enzymes are not phosphorylated in the cis Golgi network, they are not segregated by M6P receptors into the appropriate transport vesicles in the trans Golgi network. Instead, they are carried to the cell surface and secreted by a default pathway.
In I-cell disease the lysosomes in some cell types, such as hepatocytes, contain a normal complement of lysosomal enzymes, implying that there is another pathway for directing hydrolases to lysosomes that is used by some cell types but not others. The nature of this M6P-independent pathway is unknown. Similarly, the membrane proteins of lysosomes are sorted from the trans Golgi network to late endosomes by an M6P-independent pathway in all cells, and they are therefore normal in I-cell disease. These membrane proteins exit from the trans Golgi network in clathrin-coated vesicles distinct from those that transport the M6P-tagged hydrolases.
It is unclear why cells need more than one sorting pathway to construct a lysosome, although it is perhaps not surprising that different mechanisms operate for soluble and membrane-bound lysosomal proteins, especially since—unlike the M6P receptor—those membrane proteins are lysosomal residents and hence need not be returned to the trans Golgi network.
Some Lysosomes May Undergo Exocytosis
Targeting of material to lysosomes is not necessarily the end of the pathway. Lysosomal secretion (also called defecation) of their undigested content enables all cells to eliminate indigestible debris. For most cells, this seems to be a minor pathway, used only when cells are stressed. Some cell types, however, contain specialized lysosomes that have acquired the necessary machinery for fusion with the plasma membrane. Melanocytes in the skin, for example, produce and store pigments in their lysosomes. These pigment-containing melanosomes release their pigment into the extracellular space by exocytosis. The pigment is then taken up by keratinocytes, leading to normal skin pigmentation. In some genetic disorders, this transfer process is blocked owing to defects in melanosome exocytosis, leading to forms of hypopigmentation (albinism).
Summary
Lysosomes are specialized for the intracellular digestion of macromolecules. They contain unique membrane proteins and a wide variety of hydrolytic enzymes that operate best at pH 5, which is the internal pH of lysosomes. This low pH is maintained by an ATP-driven H+ pump in the lysosomal membrane. Newly synthesized lysosomal proteins are transferred into the lumen of the ER, transported through the Golgi apparatus, and then carried from the trans Golgi network to late endosomes by means of clathrin-coated transport vesicles.
The lysosomal hydrolases contain N-linked oligosaccharides that are covalently modified in a unique way in the cis Golgi network so that their mannose residues are phosphorylated. These mannose 6-phosphate (M6P) groups are recognized by an M6P receptor protein in the trans Golgi network that segregates the hydrolases and helps package them into budding transport vesicles that deliver their contents to late endosomes (the organelle that matures into lysosomes). The M6P receptors shuttle back and forth between the trans Golgi network and these endosomes. The low pH in the late endosome dissociates the lysosomal hydrolases from these receptors, making the transport of the hydrolases unidirectional. A separate transport system uses clathrin-coated vesicles to deliver resident lysosomal membrane proteins from the trans Golgi network.